Paper Menu >>
Journal Menu >>
 Vol.2, No.9, 1072-1077 (2010) Health doi:10.4236/health.2010.29157 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ Reduced bile duct contractile function in rats with chronic hyperglycemia Chi-Ming Liu1, Hui-Chen Su2, Yen-Ting Wang1, Tao-Hsin Tung1, Pesus Chou3, Yiing-Jeng Chou3, Jorn-Hon Liu1, Jan-Kan Chen4* 1Cheng-Hsin General Hospital, Taipei, Taiwan, China 2Chi-Mei Medical Center, Tainan, Taiwan, China 3Institute of Public Health, National Yang Ming University, Taipei, Taiwan, China 4Department of Physiology, College of Medicine, Chang Gung University, Taipei, Taiwan, China; *Corresponding Author: jkc508 @ mail. cgu.edu.tw Received 27 November 2009; revised 11 January 2010; accepted 3 March 2010. ABSTRACT The incidence of gallstone is higher in patients with diabetes mellitus than in general popula- tion. It is generally attributed to hypomotility and lowered emptying function of the gallblad- der. In this study, we investigate if chronic hy- perglycemia is correlated with reduced contrac- tile function of the bile ducts in rat. Hypergly- cemic rats were induced by streptozotocin-nic- otinamide treatment. Hyperglycemic rats were sacrificed eight months after induction and bile ducts were removed for the subsequent studies. The bile duct contractility of the normal rats is consistently higher than that of the hypergly- cemic rats. The contractities were measured to be 5.5 ± 0.2 mg vs. 4.2 ± 0.1 mg without CCK stimulation, and 5.5 ± 0.3 mg vs. 7.9 ± 0.4 mg with CCK stimulation, respectively for hypergly- cemic and normal rats. There was no significant difference in plasma CCK concentration in hy- perglycemic rats and normal rats. The expres- sion of CCK-A receptor protein in the bile duct tissue was decreased in hyperglycemic rats compared with that of the normal rats, and it may, at least in part, responsible for a reduced contractility. A reduced bile duct motility may cause bile retention, and may be one of the factors predispose to gallstone formation in type 2 diabetes patients, which is characterized with chronic hyperglycemia. Keywords: Hyperglycemia; Bile Duct; Contractile 1. INTRODUCTION Cholelithiasis is one of the most prevalent gastroentero- logic diseases in humans. It is a complex metabolic dis- orders, and its exact pathogenic mechanisms have not been fully elucidated. Gallstones represent a serious burden for the health care systems: over 10% of Euro- peans and Americans carry gallbladder stones [1], and the prevalence of gallstone disease seems to be rising as a result of longer life expectancy [2]. Many gallstones are silent, but symptoms and severe complications en- sure in around 25% of the cases, necessitating surgical removal of the gallbladder [3]. Mortality rates following cholecystectomy range from less than 0.1% in clinical studies [4] to 0.8% (as documented for all cholecystec- tomies performed in Germany in 2002) [5]. In the US about 3,000 deaths (0.12% of all deaths) per year are attributed to complications of cholelithiasis and gall- bladder disease [6]. Nonsurgical approaches, including gallstone dissolution by ursodeoxycholic acid and ex- tracorporeal shockwave lithotripsy, have increasingly lost their impact on therapy and are performed only for uncomplicated symptomatic cholecystolithiasis in a very small number of selected patients. Bile formation enables the removal of excess choles- terol, either directly or after catabolism to bile salts, and it is a key function of the liver. Bile is an aqueous solu- tion of lipids, in which bile salts (67% of solutes by weight), phospholipids (22%) and cholesterol (4%) rep- resenting the three main lipid species [7]. More than 80% of gallstones consist mainly of cholesterol and are formed within the gallbladder [8]. Three major mecha- nisms contribute to the formation of cholesterol gall- bladder stones: cholesterol supersaturation of bile, gall- bladder hypomotility and destabilization of bile by ki- netic protein factors. Cholesterol-supersatu-rated bile contains more cholesterol than can be solubilized by mixed micelles (cholesterol saturation index > 1). It contains multilamellar vesicles (liquid crystals), whose *Chi-Ming Liu and Hui-Chen Su contributed equally in this study.  C.-M. Liu et al. / HEALTH 2 (2010) 1072-1077 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 1073 fusion and aggregation precede the formation of solid cholesterol crystals. As illustrated in the classic triangu- lar phase diagram, solid crystals occur in bile at high relative bile salt and low phospholipids concentrations and at cholesterol: phospholipids ratios > 1 [7]. An ex- cess of biliary cholesterol in relation to bile salts and phospholipids can result from hypersecretion of choles- terol, or from hyposecretion of bile salts or phospholip- ids. Cholesterol hypersecretion is the most common cause of supersaturation [8]. It might be caused by in- creased hepatic uptake or synthesis of cholesterol, de- creased hepatic synthesis of bile salts, or decreased he- patic synthesis of cholesteryl esters for incorporation in VLDL. Accordingly, any enzyme, transporter or regula- tor involved in hepatic cholesterol metabolism could potentially affect the formation of cholesterol gallstones [9]. In humans, most gallstone cholesterol is of dietary origin, consistent with the observation that hepatic bio- synthesis contributes less than 20% of the biliary cho- lesterol [2]. The hepatic uptake of cholesterol is medi- ated by the scavenger receptor B1 for HDL, which con- tributes most of the biliary cholersterol under physiol- ogic conditions. The inverse correlation between serum HDL levels and gallstones suggests that cholesterol cholelithiasis is associated with an induced reverse cho- lesterol transport and hepatic catabolism of HDL [2]. The rate-limiting enzymes of hepatic cholesterol and bile salt synthesis are 3-hydroxy-3-methylglutary-coenzyme A reductase and cholesterol 7α-hydroxylase, respectively. These enzymes are regulated by the sterol-regulatory- lement-binding protein (SRBP) and nuclear receptor signaling pathways [10,11]. Stasis of bile in the gall- bladder favours stone formation, as indicated by stone formation during pregnancy, rapid weight loss or total parenteral nutrition. Postprandial gallbladder volumes are increased and gallbladder emptying in response to cholecystokinin (CCK) is impaired in patients with gall- stones [8], probably as a result of absorption of choles- terol from supersaturated bile by gallbladder wall. Ex- cess cholesterol in smooth-muscle cells stiffens sar- colemmal membranes and decouples the G-protein-me- diated signal transduction of the CCK, thereby paralyz- ing gallbladder contractile function [12]. Among the diabetes patients in China, about 10% of them also bear gallstones. The impaired emptying func- tion owing to hypomotility of the gallbladder is consid- ered an important factor for the development of chole- lithiasis [13-17]. Because of hypomotility and lowered emptying function, the incidence of gallstone is higher in patients with diabetes mellitus than in general population, however, its underlying mechanism has not been well understood. Gallbladder mortility is regulated by cholinergic and gastrointestinal hormone [18,19]. Postprandial gallblad- der emptying is triggered mainly by plasma CCK from small intestine. CCK interacts with CCK receptor-1 (CCK-R) in gallbladder smooth muscle cells, which in turn elicits the contraction of gallbladder by the activa- tion of post-membrane signaling passway [20]. We hy- pothesize that abnormal gallbladder contraction in re- sponse to CCK, may play an important role in the deve- lopment of cholesterol gallstone in hyperglycemic pa- tient. The purpose of this study was to examine if there are differences in gallbladder motor function, plasma CCK concentration, and the CCK-R activity in response to CCK in hyperglycemic and normal rats. 2. MATERIALS AND MATHODS 2.1. Animals Male SD rats, age 8-10 week, obtained from the Na- tional Laboratory Animal Center, Taiwan, were used for the study. Streptozotosine (STZ), which selectively de- stroys the pancreatic β-cells that secrete insulin, was used to induce insulin dependent DM, and nicotinamide was used to attenuate STZ effect to induced insulin-de- ficient diabetic rats [21]. STZ-nicotinamide DM rats were induced in overnight fasted animals by a single intravenous injection of STZ (60 mg/kg body weight), and nicotinamide (120 mg/kg body weight) (Sigma Chemical Co., St Louis, MO) was administered intrap- eritoneally 15 min after STZ. Combined administration of STZ and nicotinamide leads to the development of a diabetic syndrome, which is characterized by moderate and stable hyperglycemia and reduced pancreatic insulin stores [22]. Hyperglycemia was confirmed by elevated plasma glucose levels, measured on days 3 and 7 after drug in- jection. STZ-nicotinamide treated rats exhibited a fasting plasma glucose concentration of 13.4 ± 0.8 mmol/L and a plasma insulin level of 95.0 ± 0.2 pmol/L (n = 24). In contract, the fasting plasma glucose and insulin levels of the normal rats were 4.8 ± 0.05 mmol/L and 168.5 ± 4.8 pmol/L, respectively (n = 24). All studies were carried out with animals two weeks after the induction of diabe- tes. Blood samples were taken from overnight fasted rats at 0, 2, 4, 6 and 8 months after the confirmation of hy- perglycemia. 2.2. Bile Duct Contractile Response Bile ducts were isolated from hyperglycemic rats and normal rats, respectively, and placed in Kreb’s solution containing (in mmol/L) 118.4 NaCl, 25 NaHCO3, 11.66 glucose, 4.75 KCl, 1.18 MgSO4·7H2O, 2.5 CaCl2·2H2O, 1.19 KH2PO4, 0.02 EDTA. The solution was maintained  C.-M. Liu et al. / HEALTH 2 (2010) 1072-1077 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 1074 at pH 7.4 and continuously bubbled with 95%O2-5%CO2. Bile ducts were carefully mounted on the isometric force transducer in the organ chamber (95%O2-5%CO2, at 37˚C), and were equilibrated for 90 minutes in an organ bath with a resting tension of 1.8 mg. CCK (from 10-9- 10-5 M) was used to induce bile duct contraction. 2.3. Determination of Plasma CCK Concentration Animals were fasted overnight and anesthetized by pen- tobarbital (30 mg kg-1 body weight, i.p.). Blood samples (0.1 mL) were collected from femoral vein using a chilled syringe that contained 10 IU heparin. Plasma CCK levels were measured by enzyme immunoassay of 50 μL aliquots of plasma with a CCK octapeptiode rat ELISA kit (Harbor Boulevard, Belmont, California). The immunoplate in this kit is pre-coated with secondary antibody and the nonspecific binding sites are blocked. The secondary antibody binds to the Fc fragment of the primary antibody (peptide antibody) whose Fab frag- ment will be competitively bound by both biotinylated peptide and peptide standard or targeted peptide in sam- ple. The biotinylated peptide is able to interact with streptavidin-horseradish peroxidase (SA-HRP) which catalyzes the conversion of 3,3’,5,5’-tetramethylbenzi- dine (TMB) to produce a blue colored solution. The re- action was stopped by adding acid and the reaction solu- tion turned yellow and was read spectrophotometrically. 2.4. Determination of Plasma Glucose Animals were fasted overnight and anesthetized by pen- tobarbital (30 mg kg-1 body weight, i.p.). Blood samples (0.1 mL) were collected from femoral vein using a chilled syringe that contained 10 IU heparin. The sam- ples were centrifuged at 13,000 rpm for 3 min, and ali- quot (15 μl) of plasma was added to 1.5 ml of a Glucose Kit Reagent (Biosystems S.A., Barcelona, Spain) and incubated at 37˚C in a water bath (Yamato-BT-25, Tokyo, Japan) for 10 min. Plasma glucose was determined by a glucose analyzer (Quik-Lab, Ames, Miles Inc., Elkhart, IN). 2.5. Determination of Plasma Insulin Plasma insulin levels were measured by enzyme immu- noassay of 25 μL aliquots of plasma with a Rat Insulin ELISA kit (Mercodia, Uppsala, Sweden). During incu- bation, insulin in the sample reacted with peroxidase- conjugated anti-insulin antibodies which were bound to the plastic surface of the microtitration well. The bound conjugate was detected by reaction with 3,3’,5,5’-tetra- methylbenzidine. The reaction was stopped by adding acid to give a colorimetric endpoint that was read spec- trophotometrically. 2.6. Western Blot Analysis Bile ducts were homogenized by mechanical homogeni- zation using a glass/Teflon homogenizer. Protein content was determined using the BCATM protein assay kit (Rockford, USA). A total of 50 μg of tissue protein was fractionated on 10% sodium dodecyl sulfate/polyacryla- mide gel electrophoresis (SDS/PAGE). Following trans- fer, the membrane was washed with phosphate-buffer saline (PBS) and blocked for 1 h at room temperature with 5% (w/v) skimmed milk in PBS. Blots were then incubated overnight at 4˚C with the polyclonal antibody- ies against rat bile duct CCK-A or CCK-B (both at 1:1000) (abcam Littleton, CO). β-actin was also blotted with mouse monoclonal antibody (1:1000) against β-actin and served as internal reference. After removing the primary antibody, the blots were extensively washed with PBS/Tween 20, and incubated for 1 h at room tem- perature with the appropriate peroxidase-conjugated secondary antibody. Following removal of the secondary antibody, blots were washed as described and developed by autoradiography using the ELC-Western blotting sys- tem (Amersham Corp.). Densities of the obtained im- munoblots at 50 KDa for CCK-B, 48 KDa for CCK-A and 42 KDa for β-actin were quantified using a laser densitometer. 2.7. Statistical Analysis Data are expressed as mean ± SEM (standard error of the mean). Repeated measures of analysis of variance (ANOVA) were used to analyze the changes in plasma glucose and other parameters. The Dunnett range of post hoc comparisons was used to determine the source of significant differences where appropriate. A p value of 0.05 or less was considered as significant. 3. RESULTS 3.1. Measurement of the Plasma Glucose and Insulin Levels Plasma glucose and insulin levels were measured from blood samples taken from overnight fasted rats. The blood glucose concentrations measured at 0, 2, 4, 6 and 8 months after confirmation of the hyperglycemia were 13.4 ± 0.8, 14.4 ± 0.6, 15.6 ± 0.2, 16.2 ± 0.1, 16.6 ± 0.2 mmol/L, respectively (Figure 1). The plasma insulin concentrations measured at the same time intervals after confirmation of hyperglycemia were 95.0 ± 0.2, 99.5 ± 0.4, 115.2 ± 0.3, 125.6 ± 0.6 and 128.4 ± 0.8 pmol/L, respectively (Figure 1). During the same period of time, the plasma glucose and insulin levels of the normal rats were 4.8 ± 0.5 mmol/L and 168.5 ± 4.8 pmol/L, respec- tively (n = 24). 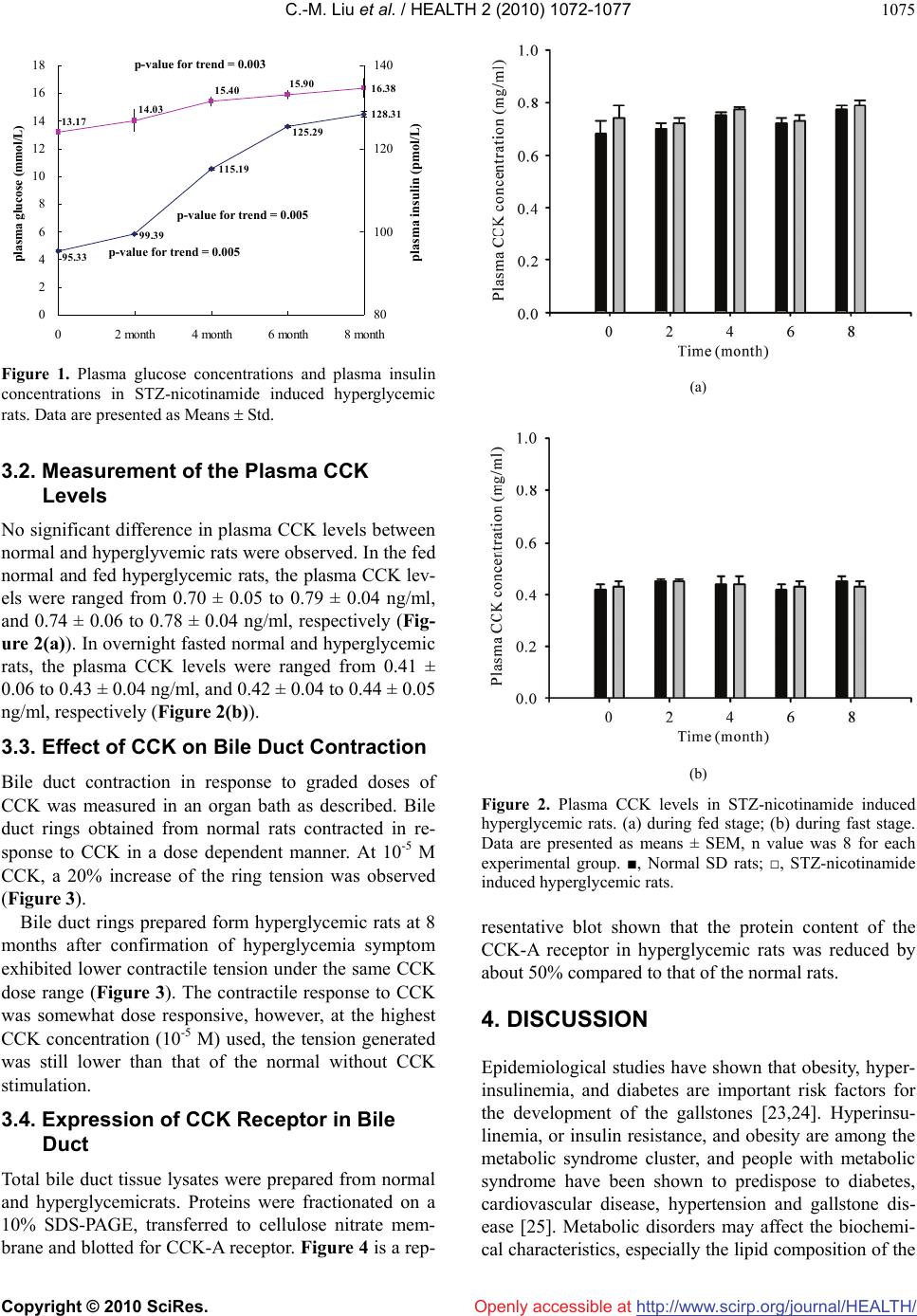 C.-M. Liu et al. / HEALTH 2 (2010) 1072-1077 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 1075 16.38 13.17 14.03 15.40 15.90 115.19 128.31 95.33 99.39 125.29 0 2 4 6 8 10 12 14 16 18 02 month4 month6 month8 month plasma glucose (mmol/L ) 80 100 120 140 plasma insulin (pmol/L ) plasma insulin (pmol/L) p-value for trend = 0.003 p-value for trend = 0.005 p-value for trend = 0.005 Figure 1. Plasma glucose concentrations and plasma insulin concentrations in STZ-nicotinamide induced hyperglycemic rats. Data are presented as Means Std. 3.2. Measurement of the Plasma CCK Levels No significant difference in plasma CCK levels between normal and hyperglyvemic rats were observed. In the fed normal and fed hyperglycemic rats, the plasma CCK lev- els were ranged from 0.70 ± 0.05 to 0.79 ± 0.04 ng/ml, and 0.74 ± 0.06 to 0.78 ± 0.04 ng/ml, respectively (Fig- ure 2(a)). In overnight fasted normal and hyperglycemic rats, the plasma CCK levels were ranged from 0.41 ± 0.06 to 0.43 ± 0.04 ng/ml, and 0.42 ± 0.04 to 0.44 ± 0.05 ng/ml, respectively (Figure 2(b)). 3.3. Effect of CCK on Bile Duct Contraction Bile duct contraction in response to graded doses of CCK was measured in an organ bath as described. Bile duct rings obtained from normal rats contracted in re- sponse to CCK in a dose dependent manner. At 10-5 M CCK, a 20% increase of the ring tension was observed (Figure 3). Bile duct rings prepared form hyperglycemic rats at 8 months after confirmation of hyperglycemia symptom exhibited lower contractile tension under the same CCK dose range (Figure 3). The contractile response to CCK was somewhat dose responsive, however, at the highest CCK concentration (10-5 M) used, the tension generated was still lower than that of the normal without CCK stimulation. 3.4. Expression of CCK Receptor in Bile Duct Total bile duct tissue lysates were prepared from normal and hyperglycemicrats. Proteins were fractionated on a 10% SDS-PAGE, transferred to cellulose nitrate mem- brane and blotted for CCK-A receptor. Figure 4 is a rep- (a) (b) Figure 2. Plasma CCK levels in STZ-nicotinamide induced hyperglycemic rats. (a) during fed stage; (b) during fast stage. Data are presented as means ± SEM, n value was 8 for each experimental group. ■, Normal SD rats; □, STZ-nicotinamide induced hyperglycemic rats. resentative blot shown that the protein content of the CCK-A receptor in hyperglycemic rats was reduced by about 50% compared to that of the normal rats. 4. DISCUSSION Epidemiological studies have shown that obesity, hyper- insulinemia, and diabetes are important risk factors for the development of the gallstones [23,24]. Hyperinsu- linemia, or insulin resistance, and obesity are among the metabolic syndrome cluster, and people with metabolic syndrome have been shown to predispose to diabetes, cardiovascular disease, hypertension and gallstone dis- ease [25]. Metabolic disorders may affect the biochemi- cal characteristics, especially the lipid composition of the  C.-M. Liu et al. / HEALTH 2 (2010) 1072-1077 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 1076 Figure 3. Dose effect of CCK on the contraction of bile duct isolated from normal (■) and STZ-nicotinamide induced hy- perglycemic rats (▲). Bile duct of the STZ-nicotinamide in- duced hyperglycemic rats was isolated at 8 months post iduc- tion. Figure 4. CCK-A receptor protein expression in the bile duct of the normal and hyperglycemic rats. bile. In fact, cholesterol supersaturation or an increase cholesterol to phospholipids ratio has been suggested to be one of the determinants for cholesterol gallstone for- mation [26]. One other possible influencing factor is the contractile function of the gallbladder. The major hor- monal regulator of gallbladder contraction in the intesti- nal phase of digestive function is CCK [27,28]. Reduced gallbladder contraction function due to defect in CCK signaling has been shown to impair gallbladder empting and enhance gallstone formation [29]. From a recent study we know, gallbladder dysmotility may have accel- erated sludge and gallstone formation in male and fe- male CCK-1(A) receptor-deficient mice, but its contri- bution was limited [30]. Since diabetes patients, which are characterized by chronic hyperglycemia have been reported to have a higher incidence of gallbladder stone disease, we therefore, sought to examine the possible effects of the hyperglycemia on the gallbladder contrac- tile function in response to CCK in rats. We found that bile duct contractility of the chronically hyperglycemia rats with and without CCK stimulation was consistently lower that of the normal rats. Further examination by Western blotting showed that the ex- pression of CCK-A receptor protein in the bile duct of the hyperglycemic rats was reduced by 50% compared with that of the normal rats. It is therefore concluded that reduced bile duct response to CCK contractile stimula- tion in hyperglycemic, and perhaps, diabetic rats is due, at least in part, to a reduced expression of CCK-A re- ceptor. Interestingly, the plasma CCK contents of the normal and hyperglycemic rats were not significantly differed. Thus, the reduced bile duct contractility may be one of the important influencing factors of the high co- incidence of gallbladder stone disease and diabetes. REFERENCES [1] Kratzer, W., Mason, R.A. and Kächele, V. (1999) Preva- lence of gallstones in sonographic surveys worldwide. Journal of Clinical Ultrasound, 27(1), 1-7. [2] Paigen, B. and Carey, M.C. (2002) Gallstones. In: The genetic basis of common disease. Oxford University Press, London, 298-335. [3] Ransohoff, D.F., Gracie, W.A., Wolfenson, L.B. and Neu- hauser, D. (1983) Prophylactic cholecystectomy or ex- pectant management for silent gallstones: Adecision analysis to assess survival. Annals of Internal Medicine, 99(2), 199-204. [4] The Southerm Surgeons Club (1999) A prospective analysis of 1518 laparoscopic cholecystectomies. New England Journal of Traditional Chinese Medicine, 324(16), 1073-1078. [5] BQS Bundesgeschäftsstelle Qualitätssicherung (2004) Qualität Sichtbar machen. BQS-Qualitätsbericht 2003, BQS, Dűsseldorf, 31-39. [6] Liver Disease Subcommittee of the Digestive Disease Interagency Coordinating Committee (2005) Action plan for liver disease research. NIH, Bethesda, 145-150. [7] Carey, M.C. and LaMont, J.T. (1992) Cholesterol gall- stone formation. 1. Physical chemistry of bile and biliary lipid secretion. Program for Liver Diseases, 10, 136-163. [8] Paumgartner, G. and Sauerbruch, T. (1991) Gallstones: Patho-genesis. Lancet, 338(8775), 1117-1121. [9] Lammert, F., Carey, M.C. and Paigen, B. (2001) Chro- mosomal organization of candidate genes involved in cholesterol gallstone formation: A murine gallstone map. Gastroenterology, 120(1), 221-238. [10] Wittenburg, H., Lyons, M.A., Li, R., Churchill, G.A., Carey, M.C. and Paigen, B.J. (2003) FXR and ABCG5/ ABCG8 as determinants of cholesterol gallstone forma- tion from quantitative trait locus mapping in mice. Gas- troenterology, 125(3), 868-881. [11] Moschetta, A., Bookout, A.L. and Mangelsdorf, D.J. (2004) Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nature Medicine, 10 (12), 1352-1358. [12] Wang, D.Q., Schmitz, F., Kopin, A.S. and Carey, M.C. (2004) Targeted disruption of the murine cholecystokinin-1  C.-M. Liu et al. / HEALTH 2 (2010) 1072-1077 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 1077 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. Journal of Clinical Investigation, 114(4), 521-528. [13] Pagliarulo, M., Fornari, F., Fraquelli, M., Zoli, F., Giangregorio, A., Grigolon, M., Peracchi, D. and Conte, D. (2004) Gallstone disease and related risk factors in a large cohort of diabetic patients. Digestive and Liver Disease, 36(2), 130-134. [14] Fraquelli, M., Pagliarulo, M., Colucci, A., Paggi, S. and Conte, D. (2003) Gallbladder mortility in obesity, dia- betes mellitus and coeliac disease. Digestive and Liver Disease, 35(S3), s12-s16. [15] Chen, C.Y., Lu, C.L., Lee, P.C., Wang, S.S., Chang, F.Y. and Lee, S.D. (1999) The risk factors for gallstone disease among senior citizens: An Oriental study. Hepatogastroen- terology, 46(27), 1606-1612. [16] Chan, D.C., Chang, T.M., Chen, C.J., Chen, T.W., Yu, J.C. and Liu, Y.C. (2004) Gallbladder contractility and vol- ume characteristics in gallstone dyspepsia. World Journal of Gastroenterology, 10(5), 721-724. [17] Portincasa, P., Moschetta, A., Berardino, M., Di-Ciaula, A., Vacca, M., Baldassarre, G., Pietrapertosa, A., Camma- rota, R., Tannoia, N. and Palasciano, G. (2004) Impaired gallbladder motility and delayed orocecal transit con- tribute to pigment gallstone and biliary sludge formation in betathalassemia major adults. World Journal of Gas- troenterology, 10(16), 2383-2390. [18] Mawe, G.M. (1998) Nerves and hormones interact to control gallbladder function. News in Physiological Sci- ences, 13(2), 84-90. [19] Patankar, R., Ozmen, M.M., Bailiey, I.S. and Johnson, C.D. (1995) Gallbladder motility, gallstones and the sur- geon. Digestive and Liver Disease, 40(11), 2323-2335. [20] Yu, P., De Prtris, G., Biancani, P., Amaral, J. and Behar, J. (1994) Cholecystokinin-coupled intracellular signaling in human gallbladder muscle. Gastroenterology, 106(3), 763- 770. [21] Rerup, C.C. (1970) Drugs producing diabetes through damage of the insulin secreting cells. Pharmacological Reviews, 22(2), 485-518. [22] Masiello, P., Broca, C., Gross, R., Roye, M., Manteghetti, M., Hillaire-Buys, D., Novelli, M. and Ribes, G. (1998) Experimental NIDDM: Development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes, 47(2), 224-229. [23] Dieh1, A.K. (1991) Epidemiology and natural history of gallstone disease. Gastroenterology Clinics of North America, 20(1), 1-19. [24] Mendez-Sanchez, N., Vega, H., Uribe, M., Guevara, L., Ramos, M.H. and Vargas-Vorackova, F. (1998) Risk fac- tors for gallstone disease in Mexicans are similar to those found in Mexican-Americans. Digestive and Liver Dis- ease, 43(5), 935-939. [25] Ruhl, C.E. and Everhart, J.E. (2000) Association of dia- betes, serum insulin, and C-peptide with gallbladder dis- ease. Hepatology, 31(2), 299-303. [26] Férézou, J., Combettes-Souverain, M., Souidi, M., Smith, J.L., Boehler, N., Milliat, F., Eckhardt, E., Blanchard, G., Riottot, M., Sérougne, C. and Lutton, C. (2000) Choles- terol, bile acid, and lipoprotein metabolism in two strains of hamster, one resistant, the other sensitive (LPN) to su- crose-induced cholelithiasis. Journal of Lipid Research, 41(12), 2042-2052. [27] Behar, J., Lee, K.Y., Thompson, W.R. and Biancani, P. (1989) Gallbladder contraction in patients with pigment and cholesterol stones. Gastroenterology, 97, 1479-1484. [28] Grider, J.R. and Makhlouf, G.M. (1990) Distinct recep- tors for cholecystokinin and gastrin on muscle cells of stomach and gallbladder. The American Journal of Physiology, 259(2), 184-190. [29] Xiong, D., Chang-You, L., Ying, M., Chang-An, L. and Yu-Jun, S. (2005) Correlation between gene expression of CCK-A receptor and emptying dysfunction of the gall- bladder in patients with gallstones and diabetes mellitus. Hepatobiliary & Pancreatic Diseases International, 4(2), 295-298. [30] Miyasaka, K., Kanai, S., Ohta, M., Hosoya, H., Sekime, A., Akimoto, S., Takiguchi, S. and Funakoshi, A. (2007) Ageassociated gallstone formation in male and female CCK-1(A) receptor-deficient mice. Journal of Gastroen- terology, 42(6), 493-496. |