Paper Menu >>
Journal Menu >>
 Engineering, 2012, 5, 35-38 doi:10.4236/eng.2012.410B009 Published Online October 2012 (http://www.SciRP.org/journal/eng) Copyright © 2012 SciRes. ENG Anticancer Gene-engineered MSC-mediated Cancer Cell Death: An Imaging Demonstration Xu-Yong Sun1*, Zhuang Chen2*, Mani R . Moniri3, Hong Lu3, Long -Jun Dai3#, Garth L. Warnock3 1Institute of Transplant Medicine1, 303 Hospital of PLA, Nanning, Guangxi, China 2Laboratory of Infection and Immunity, Affiliated Hospital of Luzhou Medical College, Luzhou, Sichuan, China 3Department of Surgery, University of British Columbia, Vancouver, Canada Email: #ljdai@mail.ubc.ca Received 2012 ABSTRACT This study was performed to demonstrate the transportation of an engineered MSC-produced intracellular anticancer gene product between mesench ymal stem cell ( MSC) and cancer cell s. MSC -mediated anticancer s trategy has h eld great promise o wing to MS Cs’ capacity of tumor-directed migration and the availability of specific anticancer genes. All anticancer genes that have been used in previous MSC-mediated anticancer studies were limited in functioning via extracellular mechanisms, mainly because of the restric- tion b y cell membrane to macromolecules including proteins. In order to apply the majority of potent anticancer genes to the MSC- mediated antican cer syste m, a specifical ly design ed expressi on ve ctor whi ch bears an intracel lular ant icancer gen e, PTEN, is utilized to demonstrate the feasib ilit y of the system in cancer th erapies. A trans acting acti vator o f transcript ion (TAT) was introdu ced in to an expression vector followed by a segment for PTEN-RFP fusion protein. A direct demonstration of PTEN-RFP transportation bet wee n MSC and cancer cells was obtained from direct co-cultures. A marked cancer cell death was observed in indirect co-cultures with conditioned media from PTEN-transfected MSCs. The demonstration of PTEN-engineered MSC-produced PTEN transportation ind icates the feasibility of applying intracellular anticancer gene expression system in MSC-mediated strategies fo r cancer th er apy. Keywords: Genet ic Engineerin g; Mesench ymal Stem Cells; Gene Ther apy; Cancer Therapy; PTEN. 1. Introduction Cancer remai ns on e of the leading causes of mortalit y and mor- bidity throughout the world. To a significant extent, current conventional cancer therapies are symptomatic and passive in nature. The major obstacle to the development of effective cancer th erapy is believed t o be the abs ence of sufficien t tumor specificity. Since the discovery of tumor-oriented homing ca- pacity of mesenchymal stem cells (MSCs), the application of specific anticancer gene-engineered MSCs has held great promise for cancer therapies [1]. MSC-mediated anticancer therapy relies on tumor-specific selectivity provided by MSCs and MSC-carried anticancer agents. Homed directly at the tu- mor microenvironment, engineered MSCs are able to express and/or release anticancer agents to constantly act on the adja- cent tumor cells. Based on the mechanisms of tumor suppres- sion, MSC-mediated anticancer agents are classified into fol- lowing categories: (1) immunostimulation, such as CX3CL1 [2], IFN [3,4], I L2 [5] , IL7 [6] and IL12 [7]. (2) prodrug conversion, such as CD [8] and HSV-tk [9]. And (3) apoptosis induction, such as IL18 [10], NK4 [11] and TRAIL [12-14]. All of these anticancer agents used in previous studies are produced by engineered MSC s and act on targeted tumor cells ext racellular- ly. However, the majority of anticancer gene products act on tumor cells through intracellular mechanisms, such as P53, Myc and phosphatase and tensin homolog (PTEN). When MSCs are used as a vehicle to deliver intracellular anticancer genes, th e anticancer gene products must be p roduced in MS Cs and secreted into extracellular space and then penetrate into adjacent tumor cells passing through biological membrane, which is a natural barrier for most macromolecules including peptides and proteins. In this conference, we present a uniquely designed expressio n vector and use PTEN-en gineered MSCs as an example to demonstrate cancer cell death induced by intra- cellul ar anticancer genes. 2. Materials and Methods 2.1. Cells and Culture C ondit ions MSCs were isolated from human pancreas and ex vivo ex- panded as previously described [14]. Based on the minimal criteria for defining human MSCs established by the Interna- tional Society of Cellular Therapy, these MSCs were verified by both membrane biomarker determination and functional differentiation. They fulfilled the characteristics of human MSCs, exhibiting positive expression of CD44+, CD73+, CD 95+, CD105+ and negative of CD34-. The results of adipogenic and osteogenic differentiation also met the standards. MSCs were cultured in MEM with 10% FCS, 2 mM L-glutamine and 1% penicillin-streptomycin solution (all from Invitrogen, Carlsbad, CA, USA) and incubated at 37oC in a humidified, 5% CO2 atmosphere. A human pancreatic cancer cell line (Panc-1) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and used as target cell s in the present study. Panc-1 cells were maintained as suggested by ATCC and their culture *These authors contributed equally t o this work. #Corresponding author. 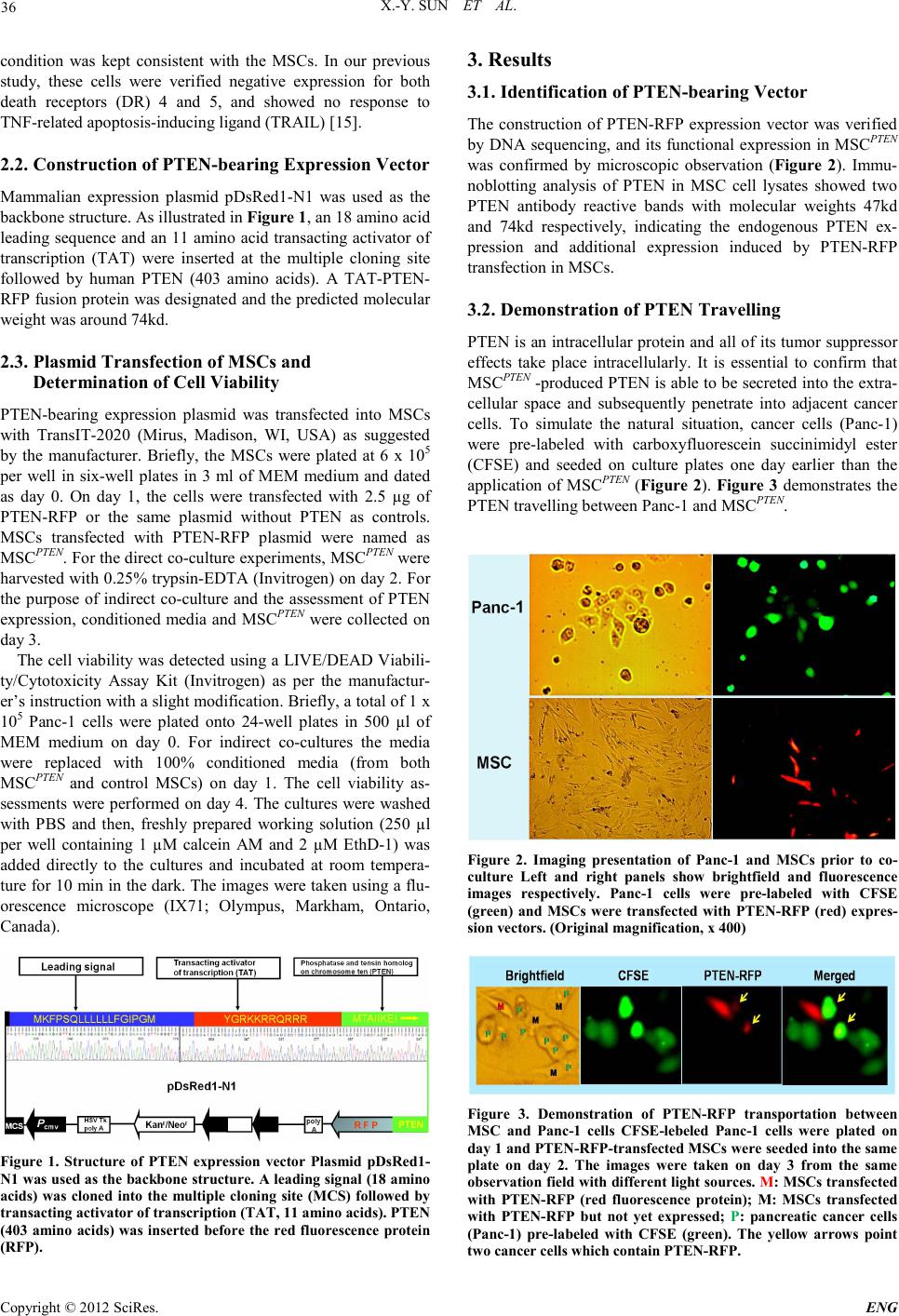 X.-Y. SUN ET AL. Copyright © 2012 SciRes. ENG 36 condition was kept consistent with the MSCs. In our previous study, these cells were verified negative expression for both death receptors (DR) 4 and 5, and showed no response to TNF-related apoptosis-inducing ligand (TRAIL) [15]. 2.2. Constru cti on of PTE N-bearing Expre s si on Vector Mammalian expression plasmid pDsRed1-N1 was used as the backbone structure. As illustrated in Figure 1, an 18 amino acid leadin g sequence and an 11 amino acid transacting activator of transcription (TAT) were inserted at the multiple cloning site followed by human PTEN (403 amino acids). A TAT-PTEN- RFP fusion protein was designated and the predicted molecu lar weight was around 74kd. 2.3. Plasmid Transfec tion of MSCs and Determination of Cell Via bility P TEN-bearing expression plasmid was transfected into MSCs with TransIT-2020 (Mirus, Madison, WI, USA) as suggested by the manufacturer. Briefly, the MSCs were plated at 6 x 105 per well in six-well plates in 3 ml of MEM medium and dated as day 0. On day 1, the cells were transfected with 2.5 µg of P TEN-RFP or the same plasmid without PTEN as controls. MSCs transfected with PTEN-RFP plasmid were named as MSCPTEN. For th e direct co-cul ture experiment s, MSC PTEN were harvested with 0.25% trypsin-EDTA (Invitrogen) on day 2. For the purpose of indirect co-culture and the assessment of PTEN expression, conditioned media and MSCPTEN were collected on day 3. The cell viability was detected using a LIVE/DEAD Viabili- ty/Cytotoxicity Assay Kit (Invitrogen) as per the manufactur- er’s instruction with a slight modification. Briefly, a total of 1 x 105 Pan c -1 cells were plated onto 24-well plates in 500 µl of MEM medium on day 0. For indirect co-cultures the media were replaced with 100% conditioned media (from both MSCPTEN and control MSCs) on day 1. The cell viability as- sessments were perfor med on day 4. The cultu res were washed with PBS and then, freshly prepared working solution (250 µl per well containing 1 µM calcein AM and 2 µM EthD-1) was added directly to the cultures and incubated at room tempera- ture for 10 min in the dar k. The images were ta ken usin g a flu- orescence microscope (IX71; Olympus, Markham, Ontario, Canada) . Figure 1. Structure of PTEN expression vector Plasmid pDsRed1- N1 was used as the backbone structure. A leading signal (18 amino acids) was cloned into the multiple cloning site (MCS) followed by transact ing ac t iv ator of tran s crip t i on (TA T , 11 ami n o aci d s). PT E N (403 amino acids) was inserted before the red fluorescence protein (RFP). 3. Results 3.1. Identification of PTEN-bearing Vector The construction of PTEN-RFP expression vector was verified by DNA sequencing, and its functional expression in MSCPTEN was confirmed by microscopic observation (Figure 2). Immu- noblotting analysis of PTEN in MSC cell lysates showed two PTEN antibody reactive bands with molecular weights 47kd and 74kd respectively, indicating the endogenous PTEN ex- pression and additional expression induced by PTEN-RFP transfection in MSCs. 3.2. Demonstration of PTEN Travelling PTEN is an intracellular protein and all of its tumor suppressor effects take place intracellularly. It is essential to confirm that MSCPTEN -produced PTEN is able to be secreted into the extra- cellular space and subsequently penetrate into adjacent cancer cells. To simulate the natural situation, cancer cells (Panc-1) were pre-labeled with carboxyfluorescein succinimidyl ester (CFSE) and seeded on culture plates one day earlier than the application of MSCPTEN (Figure 2). Figure 3 demonstrates the PTEN t r avel ling b etween Panc-1 and MSC PTEN. Figure 2. Imaging presentation of Panc-1 and MSCs prior to co- culture Left and right panels show brightfield and fluorescence images respectively. Panc-1 cells were pre-labeled with CFSE (green) and MSCs were transfected with PTEN-RFP (red) expres- sion vectors. (Original magnification, x 400) Figure 3. Demonstration of PTEN-RFP transportation between MSC and Panc-1 cells CFSE-lebeled Panc-1 cells were plated on day 1 and PTEN-RFP-transfected MSCs were seeded into the same plate on day 2. The images were taken on day 3 from the same observation field with different light sources. M: MSCs transfected with PTEN-RFP (red fluorescence protein); M: MSCs transfected with PTEN-RFP but not yet expressed; P: pancreatic cancer cells (Panc-1) pre-labeled with CFSE (green). The yellow arrows point two cance r cel ls which contain PT E N -RFP.  X.-Y. SUN ET AL. Copyright © 2012 SciRes. E NG 37 Figure 4. Panc-1 cell viability in indirect co-cultures with condi- tioned media Pan c-1 cells were plated on day 1. Conditioned media from native MSCs (top panel) and MSCPTEN (bottom panel) were replaced on day 2. The cell viability assessment was per- formed with LIVE/DEAD assay on day 4. Column 1 (brightfield): whole population of cells which were still attached to the culture surface during the assessment; column 2: live cells stained with calcein are green; column 3: dead cells stained with EthD-1 show red; column 4: merged images. (Original magnificat ion, x 400) . 3.3. MSCPTEN-mediated P anc-1 cell De ath in Indirect Co-cultures MSCPTEN –mediated P anc-1 cell death was assessed in indirect co-cultures using conditioned media from control MSCs and MSCPTEN. As shown in Figure 4, marked cell death was ob- served when Panc-1 cell were co-cultured with conditioned media from MSCPTEN. It is worth noting that LIVE/DEAD as- say only applies to the cells which remain on the culture surface during the staining. The detached cells, most of which are dead cells, are not in cluded in th e as s essment . 4. Discussion and Conclusion The tumor suppressor phosphatase and tensin homolog (PTEN) was discovered in 1997 [16,17]. The PTEN gene is located at chromosome 10q23.31. Loss of heterozygosity at 10q23 occurs frequently in many advanced-stage sporadic tumors; for exam- ple, approximately 70% in glioblastomas and 60% in advanced prostate cancers [17]. PTEN is also known as mutated in mul- tiple advanced cancer 1 (MMAC1). It functions as the central negative regulator of PI3K-AKT-mTOR path way in controlling apoptosis. PI3K-AKT-mTOR signaling pathway is the most frequently activated pathway in human cancers, because it promotes cell growth and contributes to the evasion of apopto- sis, loss of cell cycle control and genomic instability during tumorigenesis. PTEN dephosphorylates PIP3 to PIP2 thereby directly opposing the activity of PI3K. Compared to other clas- sical tumor suppressor genes, PTEN is haploinsufficient be- cause a single copy is unable to prevent cancer. Loss of its he- terozygosity or partial inhibition of its expression/activity is sufficient to promote carcinogenesis [18]. Thus, restoring PTEN function in cancer cells would break down the PTEN mutation-dependent cancer cell growth, and this holds great promise for cancer th er apy. Wid e-type PTEN can be introduced into cells by using viral vectors [19] or a non-viral vector-mediated delivery such as urocanic acid-modified chitosan-mediated PTEN delivery via aerosol [20]. However, engineering MSCs with PTEN could stron gly poten tiate the efficacy of targ eted cancer therap y [2 1]. For the first time, a cell permeable recombinant wide-type PTEN was transfected into MSCs via fusion PTEN with trans- activator of transcription (TAT). As demonstrated in Figure 3, MSC-produced PTEN-RFP appears in th e adjacen t cancer cel ls. This observation provides a direct evidence of PTEN-RFP tra- velling across multiple membranes. PTEN-engineered MSC- mediated cancer cell death was also confirmed in indirect co- cultures with Panc-1 cells which are insensitive to TRAIL (Figure 4). This concise demonstration reveals the application of intracellular anticancer gene-bearing expression system in MSC-mediated cancer therapy, which potentially broadens the spectrum of anticancer gene-r elated cancer therapy. In conclusion, the demonstration of PTEN-engineered MSC-produced PTEN transportation indicates the feasibility of applying intracellular anticancer gene expression system in MSC-mediated anticancer treatment. PTEN-induced Panc-1 cell death also suggests possible synergistic interactions of multiple anticancer genes t o highly hetero geneous car cinomas. 5. Acknowledgements This work was supported by the Guangxi Ministry of Science and Technology (0993003A, 0719002-2-7) and the VGH & UBC Hospital Foundation. The authors are gratiful to Crystal Robertson for her assistance in preparing the manuscript. REFERENCES [1] M. R. Loebinger and S. M. Janes, "Stem cells as vectors for antitumour thera py," Thorax, vol. 65, pp. 362-9, Apr 2010. [2] H. Xin, et al., "Targeted delivery of CX3CL1 to multiple lung tumors by mesenchymal stem cells," Stem Cells, vol. 25, pp. 1618-26, Jul 20 07. [3] C. Ren, et al., "Therapeutic potential of mesenchymal stem cells produc in g interfer on -alpha in a mouse melanoma lung metastasis model," Stem Cells, vol. 2 6, pp. 2332-8, Sep 2 00 8. [4] S. Kidd, et al., "Mesenchymal stromal cells alone or expressing interferon-beta suppress pancreatic tumors in vivo, an effect countered by anti-inflammatory treatment," Cytotherapy, vol. 12, pp. 615-25, Se p 2010. [5] K. Nakamura, et al., "Antitumor effect of genetically engineered mesen chymal s tem cells in a rat gli oma model, " Gene Ther, vo l. 11, pp. 1155-64, Jul 2004. [6] S. Gunnarsson, et al., "Intratumoral IL-7 delivery by mesen- chymal stromal cells potentiates IFNgamma-transduced tumor cell immunotherapy of experimental glioma," J Neuroimmunol, vol. 218, pp. 140-4, Jan 25 2010. [7] X. Chen, et al., "A tumor-selective biotherapy with prolonged impact on established metastases based on cytokine gene-engineered MSCs," Mol Ther, vol. 16, pp. 749-56, Apr 2008. [8] I. T. Cavarretta, et al., "Adipose tissue-derived mesenchymal stem cells expressing prodrug-converting enzyme inhibit human prost ate tumor growth," Mol Ther, vol. 18, pp. 223-31, Ja n 2 010. [9] H. Miletic, et al., "Bystander killing of malignant glioma by bone marrow-derived tumor-infiltrating progenitor cells expressing a suicide gene," Mol Ther, vol. 15, pp. 1373-81, J ul 2007. [10] G. Xu, et al., "Adenoviral-mediated interleu kin-18 expressi on in mesenchymal stem cells effectively suppresses the growth of glioma in rats, " Cell Biol Int, vol. 33, pp. 466-74, Apr 2009. [11] M. Kanehira, et al., "Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells," Can-  X.-Y. SUN ET AL. Copyright © 2012 SciRes. ENG 38 cer Gene Ther, vol. 14, pp. 894-903, Nov 2007. [12] L. G. Menon, et al., "Human bone marrow-derived mesenchymal stromal cells expressing S-TRAIL as a cellular delivery vehicle for human glioma therapy," Stem Cells, vol. 27, pp. 2320 -30, Sep 2009. [13] M. R. Loebinger, et al., "Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer," Cancer Res, vol. 69, pp. 4134-42, May 15 200 9. [14] X. Y. Sun, et al., "MSC(TRAIL)-mediated HepG2 cell death in direct and indirect co-cultures," Anticancer Res, vol. 31, pp. 3705-12, Nov 2011. [15] X. Y. Sun, et al., "Mesenchymal stem cell-medi at ed c anc er t her- apy: A dual-target ed st rat egy of per son al ized medici ne, " World J Stem Cells, vol. 3, pp. 96-103, Nov 26 2011. [16] P. A. Steck, et al., "Identification of a candida te tumou r suppres- sor gene, MMAC1, at chromosome 10q23.3 that is mutated in multip le advanc ed canc ers," Nat Genet, vol. 15, pp. 356-62, Apr 1997. [17] J. Li, et al., "PTEN, a putative protein tyrosine phosphat ase gen e mutated in human brain, breast, and prostate cancer," Science, vol. 275, pp. 1943-7, Mar 28 1997. [18] M. Peyrou, et al., "PTEN in liver diseases and cancer," World J Gastroenterol, vol. 16, pp. 4627-33, Oct 7 2010 . [19] G. Pappas, et al., "Adenoviral-mediated PTEN expression radi- osensi ti zes non -small cell lun g cancer c ells by sup pressin g DNA repair capacit y, " Can cer Gene Ther, vol. 14, pp. 5 4 3-9, Jun 2007. [20] H. Jin, et al., "Urocanic acid-modified chitosan-mediated PTEN deliver y via aerosol su ppressed lung tumorigen esis in K-ras(LA1) mice," Cancer Gene Ther, vol. 15, pp. 275-83, May 2008. [21] L. J. Dai, et al., "Potential implications of mesenchymal stem cells in cancer therapy," Cancer Lett, vol. 305, pp. 8-20, Jun 1 2011. |