Paper Menu >>
Journal Menu >>
 Advances in Ma terials Physics and Che mist ry, 2012, 2, 212-215 doi:10.4236/ampc.2012.24B054 Published Online December 2012 (htt p://www.SciRP.org/journal/ampc) Copyright © 2012 SciRes. AMPC Yttria Promoted Nickel Nanowire Catalyst for the Partial Oxidation of Methane to Synthesis Gas Xuebin Hong, Bingbing Li, Cong Zhang Rena i College of Tianjin University, Tianjin 301636, P. R. of China Email: hong_xuebin@yahoo.com.cn Received 2012 ABSTRACT A yttria promoted nickel nanowire catalyst was prepared by a hard templating method, and characterized by transmission electron microscopy (TEM) and N2 physical adsorption. The catalytic properties of the yttria promoted nanowire catalyst in the partial oxida- tion of methane to syngas were compared with a metallic Ni catalyst which was prepared with nickel sponge. The characterization result s showed th at the yttria promoted nickel nano wire catalyst had high specific su rface area and th ere was more NiO ph ase in the nickel nanowire cat alyst than in th e metall ic Ni catalyst. The r eaction results sho wed that the yttri a promot ed nickel nanowire catalyst had high CH4 conversion and selectivities to H2 and CO. Keywords: Yttria; Nanowire; Methane; Partial Oxidation; Syn gas 1. Introduction The conversion of natural gas into liquid fuels is commonly performed via an indirect route through synthesis gas, a mixture of H2 and CO [1-3]. Industrially, synthesis gas is mainly pro- duced from methane steam reforming process [4-6]. Such a process produces a high H2/CO ratio [7] . Furthermor e, methan e steam reforming is highly endothermic and heat-transfer limited [8]. Catalytic partial oxidation of methane (CPOM) is an attrac- tive alternative for the syngas production [9-12] as t he reaction is mildly exothermic and a H2/CO ratio of 2 can be achieved, which is desirable for Fischer-Tropsch synthesis [13-15], me- thanol synthesis, etc. The first row of transition metals (Ni, Co) and precious met- als (Ru, Rh, Pd, Pt, and Ir) have been reported as active cata- lysts for CPOM [16,17]. Among these, Ni has been intensively studied. Recently, the synthesis of nickel oxide with controlled nanostructures, such as mesoporous solids, nanotubes or nanowires, has attracted considerable attention because such material may exhibit better catalytic properties and be more read ily avail able [18-23]. Kim et al [24] synthesized a mesoporous Ni-Alumina catalyst and compared the performance with a nickel catalyst impregnated on a commercially available alumina support (Ni-IMP) in CPOM. They found that the Ni-Alumina catalyst showed a relatively high surface area with a narrow pore size distribution. And the Ni-Alumina catalyst h aving s mall er n ickel particles and lower levels of carbon deposition had a more sta- ble catalytic activity than the Ni-IMP catal yst. The reaction of CPOM over a metallic Ni catalyst prepared with nickel sponge has been studied [25]. The results showed that the metallic Ni catalyst has s ome adv antages over th e sup- ported nickel or nickel coated catalysts. For example, in the supported nickel or nickel coated catalysts, the fine nickel par- ticles ten d to aggregat e at high t emperatures and lo se the activ- ity [26,27]. However, in the metallic Ni catalyst, the nickel acts as both active component and the support, so it would not ag- gregate furt her [25]. In this work, we prepared a yttria promoted nickel nanowire catalyst by a hard templating method. The catalyst consists of nickel nanowires, which has higher specific surface area than the one prepared with nickel sponge. It is expected that the yttria promoted nickel nanowire catalyst should have higher activit y for CP OM, while keepi ng the advantages o f the metal- lic ni ckel catalyst. 2. Experimental 2.1. Catalyst Preparation Thr e e-dimensional mesoporous silica (KIT-6) was synthesized according to references [28-31], and used as the hard template for the preparation of yttria promoted nickel nanowires. For the preparation of the yttria promoted nickel nanowires, 1.5 g of Ni(NO3)2⋅6H2O (98.0%) and 1.0 g Y(NO3)3 were dissolved in 1.0 cm3 distilled water forming a saturated solution, followed by addition of 2.0 g of KIT-6, which resulted an incipient im- pregnation. After drying at 373 K until all the water had been vaporized and a dry powder obtained, the sample was heated slowly to 823 K in air and calcined in a muffle furnace at that temperature for 5 h. Then, in the presence of hydrogen, the sample was heated at 1 K/min from ambient temperature to 1123 K, kept at the final temperature for 2 h, and then cooled down to ambient temperature. The above process was repeated for four times. Then, the resulting sample was twice treated with a hot 4.0 mol/L NaOH solution to remove the silica tem- plate, followed by washing with distilled water and ethanol several t imes, and then drying at room temperature. The sample was triturated into 40-60 mesh. The preparation of the metallic Ni catalyst has been de- scribed before [25]. Briefly, a piece of metallic Ni sponge (80 % porosity, Changsha Liyuan Material Co., Ltd.) was cut into 40-60 mesh, treated with a mixture of 500 cm3 containing 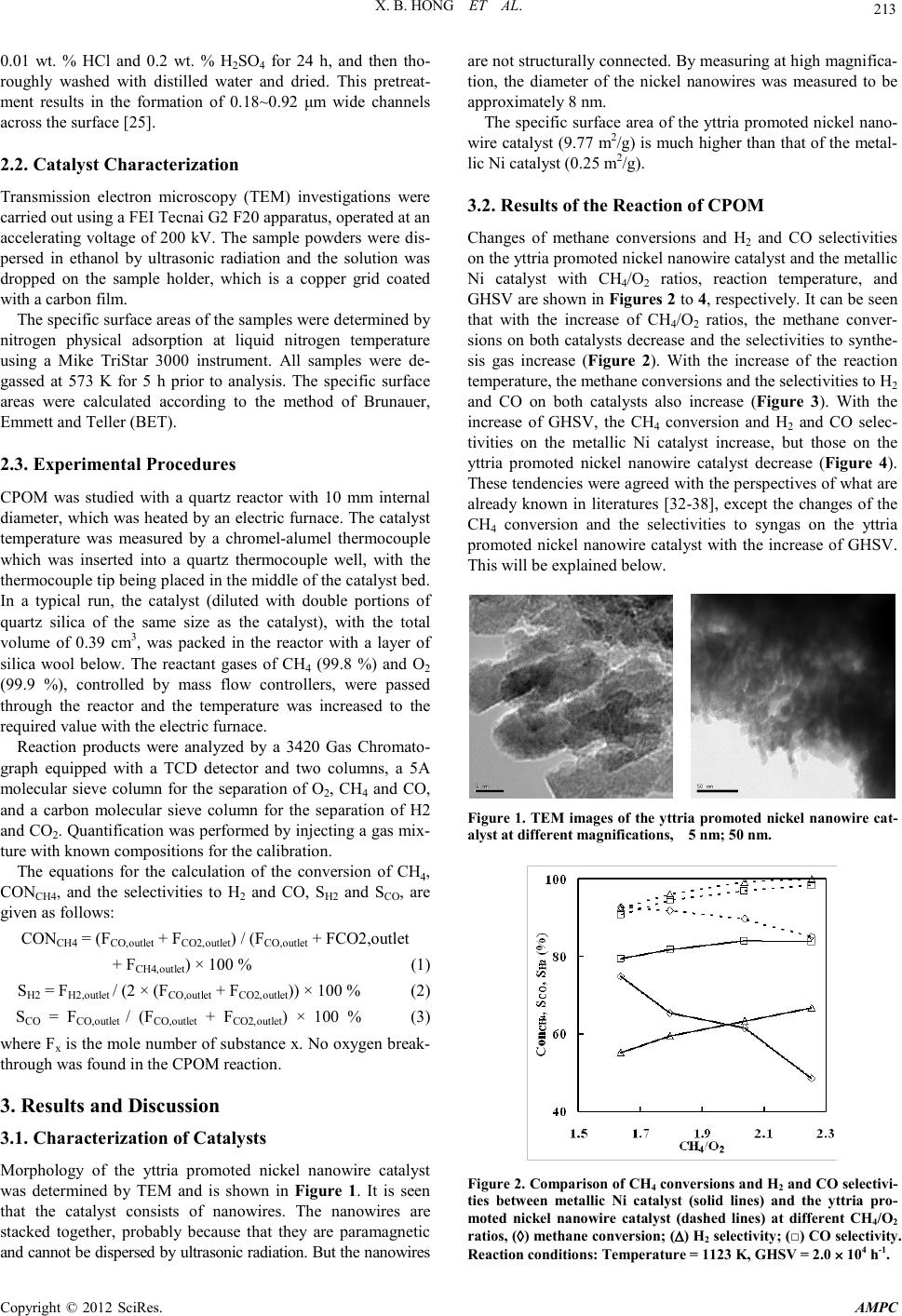 X. B. HONG ET AL. Copyright © 2012 SciRes. AMPC 213 0.01 wt. % HCl and 0.2 wt. % H2SO4 for 24 h, and then tho- roughly washed with distilled water and dried. This pretreat- ment results in the formation of 0.18~0.92 μm wide channels across the surface [25]. 2.2. Catalyst Characterization Transmission electron microscopy (TEM) investigations were carried out using a FEI Tecnai G2 F20 apparatus, operated at an accelerating voltage of 200 kV. The sample powders were dis- persed in ethanol by ultrasonic radiation and the solution was dropped on the sample holder, which is a copper grid coated with a carbon film. The specific su rface ar eas o f th e sampl es were d etermin ed b y nitrogen physical adsorption at liquid nitrogen temperature using a Mike TriStar 3000 instrument. All samples were de- gassed at 573 K for 5 h prior to analysis. The specific surface areas were calculated according to the method of Brunauer, Emmett and Teller (BET). 2.3. Experimental Procedures CPOM was studied with a quartz reactor with 10 mm internal diameter, which was heated b y an electric furn ace. The catalyst temperature was measured by a chromel-alumel thermocouple which was inserted into a quartz thermocouple well, with the thermocouple tip being placed in the middle of the catalyst bed. In a typical run, the catalyst (diluted with double portions of quartz silica of the same size as the catalyst), with the total volume of 0.39 cm3, was packed in the reactor with a layer of silica wool below. The reactant gases of CH4 (99.8 %) and O2 (99.9 %), controlled by mass flow controllers, were passed through the reactor and the temperature was increased to the required value with the electric furn ace. Reaction products were analyzed by a 3420 Gas Chromato- graph equipped with a TCD detector and two columns, a 5A molecular sieve column for the separation of O2, CH4 and CO, and a carbon molecular sieve column for the separation of H2 and CO2. Quan tification was perf ormed by injecti ng a gas mix- ture with known compositions for the calibration. The equations for the calculation of the conversion of CH4, CONCH4, and the selectivities to H2 and CO, SH2 and SCO, are given as follows: CONCH4 = (FC O ,outlet + FC O 2,outle t) / (FCO ,o u t let + FCO2 ,o ut let + FC H 4,ou tle t) × 100 % (1) SH2 = FH2,outlet / (2 × ( FC O , ou tle t + FCO2,outlet)) × 100 % (2) SCO = FCO,outlet / (FC O , ou tle t + FCO 2,o u t le t) × 100 % (3) where Fx is the mole number of substance x. No oxygen break- through was found in the CPOM reaction. 3. Results and Discussion 3.1. Characterization of Catalysts Morphology of the yttria promoted nickel nanowire catalyst was determined by TEM and is shown in Figure 1. It is seen that the catalyst consists of nanowires. The nanowires are stacked together, probably because that they are paramagnetic and cannot be dispersed by ultrasonic radiation. But the nanowires are not stru ctu ral ly conn ected . B y measuri ng at h igh magnifica- tion, the diameter of the nickel nanowires was measured to be approximately 8 nm. The specifi c surface area of th e yttria promoted nickel n ano- wire catal yst (9.7 7 m2/g) is much h igher than that of the metal- lic Ni catalyst (0.25 m2/g). 3.2. Results of the Reaction of CPOM Changes of methane conversions and H2 and CO selectivities on the yttri a pr omoted n ickel n an owire cat al yst an d t he metalli c Ni catalyst with CH4/O2 ratios, reaction temperature, and GHSV are shown in Figures 2 to 4, resp ectivel y. It can be seen that with the increase of CH4/O2 ratios, the methane conver- sions on both catalysts decrease and the selectivities to synthe- sis gas increase (Figure 2). With the increase of the reaction temperature, the methane conversions and the selectivities to H2 and CO on both catalysts also increase (Figure 3). With the increase of GHSV, the CH4 conversion and H2 and CO selec- tivities on the metallic Ni catalyst increase, but those on the yttria promoted nickel nanowire catalyst decrease (Figure 4). These tend encies were agreed with the persp ectives of what are already known in literatures [32-38], except the changes of the CH4 conversion and the selectivities to syngas on the yttria promoted nickel nanowire catalyst with the increase of GHSV. This will be explained below. Figure 1. TEM images of the yttria promoted nickel nanowire cat- alyst at different magnifications, 5 nm; 50 nm. Figure 2. Comparison of CH4 conversions and H2 and CO selecti vi- ties between metallic Ni catalyst (solid lines) and the yttria pro- moted nickel nanowire catalyst (dashed lines) at different CH4/O2 ratios, (◊) methane co nversion; (∆) H2 selectivity; (□) CO selectivity. Reaction conditions: Temperature = 1123 K, GHSV = 2.0 × 104 h-1.  X. B. HONG ET AL. Copyright © 2012 SciRes. AMPC 214 Figure 3. Compariso n o f C H4 conversions a nd H2 and CO sel ectivi- ties between metallic Ni catalyst (solid lines) and the yttria pro- moted nickel nanowire catalyst (dashed lines) at different reaction temperatures, (◊) methane conversion; (∆) H2 selectivity; (□) CO selectivity. Reaction conditions: CH4/O2 = 2.0, GHS V = 2.0 × 104 h-1. Figure 4. Comparison of CH4 conversions and H2 and CO selecti vi- ties between metallic Ni catalyst (solid lines) and the yttria pro- moted nickel nanow ir e catalyst (dashed lines) at different GHSV , (◊) methane conversio n; (∆) H2 selectivity; (□) CO selectivity. Reaction condit ions : Temperatu r e = 1 123 K , C H4/O2 = 2.0. However, it is noted that on the yttria promoted nickel nano- wire catal yst, th e methan e con ver sio n and the s electi viti es to H2 and CO are much higher than those on the metallic Ni catal yst under the same reaction conditions. For example, as shown in Figure 2, on the yttria promoted nickel nanowire catalyst, at reaction temperature 1123 K, GHSV 2.0 × 104 h-1, and CH4/O2 ratio 2.0, the conversion of methane and the selectivities to hydrogen and carbon monoxide are 90 %, 99 %, and 97 %, respecti vely, much h igher t han those on t he metallic Ni catal yst, which are 58 %, 62 %, and 82 %, respectively. The value of the conversion on the yttria promoted nickel nanowire catalyst is a little lower than the thermodynamic equilibrium value, which is 95 %, but the values of the selectivities to syngas ar e near to the thermodynamic equilibrium values, which are 98 % and 98 %, respecti vely. In general , i t is kno wn that defect s, su ch as oxygen vacan cies, are important in the surface chemistry and catalysis of metal oxides [39]. And the improved catalytic performance in oxida- tion catalysis has been attributed to a high concentration of oxygen vacan cies [ 40-43]. Lattice oxygen ions often involve in reactions over oxide catalysts. Most of the partial oxidation reactions proceed via the Mars-van Krevelen mechanism, which is a redox model [44-47]. In this model, hydrocarbons react with surface lattice oxygen ions to form oxidized products, leaving a series of oxygen vacancies which are pending to be recruited by new formed lattice oxygen ions. The cycle for catalytic partial oxidation is closed via replenishment of the extracted lattice oxygen ions through the dissociative adsorp- tion of molecular oxygen on the surface [48]. In the present work, the yttria promoted nickel nanowire cat- alyst had higher activity and select ivity. We infer that the r eac- tion might proceed through the Mars-van Kre velen mechanism. The yttria promoted nickel nanowire catalyst had higher spe- cific sur face area, which sho ws promotio n effect on the acti vity of catalyst, because the activity of catalyst was directly related to its surface area [49]. Higher surface area results in higher activity. Therefore, methane conversion and the selectivities to syngas on the yttria promoted nickel nanowire catalyst were much higher than those on the metallic Ni catalyst under the same reaction conditions. From the reaction results (Figure 4), it is seen that the con- version and the selectivities on the yttria promoted nickel na- nowire catalyst decreased with the increase of GHSV, while those on the metallic Ni catalyst increased. The difference in convecti ve heat transfer co efficients fo r the two catalysts might be the most important reason to explain the differences in cata- lytic results. When heat was removed from the surface faster than it was generated by reaction, the temperature would fall. When the temperature fell below the ignition temperature of methane oxidation, reaction no longer occurred on that portion of the catalyst. This behavior was known as blowout. Blowout would occur easi er on a catalyst geometry that had a high con- vective heat transfer coefficient [50]. Convective heat transfer occurs axially in the direction of flow, acting to transfer heat from the su rface to the coo ler gases. The convective h eat trans- fer was much more efficient at removing heat from the yttria promoted nickel nanowire catalyst because of the much higher surface area an d the tortuo us flow passages in t he catalyst [51]. With th e increase of GHSV , the reactants increased in th e feed which could result in the reaction blowout on the yttria pro- moted nickel nanowire catalyst. This led to the decrease in methane co nversion and th e selectivities t o syngas on the yttria promot ed nickel nanowire catalyst with the increase of GHSV. 4. Conclusions The yttria promoted nickel nanowire catalyst has higher BET surface area than the metallic Ni catalyst. There is more NiO phase i n the yttria p romoted nickel nanowire catal yst than in the metallic Ni catalyst, whi ch brin gs on more active cen ters in th e CPOM. The yttria promoted nickel nanowire catalyst had higher CH4 conversion and H2 and CO selectivities than the metallic Ni during CPOM. With the increase of CH4/O2 ratios, the methan e  X. B. HONG ET AL. Copyright © 2012 SciRes. AMPC 215 conversions on both catalysts decrease and the selectivities to syngas increase. With the increase of the reaction temperature, the methane conversions and the selectivities to H2 and CO on both catalysts increase. With the increase of GHSV, the me- thane con versio n and H2 and CO selectivities on the metallic Ni catalyst increase, but those on the yttria promoted nickel nano- wire catalyst decre ase. REFERENCES [1] R.C. Ramaswamy, P.A. Ramachandran, M.P. Dudukovic, Ind. Eng. Chem. Res . 46, 863 8 ( 2007). [2] C.J. Bell, C.A. Leclerc, Energy Fuels 2 1, 3548 (2007). [3] M . Fleys , W.J . Sh an , Y. S im on, P .M. M arq ua i re, Ind . Eng. Chem . Res. 46, 1063 (2007). [4] C.W. Hu, J.J. Wu, H.L. Zhang, S. Qin, AIChE J. 53, 2925 (2007). [5] T.V. Choudhary, V.R. Choudhary, Angew. Chem. Int. Ed. 47, 2 (2008). [6] K.A. Williams, R. Horn, L.D. Schmidt, AIChE J. 53, 2097 (2007). [7] J.H. Ryu, K.Y. Lee, H.J. Kim, J.I. Yang, H. Jung, Appl. Catal., B: Environ. 80, 306 (2008). [8] U. Balachandran, J.T. Dusek, P.S. Maiya, B. Ma, R.L. Mieville, M.S. Kleefisch, C.A. Udovic h, C a t al. To da y 36, 265 (19 97). [9] V.R. Choudhary, A.M. Rajput, B. Prabhakar, J. Catal. 139, 326 (1993). [10] S.J. Feng, S. Ran, D.C. Zhu, W. Liu, C.S. Chen, Energy Fuels 18, 385 (2004). [11] V.R. Choudhary, K.C. Mondal, T.V. Choudhary, Catal. Commun. 8, 561 (2007). [12] T.F. Liu, C. Snyder, G. Veser, Ind. Eng. Chem. Res. 46, 9045 (2007). [13] Z.X. Wang, T. Dong, L.X. Yuan, T. Kan, X.F. Zhu, Y. Torimoto, M. Sadakata, Q.X. Li, Energy Fuels 21, 2421 (2007). [14] P. Mukoma, D. Hildebrandt, D. Glasser, N. Coville, Ind. Eng. Chem. Res. 46, 15 6 ( 2 0 0 7) . [15] A.B. Mhadeshwar, D.G. Vlachos, Ind. Eng. Chem. Res. 46, 5310 (2007). [16] S.F. Rice, A.H. McDaniel, E.S. Hecht, A.J.J. Hardy, Ind. Eng. Chem . Res. 46, 11 14 (2007). [17] J.H. Ryu, K.Y. Lee, H. La, H.J. Kim, J. Yang, H. Jung: J. Power Sources, Available online 23 June 2007. [18] H.J. Wan, X.J. Li, S.F. Ji, B.Y. Huang, K. Wang, C.Y. Li, J. Nat. Ga s Che m . 1 6 , 139 (2007). [19] M. Rezaei, S.M. Alavi, S. Sahebdelfar, P. Bai, X.M. Liu, Z.F. Yan, Appl. Catal., B: Environ. 77, 346 (2008). [20] Y.Q. Song, D.H. He, B.Q. Xu, Appl. Catal., A: Gen. Available online 29 November 2007. [21] J.T. Hu, T.W. Odom, C.M. Lieber: Acc. Chem. Res., 32, 435 (1999). [22] A. Taguchi, F. Schuth: Micropor. Mesopor. Mater., 77, 1 (2005). [23] C.C. Wang, J.Y. Ying: Chem. Mater., 11, 3113 (1999). [24] P. Kim, Y.H. Kim, H.S. Kim, I.K. Song, J.H. Yi: Appl. Catal., A, 272, 157 (2004 ). [25] Y.H. Li, Y.Q. Wang, X.B. Hong, Z.G. Zhang, Z.P. Fang, Y. Pan, Y.B. Lu, Z.Q. Han: AIChE J., 52, 4276 (2006). [26] D.J. Mei, Y.Q. Chen, J.B. Zhong, Z.L. Wei, D. Ma, M.C. Gong: J. Rare Earth, 25, 311 (2007). [27] P.M. Torniainen, X. Chu, L.D. Schmidt: J. Catal., 146, 1 (1994). [28] F. Kleitz, S.H. Choi, R. Ryoo: Chem. Commun., 17, 2136 (2003). [29] T.W. Kim, F. Kleitz, B. Paul, R. Ryoo: J. Am. Chem. Soc., 127, 7601 (2005). [30] T.W. Kang, Y.G. Park, J.H. Yi: J. Mol. Catal. A, 244, 151 (2006). [31] M.K. Dongare, K. Malshe, C.S. Gopinath, I.K. Murwani, E. Kemnitz: J. Catal., 222, 80 (2004). [32] H. Mori, C.J. Wen, J. Otomo, K. Eguchi, H. Takahashi: Appl. Cata l. A, 245, 79 (2003). [33] S. Eriksson, S. Rojas, M. Boutonnet, J.L.G. Fierro: Appl. Catal., A, 326, 8 (2007). [34] C.T. Au, M.S. Liao, C.F. Ng: J. Phys. Chem. A, 102, 3959 (1998). [35] Y. Boucouvalas, Z.L. Zhang, X.E. Verykios: Catal. Lett., 40, 189 (1996). [36] S. Rabe, T.B. Truong, F. Vogel: Appl. Catal. A, 318, 54 (2007). [37] L. Chen, Q. Hong, J. Lin, F.M. Dautzenberg: J. Power Sources, 164, 803 (2007 ). [38] A.S. Bodke, S.S. Bharadwaj, L.D. Schmidt: J. Catal., 179, 138 (1998). [39] D.A. Hickman, L.D. Schmidt: AIChE J., 39, 1164 (1993). [40] M. Labaki, S. Siffert, J.F. Lamonier, E.A. Zhilinskaya, A. Ab- ouka is : Appl. Catal., B, 43 , 261 (2003). [41] J.J. Zhu, S. Albertsma, J.G. van Ommen, L. Lefferts: J. Phys. Chem. B, 109, 9550 (2005). [42] Li, C. Y.; Shen, Y. N.; Jia, M. L.; Sheng, S. S.; Adebajo, M. O.; Zhu, H. Y. Catal. Commun. 9, 355 (2008). [43] Durme, J. V.; Dewulf, J.; Leys, C.; Langenhove, H. V. Appl. Cata l., B: Envir on. 74, 324 (2008). [44] W.X. Kuang, Y. Fan, Y. Chen: Langmuir, 16, 1440 (2000). [45] Z.X. Liu, Q.X. Bao, N.J. Wu: J. Catal., 113, 45 (1988). [46] Galvez, M. E.; Boyano, A.; Lazaro, M. J.; Moliner, R. Chem. Eng. J. Available online 12 January 2008. [47] Baylet, A.; Royer, S.; Marecot, P.; Tatibouet, J. M.; Duprez, D. Appl. Catal., B: Environ. 77, 237 (2008). [48] J.J. Zhu, J.G. van Ommen, H.J.M. Bouwmeester, L. Lefferts: J. Catal., 233, 434 (2005). [49] S. Ozkar, M. Zahmakiran : J . Alloys. C om pd., 404, 728 (2005). [50] K.L. Hohna, L.D. Schmidt: Appl. Catal., A, 211, 53 (2001). [51] S. Whitaker: AIChE J., 18, 361 (1972). |