 Crystal Structure Theory and Applications, 2012, 1, 84-91 http://dx.doi.org/10.4236/csta.2012.13016 Published Online December 2012 (http://www.SciRP.org/journal/csta) Enhancing Lab Source Anomalous Scattering Using Cr Kα Radiation for Its Potential Application in Determining Macromolecular Structures Sibi Narayanan, Devadasan Velmurugan* Centre of Advanced Study in Crystallography and Biophysics, University of Madras, Maraimalai (Guindy) Campus, Chennai, India Email: *shirai2011@gmail.com Received October 16, 2012; revised November 22, 2012; accepted November 29, 2012 ABSTRACT Obtaining phase information for the solution of macromolecular structures is a bottleneck in X-ray crystallography. Anomalous dispersion was recognized as a powerful tool for phasing macromolecular structures. It was used mainly to supplement the isomorphous replacement or to locate the anomalous scatterer itself. The first step in solving macro- molecular structures by SAD (single-wavelength anomalous diffraction) is the location of the anomalous scatterers. The SAD method for experimental phasing has evolved substantially in the recent years. A phasing tool, 5-amino-2,4,6- triiodoisophthalic acid (I3C—magic triangle), was incorporated into three proteins, lysozyme, glucose isomerase and thermolysin using quick-soaking and co-crystallization method in order to understand the binding of metal ion with proteins. Th e high qu ality of the diffraction da ta, the us e of chro mium anode X-ray radiation and the requ ired amount of anomalous signal enabled way for successful structure determination and automated model building. An analysis and/or comparison of the sulfur and iodine anomalous signals at the Cr Kα wavelength are discussed. Keywords: Anomalous Scattering; SAD Phasing; I3C; Lysozyme; Glucose Isomerase; Thermolysin 1. Introduction Current structural genomics projects aim to solve a large number of selected protein structures as fast as possible. High degree of automation and standardization is requir- ed at every step of the whole process to speed up protein structure determination. It is not easy to obtain auto- matically the crystal d erivatives, which is appropriate for phasing. New ideas have been put forward, that aim in making the phasing of novel structures easier and more susceptible to routine and automatic treatment [1]. Phase problem is a bottleneck in macromolecular structure determination and also in model building which is a time-consuming task. Phases can be derived from some knowledge of the molecular structure. Structures of small proteins (molecular weight less than 10 kDa) can be determined in solu tion using nuclear magnetic resonance (NMR) spectroscopy and the assembly of proteins in a complex can be studied using electron microscopy, but only X-ray diffraction helps in determining the three dimensional structure of small and large proteins with a precision of about 0.1 - 0.2 Å. In macromolecular cry- stallography, the phases are derived either by Molecular Replacement (MR) method using the atomic coordinates of a structurally similar protein or by locating the posi- tions of heavy atoms that are intrinsic to the protein or that have been added (MIR, MIRAS, SIR, SIRAS, MAD and SAD) [2-5]. MIR is a classic method of solving novel crystal struc- tures of macromolecules and has been responsible for an enormous amount of success of structural biology, since the early days of protein crystallography. MR also has been widely used when appropriate models are available. Over the past decade, MAD has been a vehicle of pro- gress in phasing new crystal structures. Both MIR and MAD require the presence of either appropriate heavy atoms or anomalous scatterers, which are naturally oc- curring or specifically introduced in the macromolecule [6]. The standard method of derivatization in MIR in- volves soaking or crystallizing the native crystals in di- luted solutions or heavy metal reagents. In MAD, sele- nium derivatization is carried out by genetic engineering using which the normally occurring Methionines (Met) are replaced as Se-Met. Both the approaches have draw- backs because of heavy-atom derivatization, which re- sults in non-isomorphism between the native and deri- vatized protein crystals [7]. Sometimes, several deriva- *Corresponding a uthor. C opyright © 2012 SciRes. CSTA  S. NARAYANAN, D. VELMURUGAN 85 tives are required to achieve success. By collecting Mul- tiple wavelength anomalous diffraction (MAD) data at two or more wavelengths, the definitive phase angle can be determined using MAD technique [8,9]. In comparison, only a single set of X-ray data is re- quired by Single wavelength anomalous diffraction tech- nique (SAD) technique to provide the positions of the anomalous scatterers, which together with density modi- fication can reveal the structure of the complete protein. The sulfur SAD phasing method allows the determina- tion of protein structure de novo without reference to derivatives such as Se-Met [10,11]. For targets, with a weak MR solution from which structure cannot be de- termined, the MR solution can be incorporated into the SAD experiment and the increased number of sites iden- tified by combined SAD/MR could be used in a subse- quent SAD experiment with no MR component to calcu- late the phases [12]. The number of protein structures be- ing phased using only a single set of diffraction data by SAD method has increased due to improved in-house X-ray sources, detectors and softwares. This technique has led to the routine use of anomalous scattering to ob- tain phase information from either intrinsic sulfurs or phosphorus presented in macromolecules or by addition of heavy metal reagents by soaking /co-crystallizing method in the native protein crystal [13,14]. In its purest form, SAD can simply utilize the intrinsic anomalous scatterers presented in the macromolecule, such as the sulfur atoms of cysteine and methionine or bound ions [15]. The chal- lenge is to maximize and measure the small signal, since the Bijvoet ratio can be as low as 1% [16,17]. Both cop- per and chromium anodes (wavelengths 1.54 Å and 2.29 Å) have been increasingly employed for the same pur- pose in laboratory X-ray sources with much success [18,19]. SAD phasing has already been carried out with anomalous scatterers such as mercury [20], uranium [21], iodine [22] and a tantalum bromide cluster [23], incor- porated into the crystal lattice. However, heavy atom derivatives suffer from nonspecific binding, which re- sults in low occupancy of the heavy-atom sites, which leads to weak anomalous signal and disruption of the crystal lattice and fail in derivatization. Surprisingly, it has been observed that short halide soaks can improve the crystal diffraction [24]. Many such soak s also requ ire the use of toxic chemicals and stringent safety precau- tions [25]. Exploiting the anomalous signal already pre- sented in the native protein or in the solvent would eli- minate the extra experimental work via derivatization and would also eliminate the risk of lack of isomorphism. Phasing using the anomalous signal of sulfur alone has earlier been achieved [26]. Longer wavelengths than Cu Kα (1.54 Å) would produce a larger signal, but at the same time experimental difficulties may increase as does the noise level in the data [27]. It has been recently re- ported that data collection wavelengths in the range of λ = 1.5 - 3.0 Å are fairly easy to handle in a diffraction ex- periment and even at home sources using instant Cr Kα radiation [28,29]. The use of chromium-anode X-ray radiation is very useful for SAD experiments. The anomalous scattering signal at this wavelength is more than doubled for vari- ous metals when compared to conventional copper charac- teristic wavelength. Furthermore, naturally bound metals and atoms from crystallization solutions tend to show a significant increase in anomalous scattering with chro- mium radiation [30]. Improved data quality helps in ex- ploiting the weak anomalous signal derived only from the sulfurs or in particular from halide ions incorporated by soaking. Therefore, a new class of compound 5-ami- no-2,4,6-triiodoisophthalic acid, (I3C) that combines heavy atoms for phasing with functional groups for their spe- cific interaction(s) with biological macromolecules was used to give rise to strong anomalous signals using in- house Cr Kα radiation. The I3C consists of three iodine atoms that are arranged in an equilateral triangle (6.1 Å per side each) (Figure 1). I3C has low toxicity when compared with other heavy reagents. I3C has been incorporated into three proteins viz., lysozyme (HEWL), glucose isomerase (GI) and ther- molysin (TL) for the present study using quick-soaking and co-crystallization method. Lysozyme and glucose isomerase contain higher amount of sulfurs than most proteins in the bacterial or eukaryotic proteomes provid- ing a favorable Bijvoet ratio [31]. Thermolysin contains lesser amount of sulfurs when compared to lysozyme and glucose isomerase. I3C was derivatized successfully us- ing soaking concentrations of 500 mM for lysozyme and 150 mM for glucose isomerase. I3C was also derivatized into thermolysin using co-crystallization method. The functional groups of the compound, interacts well with the proteins via hydrogen bonds. The strong anomalous signal of iodine atoms in the I3C makes it a powerful phasing tool for in-house data. 2. Methods 2.1. Crystallization The hen egg white lysozyme crystallization droplet Figure 1. Graphical representation of I3C. Copyright © 2012 SciRes. CSTA  S. NARAYANAN, D. VELMURUGAN 86 consisted of 2 µl protein solution (20 mg/ml) and 1 µl reservoir solution [50 mM Sodium Acetate and 1 M So- dium Chloride, pH 4.7] and was equilib rated again st 1 ml well solution at 25˚C. Glucose isomerase crystallization droplet consisted of 2 µl protein solution (33 mg/ml) and 1 µl reservoir solution [200 mM Magnesium chloride and 100 mM Tris, pH 4.7] and was equilibrated against 1ml well solution at 25˚C. Thermolysin crystallizatio n drop let consisted of 2 µl protein solution (25 mg/ml), 1 µl reser- voir solution [1.4 mM Calcium Acetate, 10 mM Zinc Acetate, 1 mM Sodium Nitrate and 50 mM Tris; pH 7.3], which was equilibrated against 1 ml well solution at 20˚C. Lysozyme, Glucose Isomerase and Thermolysin crystals appeared after a day and belonged to the tetragonal P43212, orthorhombic I222 and hexagonal P6122 space groups, respectively with one molecule per asymmetric unit. Protein crystals were obtained using the hanging drop vapour-diffusion method. Crystals of each protein were harvested for collecting native and I3C quick-soak- ed and co-crystallized datasets (500 mM I3C, 150 mM I3C and 300 mM I3C). Stock solutions of I3C with 1 M concentration were obtained by dissolving so lid materials in 2 M lithium hy- droxide solution [32] to deprotonate the carboxyl groups, thereby producing a salt with high solubility. If sodium or potassium hydroxide solution or an ammonia base is used, the resulting salt will have limited solubility. Ly- sozyme was soaked for about 45 seconds in 500 mM I3C solution. Glucose isomerase crystal was soaked and tried in 500 mM, 400 mM, 300 mM and 250 mM I3C for various time-periods, but the crystal degr aded even tually. Therefore, Glucose isomerase protein crystal was soaked for about 3 minu tes 10 second s (190 seco nds) in 150 mM I3C solution. The crystals were later back-soaked for 5 seconds in a cryosolution containing the same salt and buffer concentration with 30% Glycerol and 25% MPD (2-methyl-2,4-pentanediol) respectively. Thermolysin pro- tein crystal was grown by adding 0.5 µl of 300 mM I3C in the crystallization drop itself using co-crystallization method. The grown protein crystals of thermolysin with I3C were cryo-soaked in [10 mM Calcium Acetate, 7% (v/v) DMSO, 20% (v/v) Glycerol and 10 mM Tris; pH 7.3]. The crystals were later flash cooled in liquid nitro- gen (100 K). 2.2. Data Collection and Processing Six datasets (native and 500 mM I3C for lysozyme; na- tive and 150 mM I3C for glucose isomerase; native and 300 mM I3C for thermolysin) were collected separately using Rigaku R-Axis IV++ image plate detector equip- ed with Cr Kα (2.29 Å) anode X-ray generator operated at 45 kV and 45 mA. Crystals diffracted upto 2.53 Å and 360 frames were collected with crystal to detector dis- tance being 110 mm at 0.5˚ oscillation steps and 180 seconds exposure time per frame in each case. The inten- sities were integrated with the HKL2000 [33], refining all parameters including crystal mosaicity. Scaling and mergin g we re a l s o done with the same package. 3. Results and Discussion 3.1. Substructure Solution and Data Analysis The possibility of locating the anomalous scatterers using the dual-space recycling algorithm enabled in SHELXD depends on the signif icance of the anomalous signal pre- sented in the data [34-36]. For the location of substruc- tures of anomalous scatterers with SHELXD, only the internal loop which relies on the strongest E magnitudes is used. The success rate of SHELXD solutions critically depends on data quality and redundancy of their mea- surements. Using the direct methods program SHELXD, it was possible to obtain th e positions of anomalous scat- terers from the anomalous signal contained in all the dif- fraction data. Density modification with SHELXE [37,38] resulted in high-quality starting phases. Model building was performed with ARP/wARP [39] and refinement with REFMAC [40] available in CCP4i suite [41]. Fig- ures were prepared using PyMOL software [42]. At the chromium wavelength of 2.29 Å, sulfur atom has a value of 1.14, SHELXD program found seven sul- fur atoms and eight chloride ions (weaker peaks ap- peared in substructure solution), anomalous scatterers in the native lysozyme data (Figure 2(a)). Chlorine, the halide lighter than iodine, has its K edge at a long wave- length (4.39 Å) and disp lays only a small anomalous effect [43-46]. For native glucose isomerase data, anomalous scatterers for one manganese and nine sulfur atoms were obtained using SHELXD program and treated for phas- ing (Figure 2(b)). Similarly, for native thermolysin data, anomalous scatterers for one zinc ion, four calcium ions and two sulfur atoms were obtained using SHELXD pro- gram and treated directly for phasing (Figure 2(c)). They were given as input into SHELXE for obtaining the elec- tron density maps. The density modified final maps were subjected to analysis by ARP/wARP web server [47] for automatic chain tracing and model building. The electron density map allowed ARP/wARP program to build 122 residues out of a total of 129 amino acids for native ly- sozyme with four disulfide bridges. Similarly, 388 resi- dues were automatically built o ut of a total of 389 amino acids for native glucose isomerase with a single disulfide bridge. For native thermolysin, ARP/wARP program au- tomatically built 314 residues out of a total of 316 resi- dues. The iodine absorption edges retain a significant anomalous signal (f" = 12.82 e) at the chromium charac- teristic wavelength (2.29 Å). For I3C-soaked lysozyme dataset, substructure solution determined heavy-ato m site Copyright © 2012 SciRes. CSTA  S. NARAYANAN, D. VELMURUGAN 87 for iodines of I3C that formed an equilateral triangle and anomalous scatterers for eight sulfur atoms and eight chloride ions (Figure 3(a)). For I3C-soaked glucose isomerase dataset, anomalous scatterers for twelve sulfur atoms, one manganese ion and for an equilateral triangle (a) (b) (c) Figure 2. Anomalous map at 5 sigma level. (a) the peaks of seven sulfur atoms (big) and eight chloride ions (small) with water molecules in HEWL; (b) the peaks of nine sulfur at oms and one manganese ion with water molecules in GI and (c) peaks of four calcium ions, one zinc ion and tw o sulfur atom s with water molecules in TL. (a) (b) (c) Figure 3. Anomalous map at 5 sigma level. (a) the peaks of one I3C molecule and eight sulfur atoms (big) and eight chloride ions (small) with water molecules in HEWL; (b) the peaks of one I3C molecule and twelve sulfur atoms and one manganese ion with water molecules in GI; and (c) peaks of two calcium ions, one zinc ion, two sulfur atoms and one I3C molecule with water molecules in TL. (I3C) were determined using SHELXD (Figure 3(b)). Using SHELXD program, for I3C co-crystallized ther- molysin dataset, anomalous scatterers for two calcium ions, one zinc ion, two sulfur atoms and one I3C mole- cule were determined (Figure 3(c)). Density modifica- tion was carried out using SHELXE and the obtained modified map was given as input into ARP/wARP pro- gram. The final ARP/wARP conventional and free R Copyright © 2012 SciRes. CSTA  S. NARAYANAN, D. VELMURUGAN 88 factors obtained with REFMAC were 20.3% and 22.4% (lysozyme); 21.4% and 24.9% (glucose isomerase); 20.1% and 22.4% (thermolysin) respectively, wherein 123 resi- dues of 129 amino acids for lysozyme, 386 residues of the total of 389 protein amino acids for glucose isom- erase and 312 residues out of 316 amino acids for ther- molysin were safely built by the iterative free-atom den- sity modification and model-building procedure. Only halide sites corresponding to peaks higher than 5σ in the anomalous map were included. Solvent content of lysozyme, glucose isomerase and thermolysin are 37%, 55% and 46%, respectively. In all the cases discussed above, it was possible not only to locate the anomalous scatterers, but also subsequently to solve the protein model by SAD phasing. All the col- lected datasets are of good quality and th ey have close to 100% completeness. In all the above-mentioned struc- tures, the asymmetric unit contains only a monomer. Longer wavelengths provide not only an increased ano- malous signal for phase determination, but also allow a much clearer definition of substructures; their positions and occupancies, which may turn out to be very impor- tant for elucidating the function of a molecule. 3.2. Native Sulfur Binding Sites By contrast, sulfur is presented in almost all proteins. It is heavier than any other elements (C, N, and O) found in most proteins and displays some anomalous signal. Phas- ing a protein through only the inherent anomalous signal derived from the sulfur atoms presented in both cysteines and methionines presented in the ordered solvent region was possible for lysozyme and glucose isomerase data sets with redundancy of 10 and above at wavelength of 2.29 Å. The structure of the 129 residue, tetragonal ly- sozyme (P43212) was phased using only the anomalous signal derived from seven sulfurs in the protein and eight coordinated chloride anions with high redundancy. Simi- larly, the structure of orthorhombic glucose isomerase was phased using one manganese and nine sulfur atoms from eight methionines and one cysteine from the 389 residues presented in the protein. The structure of 314 residue, hexagonal thermolysin (P6122) was phased using the anomalous signal derived from two sulfur atoms, one zinc ion and four calcium ions. The presence of metal ions originated from the crys- tallization buffer used for each protein crystallization setup. 3.3. I3C Binding Sites One binding site for I3C each was observed in lysozyme, glucose isomerase and thermolysin, respectively. The oc- cupancies for all three halogen atoms per site were (0.70, 0.66 & 0.62) for lysozyme (0.65, 0.60 & 0.56) for glu cose isomerase and (0.81, 0.77 & 0.71) for thermolysin. Interestingly, the occupancy values of both the proteins differ lightly, although similar soaking conditions were tried. The data from the I3C derivative showed signifi- cant anomalous signal to noise ratio (1.78% for lysozyme, 1.82% for glucose isomerase and 1.45% for thermolysin) throughout the entire resolution range to 2.53 Å. The interactions of the I3C in lysozyme are very similar to those previously reported [21]. They mostly replace wa- ter molecules in the crystal lattice. Inspection of the I3C sites using PyMOL software showed several Hydrogen bond interactions. The three functional groups of the phasing molecule formed hydrogen bonds with the side or the main chains of the amino acids. One carboxyl group interacts with an Arginine residue (ARG 114). The same carboxyl group interacts with oxygen and nitrogen atoms of the Asparagine residue (ASN 37) (bifurcation) via water molecules. Hydroxyl group of the I3C mole- cule also interacts with the nitrogen presented in the Ly- sine residue (L YS 33) (Figure 4(a)). (a) (b) (c) Figure 4. I3C binding site and interactions (a) HEWL; (b) GI; and (c) TL. Copyright © 2012 SciRes. CSTA 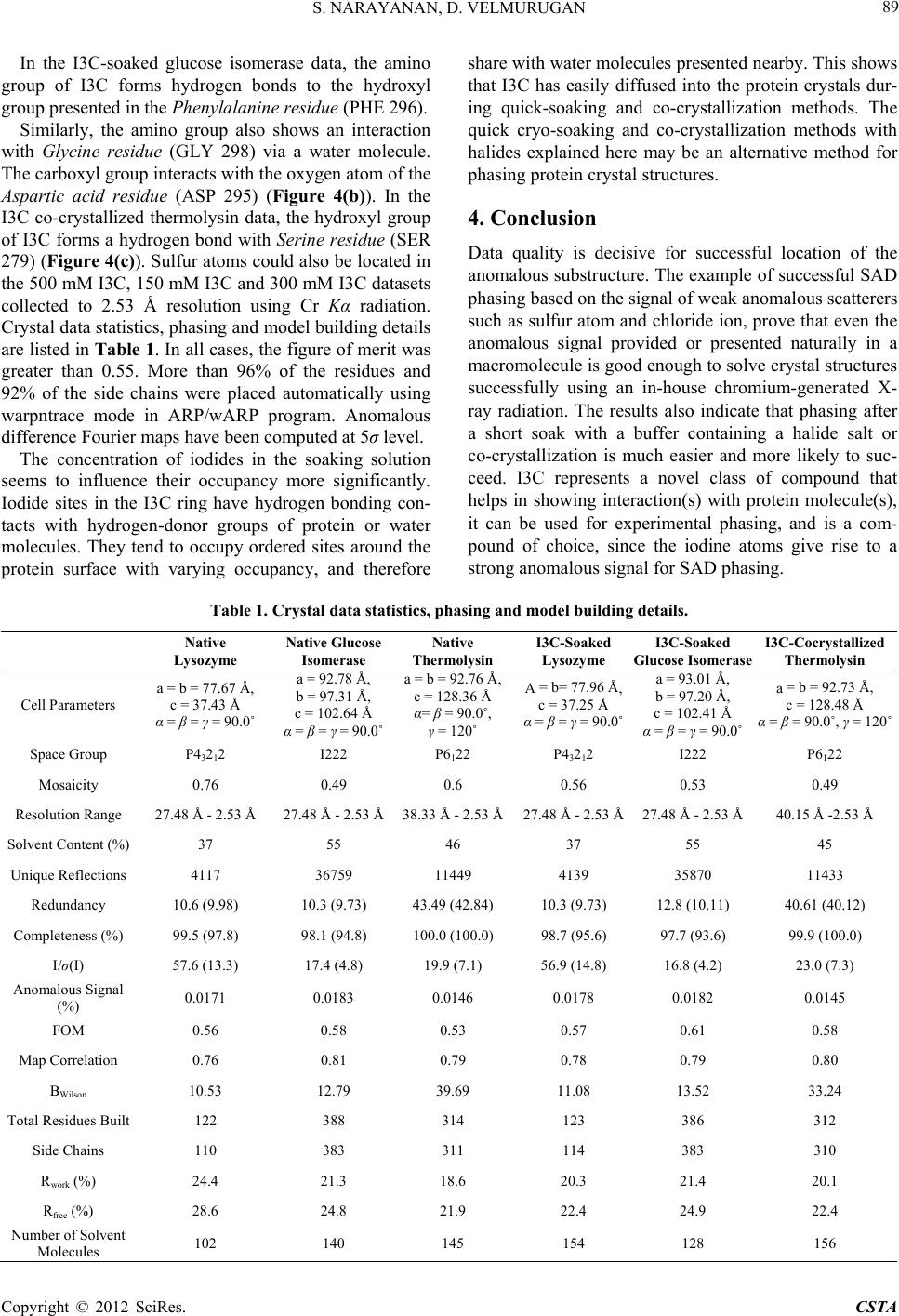 S. NARAYANAN, D. VELMURUGAN Copyright © 2012 SciRes. CSTA 89 aked glucose isomerase data, the amino gr w se Table 1. Crystal data statistics, phasing and model building details. Native L Thin ed Gl I3C-Cocrystallized In the I3C-so oup of I3C forms hydrogen bonds to the hydroxyl group presented in the Phenylalanine residue (PHE 296). Similarly, the amino group also shows an interaction ith Glycine residue (GLY 298) via a water molecule. The carboxyl group interacts with the oxygen atom of the Aspartic acid residue (ASP 295) (Figure 4(b)). In the I3C co-crystallized thermolysin data, the hydroxyl group of I3C forms a hydrogen bond with Serine residue (SER 279) (Figure 4(c)). Sulfur atoms could also be located in the 500 mM I3C, 150 mM I3C and 300 mM I3C datasets collected to 2.53 Å resolution using Cr Kα radiation. Crystal data statistics, phasing and model building details are listed in Table 1. In all cases, the figure of merit was greater than 0.55. More than 96% of the residues and 92% of the side chains were placed automatically using warpntrace mode in ARP/wARP program. Anomalous difference Fourier maps have been computed at 5σ level. The concentration of iodides in the soaking solution ems to influence their occupancy more significantly. Iodide sites in the I3C ring have hydrogen bonding con- tacts with hydrogen-donor groups of protein or water molecules. They tend to occupy ordered sites around the protein surface with varying occupancy, and therefore share with water molecules presented nearby. This shows that I3C has easily diffused into the protein crystals dur- ing quick-soaking and co-crystallization methods. The quick cryo-soaking and co-crystallization methods with halides explained here may be an alternative method for phasing protein crystal structures. 4. Conclusion Data quality is decisive for successful location of the anomalous substructure. The example of successful SAD phasing based on the signal of weak anomalous scatterers such as sulfur atom and chloride ion, prove that even the anomalous signal provided or presented naturally in a macromolecule is good enoug h to solve crystal structures successfully using an in-house chromium-generated X- ray radiation. The results also indicate that phasing after a short soak with a buffer containing a halide salt or co-crystallization is much easier and more likely to suc- ceed. I3C represents a novel class of compound that helps in showing interaction(s) with protein molecule(s), it can be used for experimental phasing, and is a com- pound of choice, since the iodine atoms give rise to a strong an omalous signal fo r S AD phasin g. ysozyme Native Glucose Native I3C-Soaked I3C-Soak Isomerase ermolys Lysozyme u cose Is omeraseThermolysin Cell Parameters a =, α ˚ α aA , α ˚α a α = 20˚ b = 77.67 Å c = 37.43 Å = β = γ = 90.0 a = 92.78 Å, b = 97.31 Å, c = 102.64 Å = β = γ = 90.0˚ = b = 92.76 Å, c = 128.36 Å α= β = 90.0˚, γ = 120˚ = b= 77.96 Å c = 37.25 Å = β = γ = 90.0 a = 93.01 Å, b = 97.20 Å, c = 102.41 Å = β = γ = 90.0˚ = b = 92.73 Å, c = 128.48 Å β = 90.0˚, γ = 1 Space Group 3 13 11 Resolution Range 27.48 Å -53 Å 27.48 Å -53 Å38.33 Å -53 Å27.48 Å -53 Å27.48 Å -53 Å 40.15 Å -.53 Å S 4117 314139 31 10. 10. 43.49 ( 10. 12 . 40. Co) Anomignal Map Cor Total Residues Built wor k Nument Molecules P4 22 I222 P6122 P422 I222 P622 Mosaicity 0.76 0.49 0.6 0.56 0.53 0.49 2. 2. 2. 2. 2.2 olvent Content (%) 37 55 46 37 55 45 Unique Reflections 6759 1449 5870 1433 Redundancy 6 (9.98)3 (9.73)42.84)3 (9.73)8 (10.11)61 (40.12) mpleteness (%99.5 (97.8) 98.1 (94.8) 100 .0 (100.0) 98.7 (95.6) 97.7 (93.6) 99.9 (100.0) I/σ(I) 57.6 (13.3) 17.4 (4.8) 19.9 (7.1) 56.9 (14.8) 16.8 (4.2) 23.0 (7.3) alous S (%) FOM 0.0171 0.0183 0.0146 0.0178 0.0182 0.0145 0.56 0.58 0.53 0.57 0.61 0.58 relation 0.76 0.81 0.79 0.78 0.79 0.80 BWilson 10.53 12.79 39.69 11.08 13.52 33.24 122 388 314 123 386 312 Side Chains 110 383 311 114 383 310 R (%) 24.4 21.3 18.6 20.3 21.4 20.1 Rfree (%) 28.6 24.8 21.9 22.4 24.9 22.4 ber of Solv102 140 145 154 128 156  S. NARAYANAN, D. VELMURUGAN 90 5 emen overnment of India for the arch. DV thanks DST-FI ST ENCES [1] W. A. HendriPhase Determina- tion from Mus Diffraction Mea- . Acknowledgts SN and DV thank UGC, G financial suppor t for this rese and UGC-SAP for funding facilities to the Centre for Advanced Study in Crystallography and Biophysics. Chromium datasets were collected at X-ray facility, CCMB, Hyderabad funded by CSIR Facility Creation Project (FAC0004) as part of Eleventh Five Year Plan. SN and DV thank Dr. R. Shankaranarayanan for extend- ing his lab facilities to collect anomalous scattering d ata- sets using Cr Kα radiation. REFER ckson and C. M. Ogata, “ ltiwavelength Anomalou surements,” Methods in Enzymology, Vol. 276, 1997, pp. 494-523. doi:10.1016/S0076-6879(97)76074-9 [2] M. F. Perutz, “Isomorphous Replacement and Phase De- termination in Non-Centrosymmetric Space Groups,” Acta Crystallographica, Vol. 9, No. 11, 1956, pp. 867-873. doi:10.1107/S0365110X56002485 [3] J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H Wyckoff and D. C. Philli ps, “A Three. -Dimensiona l Model of the Myoglobin Molecule Obtained by X-Ray Analy- sis,” Nature, Vol. 181, No. 4610, 1958, pp. 662-666. doi:10.1038/181662a0 [4] D. Blow, “Outline of Crystallography for Biologist Oxford University Press, s,” Oxford, 2002. , 1956, pp. 901- [5] F. H. C. Crick and B. S. Magdoff, “Expected Signal,” Acta Crystallographica, Vol. 9, No. 11 908. doi:10.1107/S0365110X56002552 [6] S. Trakhanov and F. A. Quiocho, “Influence of Divalent Cations in Protein Crystallization,” Protein Science, Vol. 9, No. 4, 1995, pp. 1914-1919. doi:10.1002/pro.5560040925 [7] U. A. Ramagopal, M. Dauter and Z. D Anomalous Signal of Sulfursauter, “Phasing on : What Is the Limit?” Acta Crystallographica, Vol. D59, Part 6, 2003, pp. 1020-1027. doi:10.1107/S0907444903007467 [8] J. Karle, “Some Developments in Anomalous Dispersion for the Structural Investigation of Macromolecular Sys- ination Based on Anomalous Scattering,” tems in Biology,” International Journal of Quantum Chemistry: Quantum Biology Symposium, Vol. 7, 1980, pp. 357-367. [9] W. A. Hendrickson, J. L. Smith and S. Sheriff, “Direct Phase Determ Methods in Enzymology, Vol. 115, 1985, pp. 41-55. doi:10.1016/0076-6879(85)15006-8 = [10] B.-C. Wang, “Resolution of Phase Ambiguity in Ma molecular Crystallography,” Methods in cro- Enzymology, Vo l. 115, 1985, pp. 90-111. doi:10.1016/0076-6879(85)15009-3 [11] K. D. Cowtan and K. Y for Macromolecular Phase Improve . Zhang, “Density Modification ment,” Progress in Biophysics and Molecular Biology, Vol. 72, No. 3, 1999, pp. 245-270. doi:10.1016/S0079-6107(99)00008-5 & Cognition, Vol. 35, No. 7, 2007, pp. 1801-1813. [12] A. Dieckmann and J Rieskamp, “The Influe mation R on Probabilistic Inferences,” Memory nce of Infor- edundancy doi:10.3758/BF03193511 [13] K. N. Vennila and D. Velmurugan, “In-House SAD Phasing with Surface-Bound Cerium Ions,” Acta Crys- tallographica, Vol. F67, No. 7, 2011, pp. 1662-1665. doi:10.1107/S1744309111035718 [14] M. Yogavel, et al., “Structural Analysis of Actinidin and a Comparison of Cadmium and Sulfur Anomalous Sig- nals from Actinidin Crystals Measured Using In-House Copper- and Chromium-Anode X-Ray Sources,” Acta Crystallographica, Vol. D66, No. 12, 2010, pp. 1323-1333. doi:10.1107/S0907444910040394 [15] D. Velmurugan, et al., “Ab Initio Structure Determination of the Triple Mutant (K53,56,121 M) of Bovine Pancre- atic Phospholipase A2 at Atomic and High Resolution Using ACORN,” Current Science, Vol. 90, No. 8, 2006, pp. 1091-1099. [16] W. A. Hendrickson and M. M. Teeter, “Structure of the Hydrophobic Protein Crambin Determined Directly from the Anomalous Scattering of Sulphur,” Nature, Vol. 290, No. 5802, 1981, pp. 107-113. doi:10.1038/290107a0 [17] M. Yogavel, J. Gill and A. Sharma, “Iodide-SAD, SIR and SIRAS Phasing for Structure Solution of a Nucle- osome Assembly Protein,” Acta Crystallographica, Vol. D65, 2009, pp. 618-622. doi:10.1107/S0907444909013171 [18] J. E. Debreczeni, G. Bunkóczi, B. Girmann and G. M. Sheldrick, “In-House Phase Determination of the Lima Bean Trypsin Inhibitor: A Low-Resolution Sulfur-SAD Case,” Acta Crystallograph 393-395. ica, Vol. D59, No. 2, 2003, pp. 20917doi:10.1107/S09074449020 . 8, 2007, pp. [19] M. Yogavel, J. Gill, P. C. Mishra and A. Sharma, “SAD Phasing of a Structure Based on Cocrystallized Iodides Using an In-House Cu Kalpha X-Ray Source: Effects of Data Redundancy and Completeness on Structure Solu- tion,” Acta Crystallographica, Vol. D63, No 931-934. doi:10.1107/S0907444907029174 [20] T. Beck, G. Tim and G. M. Sheldrick, “The Magic Trian- gle Goes MAD: Experimental Phasing with a Bromine Derivative,” Acta Crystallographica, Vol. D66, No. 4, 2010, pp. 374-380. doi:10.1107/S0907444909051609 [21] T. Beck, C. E. Cunha and G. M. Sheldrick, “How to Get the Magic Triangle and the MAD Triangle into Your Pro- tein Crystal,” Acta Crystallographica, Vol. F65, No. 10, 2009, pp. 1-3. doi:10.1107/S1744309109036884 [22] T. J. Boggon and L. Shapiro, “Screening for Phasing Atoms in Protein Crystallography,” Structure, Vol. 8, No. 7, 2000, pp. R143-R149. doi:10.1016/S0969-2126(00)00168-4 [23] K. Wernimont, D. L. Huffman, A. L. Lamb, T. V. ,” Nature, Vol. 7, No. 9, 2000, O’Halloran and A. C. Rosenzweig, “Structural Basis for Copper Transfer by the Metallochaperone for the Men- kes/Wilson Disease Proteins pp. 766-771. doi:10.1038/78999 [24] Z. Dauter, M. Dauter and K. R. Rajashankar, “Novel Approach to Phasing Proteins: Derivatization by Short Copyright © 2012 SciRes. CSTA  S. NARAYANAN, D. VELMURUGAN 91 Cryo-Soaking with Halides,” Acta Crystallographica, Vol. D56, No. 2, 2000, pp. 232-237. doi:10.1107/S0907444999016352 [25] R. H. Szczepanowski, R. Filipek and M. Bochtler, “Crys- tal Structure of a Fragment of Mouse Ubiquitin Acti- vating Enzyme,” The Journal of Biological Chemistry, Vol. 280, No. 50, 2005, pp. 22006-22011. doi:10.1074/jbc.M502583200 [26] M. Harrel, et al., “The Binding Site of Acetylcholin Phase: Us Structure Determination,” e Receptor as Visualized in the X-Ray Structure of a Com- plex between Alpha-Bungarotoxin and a Mimotope Pep- tide,” Neutron, Vol. 32, No. 2, 2001, pp. 265-270. [27] Z. Dauter and M. Dauter, “Entering a New Solvent Halide Ions in Proteining Structure, Vol. 9, No. 2, 2001, pp. R21-R26. doi:10.1016/S0969-2126(01)00565-2 [28] W. A. Hendrickson and M. M. Teeter, “Characterization of the Aromatic Proton Magnetic Resonance Spectrum of Crambin,” Nature, Vol. 290, No. 5802, 1981, pp. 107-113. doi:10.1038/290107a0 [29] D. K. Carugo, J. R. Helliwell, H. Stuhrmann Weiss, “Softer and Soft X-Rays in Macrand omolecular Crys- M. S. tallography,” Journal of Synchrotron Radiation, Vol. 12, No. 4, 2005, pp. 410-419. doi:10.1107/S0909049504025762 [30] M. Cianci, et al., “Structure of Lobster Apocrustacyanin A1 Using Softer X-Rays,” J. R. Acta Crystallographica, Vol. D59, No. 9, 2001, pp. 1219-1229. doi:10.1107/S0907444901009350 [31] C. Yang, J. W. Pflugarth, D. A. Courville and J. D. Ferrara, “Away from th, C. N. Stenc e Edge: SAD Phasing e from the Sulfur Anomalous Signal Measured In-House with Chromium Radiation,” Acta Crystallographica, Vol. D59, No. 11, 2003, pp. 1943-1957. doi:10.1107/S0907444903018547 [32] J. H. Naismith, et al., “Refined Structure of Cadmium- Substituted Concanavalin A at 2.0 A Resolution,” Acta Crystallographica, Vol. D49, No. 6, 1993, pp. 561-571. doi:10.1107/S0907444993006390 [33] T. Beck, A. Krasauskas, T. Gruene and “A Magic Triangle for Experimental Phasing G. M. Sheldrick, of Macro- molecules,” Acta Crystallographica, Vol. D64, No. 11, 2008, pp. 1-4. doi:10.1107/S0907444908030266 [34] G. M. Sheldrick, “Experimental Phasing with SHELXC/D/E: Combining Chain Tracing with Dens ity Modification, m International Tables for Crystallo- o. 5, 1999, pp. 643-648. ” Acta Crystallographica, Vol. D66, No. 4, 2010, pp. 479-485. [35] Z. Otwinowski and W. Minor, “Processing of X-Ray Diffraction Data Collected in Oscillation Mode,” In: C. W. Carter, Jr. and R. M. Sweet, Eds., Methods in Enzy ology, Macromolecular Crystallography, Part A, New York, 1997, pp. 307-326. [36] G. M. Sheldrick, H. A. Hauptman, C. M. Weeks, M. Miller and I. Uson, “Ab Initio Phasing,” In: E. Arnold and M. Rossman, Eds., graphy, IUCr and Kluwer Academic Publishers, Dor- drecht, 2001, pp. 333-351. [37] I. Uson and G. M. Sheldrick, “Advances in Direct Meth- ods for Protein Crystallography,” Current Opinion in Structural Biology, Vol. 9, N doi:10.1016/S0959-440X(99)00020-2 [38] T. R. Schneider and G. M. Sheldrick, “Substructure Solu- tion with SHELXD,” Acta Crystallographica, Vol. D5 No. 10, 2002, pp. 1772-1779. 8, doi:10.1107/S0907444902011678 [39] A. Perrakis, R. J. Morris and V. S. Lamzin, “Automated Protein Model Building Comb ture Refinement,” Nature Structura ined with Iterative Struc- l Biology, Vol. 6, No. 5, 1999, pp. 458-463. doi:10.1038/8263 [40] G. N. Murshudov, A. A. Vagin and E. J. Dodson, “Re- finement of Macromolecular Structures by the Maxi- mum-Likelihood Method,” Acta Crystallographica, Vol. D53, No. 3, 1997, pp. 240-255. doi:10.1107/S0907444996012255 [41] CCP4 (Collaborative Computational Project, Number 4), “The CCP4 Suite: Programs for P Acta Crystallographica, Vol. D50, rotein Crystallography,” No. 5, 1994, pp. 760- 763. doi:10.1107/S0907444994003112 [42] W. L. DeLano, “The PyMOL Molecular Graphics Sys- tem,” DeLano Scientific, San Carlos, 1998. [43] L. Q. Chen, et al., “Crystal Structure of a Bovine Neuro- ng Signal of physin II Dipeptide Complex at 2.8 A Determined from the Single-Wavelength Anomalous Scatteri an Incorporated Iodine Atom,” Proceedings of the Na- tional Academy of Sciences of the USA, Vol. 88, No. 10, 1991, pp. 4240-4244. doi:10.1073/pnas.88.10.4240 [44] L. Brady, “A Serine Protease Triad Forms the Catalytic Centre of a Triacyl glyce rol Lipase, ” Nature, Vol. 343, No. 6260, 1990, pp. 767-770. doi:10.1038/343767a0 [45] Lehmann, “Location of the Sulfur Atoms from the Phased Anomalous Map Using Native Protein Data Can Be Very Helpful in Tracing the Peptide Chain,” Ph.D. Dissertation, ol. D57, No. 7, 2001, pp. University of Gottingen, 2000. [46] P. J. Loll, “De Novo Structure Determination of Vanco- mycin Aglycon Using the Anomalous Scattering of Chlo- rine,” Acta Crystallographica, V 977-980. doi:10.1107/S0907444901007314 [47] G. G. Langer, S. X. Cohen, A. Perrakis and V. S. Lamzin, “Automated Macromolecular Model Building for X-Ray Crystallography Using ARP/wARP Version 7,” Nature Protocols, Vol. 3, No. 7, 2008, pp. 1171-1179. doi:10.1038/nprot.2008.91 Copyright © 2012 SciRes. CSTA
|