Journal of Cancer Therapy
Vol. 4 No. 1 (2013) , Article ID: 28187 , 15 pages DOI:10.4236/jct.2013.41041
The 2-Series Eicosanoids in Cancer: Future Targets for Glioma Therapy?
![]()
Division of Neurosurgery, St. Michael’s Hospital, Toronto, Canada.
Email: *DasS@smh.ca
Received December 8th, 2012; revised January 10th, 2013; accepted January 18th, 2013
Keywords: Eicosanoids; Prostaglandins; Prostacyclins; Thromboxanes; Cancer; Glioma
ABSTRACT
The 2-series eicosanoids are structurally related lipid-soluble hormones synthesized by cyclooxygenase enzymes from arachidonic acid. These compounds have well-established roles in the inflammatory response and the coagulation cascade. More recently, the eicosanoids have garnered attention for their potential roles in cancers of the lung, colon, breast, and brain. In this paper, we review the contributions of the different cyclooxygenase metabolites (i.e. prostaglandins, prostacyclins and thromboxanes) to cancer development, progression and recurrence, with special attention paid to their relevance to glioma biology. Our review suggests that 2-series eicosanoids merit further study as possible targets for therapy in patients with glioma.
1. Introduction
Despite advances in surgical technique, available chemotherapies, and radiation therapy, the prognosis for patients diagnosed with a glioma remains grim. Median survival for patients with glioblastoma treated with aggressive multi-modality therapy is fourteen months [1]. While significant attention has been given to the genetic changes that underlie gliomagenesis, recent work has also focused on the importance of signalling within the tumor milieu and intratumoural communication in glioma development, progression and recurrence. In this review, we will focus on eicosanoid signalling and its possible role in glioma biology.
Eicosanoid signaling has long been a therapeutic target in inflammatory conditions. More recent research has delineated a role for eicosanoids in the development and progression of multiple cancers, including those of the breast [2], lung [3], colon [4], kidney [5], prostate [6] and brain [7]. Eicosanoids have been proposed to activate oncogenes [8] and the epithelial-to-mesenchymal transition (EMT) [9], to inhibit tumor suppressor genes [10], to participate in tumor cell evasion of the immune response [3], and to initiate angiogenesis [4]. In Table 1, we highlight the known roles of the 2-series eicosanoids in CNS and systemic cancers.
Eicosanoid synthesis begins with phospholipase A2, which releases arachidonic acid (AA) from membranebound phospholipids. AA is subsequently converted to prostaglandin H2 (PGH2) by the cyclooxygenase enzymes (COX-1, COX-2 and COX-3), which are also known as prostaglandin H synthase (PGHS) [11]. PGH2 subsequently serves for the substrate for the 2-series eicosanoids, a group of compounds that includes prostaglandins-D2 (PGD2), E2 (PGE2), F2α (PGF2α), and J2 (PGJ2)- prostacyclin (PGI2) and thromboxane (TxA2) (Figure 1). Owing to their inherently unstable chemical structure, eicosanoids decay rapidly and are thus only able to mediate local (i.e. paracrine or autocrine) signaling. The 2-series eicosanoids signal in one of two ways: they either activate a G protein-coupled receptor (GPCR) [3]— which in turn affects the levels of second messengers like cyclic adenosine monophosphate (cAMP) or calcium (Ca2+)—or bind to nuclear receptors that alter DNA transcription [12,13]. Given the diverse roles of eicosanoids in human disease, there has been significant research in developing new drugs that can modulate eicosanoid signaling in a selective manner.
Gliomas represent a unique therapeutic challenge in part because they are chemically isolated from the rest of the body. In the context of cancer therapy, the bloodbrain barrier (BBB) is a significant obstacle as it can prevent chemotherapeutic agents that are active in the periphery from achieving therapeutic concentrations in the CNS. The drug tamoxifen, for example, is an agent that is profoundly effective in the treatment of metastatic breast cancer, but that has limited efficacy against brain
Table 1. Reported associations of eicosanoids in tumor biology.

metastases because it is excluded from the CNS by the BBB[14]. Due to their lipid-soluble structure, derivatives of 2-series eicosanoids have the potential to cross the BBB and overcome this problem. In the following sections, we discuss the signaling pathways associated with COX and each of the 2-series eicosanoids, and compare their roles in systemic cancers and glioma. In Table 2, we list pharmacologic agents that have been used to target different eicosanoid pathways.
1.1. Role of COX in Systemic Cancers
Early studies recognized that growth factors, tumor promoters, and oncogenes all induce prostaglandin synthesis [15]. More recent studies have demonstrated that COX-2 has an important role in cancer generation and progression, but that the role of prostaglandins varies in a tumor-specific manner. Analysis of normal and neoplastic human breast tissue samples has shown that COX-2 expression correlates with expression of oncogenes such as HER-2/neu [16]. COX-2 may also contribute to drug resistance in MCF-7 breast cancer cells via concomitant effects on the phosphoinositide-3-kinase (PI3K)/Akt, mitogen-activated protein kinase (MAPK), epidermal growth factor receptor (EGFR), and matrix metalloproteinase- 2(MMP2) and -9(MMP9) pathways [8]. For example, pharmacologic inhibition of COX-2 decreased invasiveness of MDA-MB-231 human breast cancer cells by preventing MMP2 release [17]. Furthermore, transgenic loss of COX-2 delayed tumor progression in a mouse mammary epithelial cell model of breast cancer. These effects were driven by COX-2 genetic deletion, which resulted in an enhanced host immune response, and could be overcome by providing exogenous PGE2 (a product of COX-2 activity) [18].
Tumor-induced immune modulation is similarly relevant incolon cancer, where inhibition of the COX-2/ PGE2 pathway decreases the levels of FoxP3+ regulatory T cells (Tregs) and results in an enhanced antitumor immune response [19]. Long-term COX-2 inhibition also appears to have protective benefits against non-small cell lung [20] and colon [21] cancers, suggesting a more general role for COX-2 in immune surveillance against neoplastic cells.
The role of COX-2 in tumor biology appears to extend beyond the immune response. COX-2 inhibitors can induce the expression of tumor-suppressors such as MAGI1 [22] in colorectal cancer cells (SW480 and HCT116) and 15-hydroxyprostaglandin dehydrogenase (15-PGDH) in

Figure 1. The 2-Eicosanoid signaling family.
colon (HT29), lung (A549) and glioblastoma (T98G) cancer cells [23]. In addition, COX-2 has a direct effect on cell proliferation and survival in SGC-7901 and AGS gastric cancer cells [24]. In HepG2 human hepatocellular carcinoma cells, COX-2 inhibition also results in decreased expression of the drug efflux pumps P-glycoprotein and MRP1 [25]. Thus, COX-2 inhibitors might render tumor cells more sensitive to chemotherapy. Further, in HepG2 cells, COX-2 inhibition with nonsteroidal anti-inflammatory drugs (NSAIDs) can induce apoptosis through oxidative stress and mitochondrial toxicity [26].
The pro-apoptotic effect of COX-2 inhibition on hepatocellular carcinoma cells does not, however, hold true for all cancers. Treatment with NSAIDs appeared to protect U937 human hematopoietic cancer cells from apoptosis [27]. Furthermore, it is not universally true that COX-2 expression correlates with cell proliferation. In a T24 bladder cancer cell model of interstitial cystitis, anti-proliferative factor (APF) inhibits cell proliferation by decreasing β-catenin expression, which results in increased COX-2 expression [28]. The data from T24 cells provides the first example of a cancer cell line where increased COX-2 expression is associated with decreased proliferation. This observation stands in stark contrast to the cases of breast and colon cancers previously discussed where COX-2 activity drives cellular proliferation.
1.2. Role of COX in Glioma
Both COX-1 and COX-2 are expressed in glioma cells
Table 2. Therapeutic modulation of eicosanoid signaling.
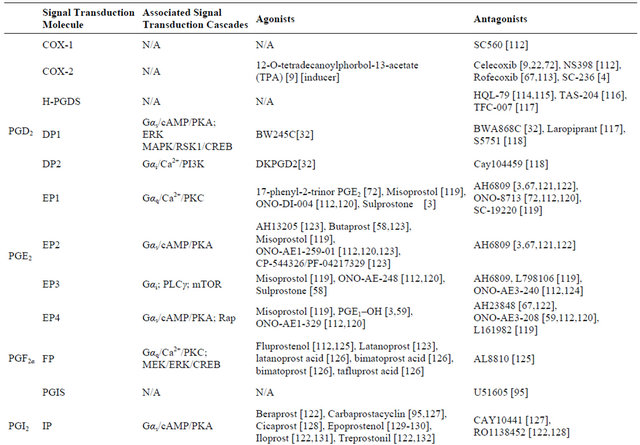

and expression levels of COX-2 increase with glioma grade [29]. COX-2 expression has been found to correlate negatively with survival in human astrocytomas and can thus be considered a poor prognostic indicator [29]. A phase II study of temozolomide, thalidomide and celecoxib combination therapy in glioblastoma patients unfortunately failed to demonstrate a statistically significant improvement in patient survival [30]. One reason why this study may not have shown a survival benefit is that patients did not receive temozolomide during radiotherapy, which is now considered the standard of care. It remains to be determined whether the combination of temozolomide, concomitant radiation therapy and a prostaglandin signaling modulator would confer a survival benefit over the current standard-of-care.
2. PGD2 Signaling
PGD2 is derived from PGH2 via the action of prostaglandin D synthases (PGDS), of which there are lipocalin (L-PGDS) and hematopoietic (H-PGDS) subtypes. L-PGDS shares a structural homology with other members of the lipocalin family, which are extracellular proteins that bind to a lipophilic substrate. Interestingly, H-PGDS is a sigma-class glutathione transferase [31], suggesting that PGD2 synthesis could be regulated by environmental oxidative stress through depletion of glutathione (GSH). PGD2 can either signal directly by binding to its cognate receptor or by being converted to PGJ2, the actions of which will be discussed later. PGD2 signaling is mediated by two receptors: DP1 (or DP) and DP2 (also called CRTH2), which are both GPCRs. DP1 initiates a Gαs/cAMP/protein kinase A (PKA) cascade, while DP2 signals through Gαi proteins that mobilize Ca2+ store and actives PI3K [32].
2.1. PGD2 in Systemic Cancers
In mice implanted with Lewis lung cancer cells, administration of a synthetic DP1 agonist impairs angiogenesis [33]. PGD2 appears to inhibit prostate cancer cell proliferation in tumors expressing aldoketoreductase 1C3 (AKR1C3, also called 17β-hydroxysteroid dehydrogenase (17β-HSD) type 5) [34]. Likewise, a study of tumor-prone ApcMin/+ mice showed that high levels of HPGDS can suppress colon tumorigenesis [35]. Together, these observations establish PGD2 as a tumor-suppressing molecule that could be exploited as an anti-cancer and anti-angiogenesis therapeutic.
2.2. PGD2 in Glioma
PGD2 has long been known to inhibit cell proliferation in glioma cell lines (NCE-G 2,3,7,8,17) in vitro [7] . More recently, loss of L-PGDS expression was found to be a defining event in the progression of Grade II to Grade III astrocytomas; further, PDGS expression was noted to vary inversely with survival across all glioma grades [36]. Treatment of A172 glioma cells in culture with PGD2 inhibits cell proliferation and induces apoptosis; these effects were amplified by concomitant inhibition of COX-2 [36]. Interestingly, PKC, which is driver of EMT [37], activates PGDS in TE671 medulloblastoma cells [38]. Thus, the effect of PGD2 appears to be tumor-specific even among tumors of the CNS. Regardless, these findings establish a potential role for exogenous PGD2 analogues in the treatment of glioma.
3. PGJ2 Signaling
15-deoxy-Δ12,14-prostaglandin J2 (15-d-PGJ2) is produced in vivo by the metabolism of PGD2. 15-d-PGJ2 signals via the DP2 and PPARγ receptors, but also has direct effects on glycolytic enzymes, molecular chaperones and cytoskeletal proteins in neuronal membranes [39,40]. An unusual aspect of 15-d-PGJ2 signaling is its ability to signal by redox reactions. 15-d-PGJ2 induces Akt and Nrf2 signaling by forming a covalent adduct with GSH [41]. This does not appear to be an isolated phenomenon, but a normal process in 15-d-PGJ2 signaling. In addition, 15-d-PGJ2 oxidizes p38 at cysteine residues near the protein surface resulting inp38 inactivation [42]. Similarly, Δ12-PGJ2 reacts with human serum albumin to form a covalent adduct with histidine-146, a reaction which chemically stabilizes the bound prostaglandin [43].
3.1. PGJ2 in Systemic Cancers
Of all the prostaglandins, PGJ2 exerts the broadest range of effects in cancer, echoing its biochemical diversity. 15-d-PGJ2 induces expression of the tumor suppressor, HtrA3, in 786-O and RCC4 renal cell carcinoma lines through a mechanism dependent on MEK/ERK signaling, but not PPARγ [12]. 15-d-PGJ2 also augments the antitumor activity of the alkylating agent, camptothecin, against Caki-2 renal cell carcinoma cellsin a manner independent of topoisomerase-II and PPARγ signaling pathways [44].
In addition to its effect on tumor suppressor pathways, 15-d-PGJ targets molecular drivers of stem cell identity and proliferation. In 786-O cells, 15-d-PGJ2 inhibits expression of hypoxia-inducible factor 2α (HIF2α)—which has been implicated in modulating cancer stem cell identity [45]—by binding to iron regulatory protein-1 (IRP1) [46]. In a mouse model, 15-d-PGJ2 inhibited proliferation of embryonic stem cells by antagonizing the leukemia inhibitory factor (LIF)-Tyk2-Stat3 signal transduction pathway [47].
Moreover, 15-d-PGJ2 signaling appears to modulate cell survival and apoptosis pathways in multiple cancer types.15-d-PGJ2 signaling induces expression of EGFR and COX-2 in MG-63 osteosarcoma cells via reactive oxygen species (ROS) and the p38 and p42/p44 MAPK pathways [48]. On the one hand, PGJ2-dependent inflammation and induction of EGFR could promote cancer genesis and survival. Conversely, ROS formation by 15-d-PGJ2 has been shown to induce apoptosis in A549 lung cancer cells [49] and synergistically enhance histone deacetylase inhibitor-driven apoptosis in DLD-1 colon cancer cells [50]. In MCF-7 breast cancer cells, 15-dPGJ2 activates a Ca2+-ERK1/2 signal transduction cascade that increases levels of the transcription factor, EGR1, which acts as an inhibitor of breast cancer cell proliferation [51]. EGR1 is also a positive regulator of the tumor suppressor gene, PTEN [52]. In MCF-7 cancer cells, 15-d-PGJ2 also reacts with GSH to form a 15-dPGJ2-GSH conjugate, which subsequently activates Akt and Nrf2 and results in MRP1-dependent export of the 15-d-PGJ2-GSH molecules [41]. Depletion of the intracellular GSH pool is in turn believed to trigger apoptosis, while relatively moderate depletion of GSH stores is thought to augment adaptation of cancer cells to external stresses. This mechanism suggests that one could exploit the anti-cancer effects of 15-d-PGJ2 by concomitant administration of selective inhibitors of MRP1.
Studies examining the function of 15-d-PGJ2 in normal cells have revealed other novel effects of this molecule on cellular physiology that could be relevant to human disease and cancer therapy. For example, 15-d-PGJ2 signaling has been found to modulate CRM1 transporterdependent nuclear protein export [53]. Altering trafficking between the nucleus and cytosol using modulators of 15-d-PGJ2 signaling could be used to perturb the actions of oncogenes by limiting their access to their nuclear targets. Also interesting, in murine cell lines 15-d-PGJ2 signaling inhibits mitochondrial fission activities, which leads to subsequent remodeling of mitochondrial proteins [54] and results in increased ROS formation. Conceivably, 15-d-PGJ2 signaling could be used to therapeutically trigger apoptosis in malignant cells.
3.2. PGJ2 in Glioma
Like PGD2, Δ12-PGJ2 alters the morphology of C6BU-1 rat glioma cells and substantially slows their proliferation [55]. Furthermore, 15-d-PGJ2 has been shown to induce caspase-independent apoptosis in human A172 glioma cells through ROS formation and mitochondrial damage [56]. Curiously, 15-d-PGJ2 was found to protect rat C6 glioma cells from methylmercury toxicity-precisely by preventing damage to the mitochondrial membrane [57]. Further study is needed to determine under what circumstances PGJ2 signaling is cytotoxic or cytoprotectivebefore modulators of PGJ2 signaling can be considered for treatment of human gliomas.
4. PGE2 Signaling
PGE2 signals through four receptors—EP1, EP2, EP3 and EP4—which are all GPCRs. EP2 and EP4 are linked to Gαs and activate a cAMP/PKA pathway, EP1 is thought to be linked to Gαq, and EP3 to Gαi3. EP3 also appears to be linked to PLCγ signaling [58]. Not surprisingly, PGE2 has the most diverse physiologic effects of any of the prostaglandins. Its activities include effects on the inflammatory response, lipid metabolism, tumorigenesis, neurotransmission (via GABA and dopamine), and B cell survival [59].
4.1. PGE2 in Systemic Cancers
Inhibitors of PGE2 synthesis, such as curcumin, are being tested in clinical trials as agents for cancer prevention. Phase IIa studies have shown that curcumin can prevent the formation of aberrant crypt foci in the colon, which are thought to precede development of colon cancer [60].
Treatment of the human colon cancer cell line HT-29 with epinephrine stimulated cell proliferation and increased PGE2 synthesis and release, which in turn increased vascular endothelial growth factor (VEGF) secretion (likely via EP4 signaling [61]) and MMP9 activity4. Protein kinase CK2 and the Wnt/β-catenin pathway also activate production of PGE2, with subsequent proliferation of human colon (HT29-US and DLD-1) and breast (ZR-75) cancer cells. In addition to its effects as a promoter of cell proliferation and angiogenesis in colon cancer, PGE2 has also been shown to help colon cancer cells evade the immune response and advantageously alter their energy metabolism. In LS-174T and HCT-116 colon cancer cells, PGE2 signaling activates the nuclear orphan receptor NR4A2, which increases fatty acid oxidation as an alternative fuel source to glucose [62]. This activity could promote tumor survival under conditions of starvation. Inhibition of PGE2 synthesis has also been associated with decreased incidence of colon cancer in murine studies and decreased levels of tumor-cell protective FoxP3+Tregs_ENREF_19 [19].
Interestingly, NR4A2, under the regulation of PGE2, is involved in the development of drug resistance in human oral squamous cell carcinoma. In HSC3, HSC4, Ho-1- u-1 and Ca9-22 lines, PGE2 was shown to promote 5- fluorouracil resistance of EGFR-dependent tumors by induction of NR4A2 [63].
As in colon cancer, PGE2 is a stimulator of angiogenesis in breast cancer. Apoptotic MCF-7 breast cancer cells were shown to signal via the sphingosine-1-phosphate (S1P) S1P1 and S1P3 receptors in order to induce PGE2 production in macrophages. The activated macrophages then released PGE2, thereby triggering vascular endothelial cell migration and subsequent angiogenesis in breast tumors [64]. As is the case in colon cancer, PGE2 also appears to plays a role in immune evasion in breast cancer. PGE2 produced ad secreted by breast cancer cells suppresses NK cell function through activation of the EP4 receptor. PGE2 also induces expression of the oncogene aromatase in breast adipose fibroblasts in a pathway that involves JunD and JunB [65]. Aromatase activity subsequently results in elevated levels of estradiol. This finding could explain why clinical studies have shown that COX-1 levels correlate with high levels of serum estradiol in patients with breast cancer [66]. In fact, therapeutic agents targeting the PGE2 signaling pathway have been studied as potential adjuncts for breast cancer treatment. The natural PGE2 antagnoists frondoside A (which inhibits EP2 and EP4 receptors) [67] and saponin (which also acts via an AMP-activated protein kinase pathway to inhibit COX-2) [68] have been shown to slow breast cancer progression and induce apoptosis in Balb/ cfC3H mouse and MCF-7 human breast cancer cells, respectively.
Current understanding of the role of PGE2 in other cancer types is more fragmented, but several recent findings are worth noting. PGE2 expression by blood mononuclear cells induced by AsPC-1 and MiaPaCa-2 pancreatic cancer cell lines is critical to generate a supportive tumor microenvironment [69]. PGE2 has also been shown to induce the crucial oncogene telomerase (hTERT) in a signaling cascade dependent on EP4 and Sp1 in both lung (H1838 and H1792) [70] and cervical (HeLa, SiHa, Caski) [71] cancer cell lines. In renal cell carcinoma cell lines (RCC7 and Caki-1), PGE2 stimulates tumor cell migration and invasion via the EP4-Rap pathway [5]. Microsomal prostaglandin E synthase-1 (mPGES-1) is known to inhibit the tumor suppressor protein phosphate and tensin homolog (PTEN), which supports biliary tract cancer progression [10]. This effect could be an extension of the normal function of EP1, which signals via Gαq, leading to Ca2+ mobilization [3] and PI3K activation, which directly antagonizes PTEN. A similar role for EP1 signaling through Ca2+ mobilization has been postulated inthe development of melanoma [72]. Importantly, the biological effects of PGE2 signaling on cancer progression do not appear to be isolated from one another; rather, they appear to be related by a more fundamental process, the EMT. Vaid et al. demonstrated that natural products isolated from grape seeds can reverse EMT in melanoma cell lines (A375 and Hs294) and that this effect was duplicated by inhibiting PGE2 with celecoxib [9]. Thus, one can conclude that PGE2 signaling affects all the essential processes of tumor generation and malignant progression–from antagonizing tumor suppressor genes and activating oncogenes, to stimulating immune system evasion, angiogenesis, cell migration and the EMT.
4.2. PGE2 in Glioma
As in colon cancer, PGE2 released by glioma cells has an inhibitory effect on host immunity. Release of PGE2 by glioblastoma cells decreases induction and cytotoxicity of anti-tumor lymphocytes [73]. PGE2 secretion by MG- 377 glioblastoma cells can also stimulate CD11c+ dendritic cells to induce CD4+Tregs, which again results in suppression of the host immune response [74]. It also appears that human U251 and T98G glioblastoma cells secrete soluble factors that drive macrophages to produce PGE2 [75]. In an induced glioma mouse model, blockade of systemic PGE2 synthesis using COX-2 inhibitors or knock-out of COX-2 suppressed gliomagenesis, possibly due to an increase in host immune surveillance [76]. Interestingly, macrophages that are capable of killing T9 rat glioma cells are resistant to the immunosuppressive effects of PGE2 [77].
The role of PGE2 inglioma biology extends beyond its effects as an immunomodulator. In U87-MG glioma xenografts, mPGES-1 drives tumor cell proliferation and tumor growth via activation of type II PKA [78], which in turn inhibits ERK and increases CREB transcriptional activity [79]. This mechanism mirrors the inhibitory effect of mPGES-1on PTEN seen in biliary tract cancers [10].There also seems to be a role for PGE2 in glioma cell invasion via its activation of PKC [80].
Thus far, it appears that PGE2 signaling almost universally drives cancer proliferation and migration, but this may not uniformly be the case. In one study, PGE2 was found to induce Bax-dependent apoptosis in primary glioblastoma cells, and patients expressing a high level of mPGES-1 were found longer survival times than those with low levels of mPGES-1 [81]. In principle, this trend could be explained by pro-apoptotic PGE2 signaling through the EP4 receptor [59]. Whereas the EP4 receptor may mediate the clinically important effects of PGE2 in gliomas, in medulloblastoma it appears that the EP1 and EP3 receptors are more crucial. Specifically, EP1 and EP3 drive proliferation of medulloblastoma cells [82]. These differing roles of PGE2 in different cancers and contexts speak to the intricacy of prostaglandin signaling, and how development of prostaglandin-based therapies will require an appreciation of this complexity.
Finally, these observations suggest that EP1 and EP3 modulators could provide a novel means of treating gliomas by augmenting the host anti-tumor immune response, similar to the use of ipilimumab in the treatment of metastatic melanoma [83].
5. PGF2α Signaling
In humans, aldoketoreductase (AKR) 1B1 is the primary enzyme that produces PGF2α from PGH2 [84], while AKR1C3 plays a minor role in PGF2α synthesis [34]. The PGF2α receptor FP is a GPCR linked to Gαq, which affects Ca2+ homeostasis and regulates smooth muscle cell contraction, most notably in the intestines [85] and uterus [86]. PGF2α analogues (e.g. latanoprost) are also used clinically to lower intraocular pressure in the treatment of glaucoma [87].
5.1. PGF2α in Systemic Cancers
The PGF2α metabolite 8-isoPGF2α has been found to be a reliable marker of cancer progression in a rat breast cancer model [88]. Levels of 8-isoPGF2α have also been monitored to track cellular damage associated with renal oxidative stress [89] and bladder obstruction [90] ,which are considered risk factors for the development of renal cell and uroepithelial cancers, respectively.
5.2. PGF2α in Glioma
In NG108-15 hybrid neuroblastoma-glioma cells, PGF2α (and also PGD2 and PGE2) raises intracellular Ca2+ levels via a cGMP-dependent mechanism [91]. Since these early studies, very little attention has been given to the role of PGF2α in CNS cancers. The few described effects of PGF2α in glioblastoma have focused on its roleon tumor-associated vasculature. Glioma cells appear to synthesize high levels of both PGF2α and TxA2, but the disproportionate increase in TxA2 synthesis over PGF2α synthesis is believed to contribute to the changes seen in vascular permeability and the resulting cerebral edema [92]. It appears that PGF2α signaling is also involved in the remodeling of cerebral vascular architecture: in SV40-transfected microglial cells, PGF2α acts via a Ras/Rafand Tcf-pathway dependent to increase production of the CYR61 protein [93], which induces physiologic angiogenesis in the corpus luteum [94].Thus, building on the work of Kesari et al. [30], modulators of PGF2α signaling might be good targets as angiogenesis inhibitors in the treatment of glioblastoma multiforme.
6. PGI2 Signaling
PGI2 is synthesized in vascular endothelial cells by prostacyclin synthase (PGIS) through the catalysis of PGH2. The PGI2 receptor (IP) is a rhodopsin-like GPCR that signals via Gαs to activate cAMP synthesis. PGI2 may also signal through Gαq, Gαi and the PPARδ pathways [13]. A recent mouse model has shown that maternal PGI2 signaling through fetal PPARδ is key for blastocyst hatching and subsequent implantation [95].
6.1. PGI2 in Systemic Cancers
The PGI2 analogue iloprost has been investigated as a potential agent for lung cancer prevention. A phase II placebo-controlled randomized study showed that iloprost significantly reduced dysplasia in lung tissue biopsies obtained from former smokers [96]. Whether PGI2 signaling levels have a meaningful effect on cancer survival remains unclear. An observational study of patients in Ireland with various forms of lung cancer showed that overall PGI2 synthase (PGIS) expression was decreased in lung cancer, but PGIS expression levels did not correlate with survival [97]. On the other hand, a recent case study showed that a patient with lung cancer treated with iloprost showed no evidence of cancer progression in the absence of conventional chemotherapy [98].
6.2. PGI2 in Glioma
The role of PGI2 signaling in gliomas is not well understood. Many CNS tumors express endogenous PGI2 receptors (IP). PGI2 signaling results in cAMP and cGMP accumulation in N4TG3 murine neuroblastoma cells, but notin 1321N1 human astrocytoma cells [99]. Angiotensin is known to induce release of PGI2 from rat C6 glioma cells [100] and the IP receptor can generate inward Ca2+ currents in hybrid rodent NG108-15 glioma-neuroblastoma cells [101]. Activation of the IP receptor, however, is subject to desensitization in response to prolonged stimulation in these cells [102]. Surprisingly, the IP receptor in NG108-15 cells does not activate the ERK1/2 pathways, as would be expected from IP-driven cAMP production [103]. In these experiments, PI3Kand PKCdependent currents were observed in CHO but not glioma cells. Further work is needed to determine whether PGI2 signaling has functional relevance in glioblastoma.
7. TxA2 Signaling
Thromboxane A2 (TxA2) is produced by the thromboxane synthesis enzyme (TXAS) and signals via the TP receptor, a GPCR linked to Gαq. Like some of the other prostaglandin receptors, TP exerts its effects by mobilizing Ca2+ stores, most notably in the processes of coagulation and regulation of vascular smooth muscle tone [104].
7.1. TxA2 in Systemic Cancers
Binding of TxA2 to the TP receptor results in enhanced activity of the protein kinase C-related kinase (PRK)1. PRK1 signals downstream of RhoA and is implicated in the development and progression of prostate cancer [6]. Though not as extensively described as a therapeutic target as PGE2, TxA2 has shown promise for as a target for investigation in lung [105] and prostate [6] cancers.
7.2. TxA2 in Glioma
TxA2 synthesis is known to be elevated in gliomas, and increases with increasing tumor grade [106]. TxA2 signaling results in CREB-dependent induction of IL-6 by human 1321N1 astrocytoma cells through activation of the p38 MAPK and PKA pathways [107]. Furthermore, TxA2 induces cell swelling in 1321N1 cells in a mechanism dependent on Gαq, RhoA, the Na+/H+ exchange pump, and aquaporins [108]. Since aquaporins are known to be involved in migration and proliferation of human glioma cell lines (D54, D65, STTG1, U87, U251) [109], this discovery regarding the role of TxA2 in astrocytoma cells raises the question of whether TxA2 inhibition could be used to inhibit glioma invasion. Inhibition of TxA2 signaling by blocking TXAS also renders glioma cells more sensitive to apoptosis when subjected to g-radiation [110] or alkylating chemotherapy (U87 glioblastoma cells) [111]. Thus, inhibition of TxA2 signaling may be a valuable adjunct to radiation therapy in glioma.
8. Conclusion
The 2-series eicosanoids have diverse roles in the biology of systemic cancers and glioma. These effects vary drastically in a cancerand context-specific manner. The list of cancer cell processes affected by prostaglandin signaling is impressive and includes modulation of the immune system, induction of the EMT, modulation of tumor cell migration and invasion, changes in the cell metabolic state, and alterations in the balance between oncogene and tumor-suppressor activity. New therapies based on eicosanoid biology could provide valuable therapies to currently intractable cancers. One possible strategy would be to combine the purported anti-proliferative and pro-apoptotic effects of PGD2, PGI2 and PGJ2 while inhibiting the oncogenic effects of the other 2-series eicosanoids. Our current knowledge suggests that the 2-series eicosanoids merit further study as possible therapeutic targets in patients with glioblastoma and other cancers.
REFERENCES
- R. Stupp, et al., “Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma,” The New England Journal of Medicine, Vol. 352, No. 10, 2005, pp. 987-996.
- R. Yefi, et al., “Protein Kinase CK2 Promotes Cancer Cell Viability via Up-Regulation of Cyclooxygenase-2 Expression and Enhanced Prostaglandin E2 Production,” Journal of Cellular Biochemistry, Vol. 112, No. 11, 2011, pp. 3167-3175. doi:10.1002/jcb.23247
- D. Holt, X. Ma, N. Kundu and A. Fulton, “Prostaglandin E(2) (PGE (2)) Suppresses Natural Killer Cell Function Primarily through the PGE(2) Receptor EP4,” Cancer Immunology, Immunotherapy: CII, Vol. 60, No. 11, 2011, pp. 1577-1586.
- H. P. Wong, et al., “Effects of Adrenaline in Human Colon Adenocarcinoma HT-29 Cells,” Life Sciences, Vol. 88, No. 25-26, 2011, pp. 1108-1112.
- J. Wu, et al., “Prostaglandin E2 Regulates Renal Cell Carcinoma Invasion through a EP4-Rap Signal Transduction Pathway,” The Journal of Biological Chemistry, Vol. 286, No. 39, 2011, pp. 33954-33962.
- E. C. Turner, et al., “Identification of an Interaction between the TPalpha and TPbeta Isoforms of the Human Thromboxane A2 Receptor with Protein Kinase C-Related Kinase (PRK) 1: Implications for Prostate Cancer,” The Journal of Biological Chemistry, Vol. 286, No. 17, 2011, pp. 15440-15457. doi:10.1074/jbc.M110.181180
- M. Westphal, M. Neuss and H. D. Herrmann, “Prostaglandins: Antiproliferative Effect of PGD 2 on Cultured Human Glioma Cells,” Acta Neurochirurgica (Wien), Vol. 83, No. 1-2, 1986, pp. 56-61. doi:10.1007/BF01420509
- J. H. Kang, et al., “Involvement of Cox-2 in the Metastatic Potential of Chemotherapy-Resistant Breast Cancer Cells,” BMC Cancer, Vol. 11, 2011, p. 334. doi:10.1186/1471-2407-11-334
- M. Vaid, T. Singh and S. K. Katiyar, “Grape Seed Proanthocyanidins Inhibit Melanoma Cell Invasiveness by Reduction of PGE(2) Synthesis and Reversal of Epithelial-to-Mesenchymal Transition,” PloS One, Vol. 6, No. 6, 2011, p. e21539. doi:10.1371/journal.pone.0021539
- D. Lu, C. Han and T. Wu, “Microsomal Prostaglandin E Synthase-1 Inhibits PTEN and Promotes Experimental Cholangiocarcinogenesis and Tumor Progression,” Gastroenterology, Vol. 140, No. 7, 2011, pp. 2084-2094. doi:10.1053/j.gastro.2011.02.056
- E. V. Hersh, E. T. Lally and P. A. Moore, “Update on Cyclooxygenase Inhibitors: Has a Third COX Isoform Entered the Fray?” Current Medical Research & Opinion, Vol. 21, No. 8, 2005, pp. 1217-1226. doi:10.1185/030079905X56367
- S. Theoleyre, S. Mottier, D. Masson and M. G. Denis, HtrA3 is Regulated by 15-Deoxy-Delta12,14-Prostaglandin J2 Independently of PPARgamma in Clear Cell Renal Cell Carcinomas,” Biochemical and Biophysical Research Communications, Vol. 394, No. 3, 2010, pp. 453-458. doi:10.1016/j.bbrc.2009.11.163
- J. Stitham, C. Midgett, K. A. Martin and J. Hwa, “Prostacyclin: An Inflammatory Paradox,” Frontiers in Pharmacology, Vol. 2, 2011, p. 24.
- D. Iusuf, et al., “P-Glycoprotein (ABCB1) Transports the Primary Active Tamoxifen Metabolites Endoxifen and 4-Hydroxytamoxifen and Restricts Their Brain Penetration,” Journal of Pharmacology and Experimental Therapeutics, Vol. 337, No. 3, 2011, pp. 710-717. doi:10.1124/jpet.110.178301
- L. Levine, “Arachidonic Acid Transformation and Tumor Production,” Advances in Cancer Research, Vol. 35, 1981, pp. 49-79. doi:10.1016/S0065-230X(08)60908-2
- I. M. Ciris, K. K. Bozkurt, S. Baspinar and F. N. Kapucuoglu, “Immunohistochemical COX-2 Overexpression Correlates with HER-2/neu Overexpression in Invasive Breast Carcinomas: A Pilot Study,” Pathology, Research and Practice, Vol. 207, No. 3, 2011, pp. 182-187. doi:10.1016/j.prp.2011.01.005
- B. Paquette, et al., “Radiation-Enhancement of MDAMB-231 Breast Cancer Cell Invasion Prevented by a Cyclooxygenase-2 Inhibitor,” British Journal of Cancer, Vol. 105, 2011, pp. 534-541. doi:10.1038/bjc.2011.260
- N. Markosyan, et al., “Deletion of Cyclooxygenase 2 in Mouse Mammary Epithelial Cells Delays Breast Cancer Onset through Augmentation of Type 1 Immune Responses in Tumors,” Carcinogenesis, Vol. 32, No. 10, 2011, pp. 1441-1449. doi:10.1093/carcin/bgr134
- M. Nakanishi, et al., “Selective PGE2 Suppression Inhibits Colon Carcinogenesis and Modifies Local Mucosal Immunity,” Cancer Prevention Research, Vol. 4, 2011, pp. 1198-1208. doi:10.1158/1940-6207.CAPR-11-0188
- J. T. Mao, et al., “Lung Cancer Chemoprevention with Celecoxib in Former Smokers,” Cancer Prevention Research, Vol. 4, 2011, pp. 984-993. doi:10.1158/1940-6207.CAPR-11-0078
- A. M. Algra and P. M. Rothwell, “Effects of Regular Aspirin on Long-Term Cancer Incidence and Metastasis: A Systematic Comparison of Evidence from Observational Studies versus Randomised Trials,” The Lancet Oncology, Vol. 13, No. 5, 2012, pp. 518-527. doi:10.1016/S1470-2045(12)70112-2
- J. Zaric, et al., “Identification of MAGI1 as a TumorSuppressor Protein Induced by Cyclooxygenase-2 Inhibitors in Colorectal Cancer Cells,” Oncogene, Vol. 31, No. 1, 2011, pp. 48-59.
- H. H. Tai, X. Chi and M. Tong, “Regulation of 15-Hydroxyprostaglandin Dehydrogenase (15-PGDH) by NonSteroidal Anti-Inflammatory Drugs (NSAIDs),” Prostagndins & Other Lipid Mediators, Vol. 96, No. 1-4, 2011, pp. 37-40. doi:10.1016/j.prostaglandins.2011.06.005
- D. Ma, M. Liu, A. P. Wang and H. Yang, “Cycloxygenase-2 is Essential for the Survival and Proliferation of Gastric Cancer Cells,” Cell Biochemistry and Biophysics, Vol. 61, No. 3, 2011, pp. 637-641. doi:10.1007/s12013-011-9249-6
- C. G. Ye, et al., “Indomethacin and SC236 Enhance the Cytotoxicity of Doxorubicin in Human Hepatocellular Carcinoma Cells via Inhibiting P-Glycoprotein and MRP1 Expression,” Cancer Letters, Vol. 304, No. 2, 2011, pp. 90-96. doi:10.1016/j.canlet.2011.01.025
- H. Raza, A. John and S. Benedict, “Acetylsalicylic AcidInduced Oxidative Stress, Cell Cycle Arrest, Apoptosis and Mitochondrial Dysfunction in Human Hepatoma HepG2 Cells,” European Journal of Pharmacology, Vol. 668, No. 1-2, 2011, pp. 15-24. doi:10.1016/j.ejphar.2011.06.016
- C. Cerella, et al., “COX-2 Inhibitors Block Chemotherapeutic Agent-Induced Apoptosis Prior to Commitment in Hematopoietic Cancer Cells,” Biochemical Pharmacology, Vol. 82, No. 10, 2011, pp. 1277-1290. doi:10.1016/j.bcp.2011.06.028
- W. Yang, et al., “Quantitative Proteomics Identifies a Beta-Catenin Network as an Element of the Signaling Response to Frizzled-8 Protein-Related Antiproliferative Factor,” Molecular & Cellular Proteomics: MCP, Vol. 10, No. 6, 2011.
- T. Shono, P. J. Tofilon, J. M. Bruner, O. Owolabi and F. F. Lang, “Cyclooxygenase-2 Expression in Human Gliomas: Prognostic Significance and Molecular Correlations,” Cancer Research, Vol. 61, No. 11, 2001, pp. 4375-4381.
- S. Kesari, et al., “Phase II Study of Temozolomide, Thalidomide, and Celecoxib for Newly Diagnosed Glioblastoma in Adults,” Journal of Neuro-Oncology, Vol. 10, No. 3, 2008, pp. 300-308. doi:10.1215/15228517-2008-005
- J. U. Flanagan and M. L. Smythe, “Sigma-Class Glutathione Transferases,” Drug Metabolism Reviews, Vol. 43, No. 2, 2011, pp. 194-214. doi:10.3109/03602532.2011.560157
- F. P. Mesquita-Santos, et al., “Co-Operative Signalling through DP(1) and DP(2) Prostanoid Receptors is Required to Enhance Leukotriene C(4) Synthesis Induced by Prostaglandin D(2) in Eosinophils,” British Journal of Pharmacology, Vol. 162, No. 8, 2011, pp. 1674-1685. doi:10.1111/j.1476-5381.2010.01086.x
- T. Murata, et al., “Role of Prostaglandin D2 Receptor DP as a Suppressor of Tumor Hyperpermeability and Angiogenesis in Vivo,” Proceedings of the National Academy of Sciences of USA, Vol. 105, No. 50, 2008, pp. 20009- 20014. doi:10.1073/pnas.0805171105
- T. M. Downs, D. W. Burton, F. L. Araiza, R. H. Hastings and L. J. Deftos, “PTHrP Stimulates Prostate Cancer Cell Growth and Upregulates Aldo-Keto Reductase 1C3,” Cancer Letters, Vol. 306, No. 1, 2011, pp. 52-59. doi:10.1016/j.canlet.2011.02.027
- B. L. Tippin, et al., “Hematopoietic Prostaglandin D Synthase (HPGDS): A High Stability, Val187Ile Isoenzyme Common among African Americans and Its Relationship to Risk for Colorectal Cancer,” Prostaglandins & Other Lipid Mediators, Vol. 97, No. 1-2, 2011, pp. 22-28.
- C. A. Payne, et al., “Loss of Prostaglandin D2 Synthase: A Key Molecular Event in the Transition of a Low-Grade Astrocytoma to an Anaplastic Astrocytoma,” Molecular Cancer Therapeutics, Vol. 7, 2008, pp. 3420-3428. doi:10.1158/1535-7163.MCT-08-0629
- M. Sabbah, et al., “Molecular Signature and Therapeutic Perspective of the Epithelial-To-Mesenchymal Transitions in Epithelial Cancers,” Drug Resistance Updates, Vol. 11, No. 4, 2008, pp. 123-151. doi:10.1016/j.drup.2008.07.001
- K. Fujimori, K. Kadoyama and Y. Urade, “Protein Kinase C Activates Human Lipocalin-Type Prostaglandin D Synthase Gene Expression through De-Repression of NotchHES Signaling and Enhancement of AP-2 Beta Function in Brain-Derived TE671 Cells,” The Journal of Biological Chemistry, Vol. 280, 2005, pp. 18452-18461. doi:10.1074/jbc.M411755200
- Y. Yamamoto, et al., “Proteomic Identification of Protein Targets for 15-Deoxy-Delta(12,14)-prostaglandin J2 in Neuronal Plasma Membrane,” PloS One, Vol. 6, No. 3, 2011, p. e17552. doi:10.1371/journal.pone.0017552
- A. N. Hata, R. Zent, M. D. Breyer and R. M. Breyer, “Expression and Molecular Pharmacology of the Mouse CRTH2 Receptor,” Journal of Pharmacology and Experimental Therapeutics, Vol. 306, No. 2, 2003, pp. 463- 470. doi:10.1124/jpet.103.050955
- N. Y. Song, et al., “Multidrug Resistance-Associated Protein 1 Mediates 15-Deoxy-Delta(12,14)-prostaglandin J(2)-Induced Expression of Glutamate Cysteine Ligase Expression via Nrf2 Signaling in Human Breast Cancer Cells,” Chemical Research in Toxicology, Vol. 24, No. 8, 2011, pp. 1231-1241. doi:10.1021/tx200090n
- D. J. Templeton, M. S. Aye, J. Rady, F. Xu and J. V. Cross, “Purification of Reversibly Oxidized Proteins (PROP) Reveals a Redox Switch Controlling p38 MAP Kinase Activity,” PloS One, Vol. 5, No. 11, 2010, p. e15012. doi:10.1371/journal.pone.0015012
- S. Yamaguchi, et al., “Delta12-Prostaglandin J2 as a Product and Ligand of Human Serum Albumin: Formation of an Unusual Covalent Adduct at His146,” Journal of the American Chemical Society, Vol. 132, No. 2, 2010, pp. 824-832. doi:10.1021/ja908878n
- Y. Yamamoto, et al., “15-Deoxy-Delta(12,14)-prostaglandin J(2) Enhanced the Anti-Tumor Activity of Camptothecin against Renal Cell Carcinoma Independently of Topoisomerase-II and PPARgamma Pathways,” Biochemical and Biophysical Research Communications, Vol. 410, No. 3, 2011, pp. 563-567. doi:10.1016/j.bbrc.2011.06.026
- K. De Bock, M. Mazzone and P. Carmeliet, “Antiangiogenic Therapy, Hypoxia, and Metastasis: Risky Liaisons, or Not? Nature Reviews,” Clinical Oncology, Vol. 8, 2011, pp. 393-404.
- M. Zimmer, et al., “The Connectivity Map Links Iron Regulatory Protein-1-Mediated Inhibition of HypoxiaInducible Factor-2a Translation to the Anti-Inflammatory 15-Deoxy-delta12,14-prostaglandin J2,” Cancer Research, Vol. 70, 2010, pp. 3071-3079. doi:10.1158/0008-5472.CAN-09-2877
- C. Mo, W. Chearwae and J. J. Bright, “PPARgamma Regulates LIF-Induced Growth and Self-Renewal of Mouse ES Cells through Tyk2-Stat3 Pathway,” Cellular Signalling, Vol. 22, No. 3-4, 2010, pp. 495-500. doi:10.1016/j.prostaglandins.2011.01.004
- K. Kitz, et al., “15-Deoxy-Delta12,14-prostaglandin J2 Induces Cox-2 Expression in Human Osteosarcoma Cells through MAPK and EGFR Activation Involving Reactive Oxygen Species,” Free Radical Biology & Medicine, Vol. 50, 2011, pp. 854-865.
- J. J. Wang and O. T. Mak, “Induction of Apoptosis by 15d-PGJ2 via ROS Formation: An Alternative Pathway without PPARgamma Activation in Non-Small Cell Lung Carcinoma A549 Cells,” Prostaglandins & Other Lipid Mediators, Vol. 94, No. 3-4, 2011, pp. 104-111.
- M. Koyama, et al., “Histone Deacetylase Inhibitors and 15-Deoxy-Delta12,14-prostaglandin J2 Synergistically Induce Apoptosis,” Clinical Cancer Research, Vol. 16, No. 8, 2010, pp. 2320-2332.
- S. Chbicheb, et al., “EGR1 Expression: A Calcium and ERK1/2 Mediated PPARgamma-Independent Event Involved in the Antiproliferative Effect of 15-DeoxyDelta12,14-prostaglandin J2 and Thiazolidinediones in Breast Cancer Cells,” Biochemical Pharmacology, Vol. 81, No. 9, 2011, pp. 1087-1097. doi:10.1016/j.bcp.2011.02.006
- T. Virolle, et al., “The Egr-1 Transcription Factor Directly Activates PTEN during Irradiation-Induced Signalling,” Nature Cell Biology, Vol. 3, No. 12, 2001, pp. 1124-1128. doi:10.1038/ncb1201-1124
- M. Hilliard, et al., “The Anti-Inflammatory Prostaglandin 15-Deoxy-delta(12,14)-PGJ2 Inhibits CRM1-Dependent Nuclear Protein Export,” The Journal of Biological Chemistry, Vol. 285, 2010, pp. 22202-22210. doi:10.1074/jbc.M110.131821
- R. Kar, N. Mishra, P. K. Singha, M. A. Venkatachalam and P. Saikumar, “Mitochondrial Remodeling Following Fission Inhibition by 15d-PGJ2 Involves Molecular Changes in Mitochondrial Fusion Protein OPA1,” Biochemical and Biophysical Research Communications, Vol. 399, No. 4, 2010, pp. 548-554. doi:10.1016/j.bbrc.2010.07.108
- H. Sugino, T. Miwa, R. Ueno and O. Hayaishi, “Alteration in Morphology and Induction of Glutamine Synthetase in Rat Glioma C6BU-1 Cells Cultured with Prostaglandin D(2)-Supplemented Media,” Neurochemistry International, Vol. 12, No. 1, 1988, pp. 5-12. doi:10.1016/0197-0186(88)90141-6
- W. H. Cho, C. H. Choi, J. Y. Park, S. K. Kang and Y. K. “Kim, 15-Deoxy-(Delta12,14)-prostaglandin J2 (15dPGJ2) Induces Cell Death through Caspase-Independent Mechanism in A172 Human Glioma Cells,” Neurochemical Research, Vol. 31, No. 10, 2006, pp. 1247-1254. doi:10.1007/s11064-006-9157-0
- J. Y. Chang and P. F. Tsai, “Prevention of Methylmercury-Induced Mitochondrial Depolarization, Glutathione Depletion and Cell Death by 15-Deoxy-delta-12,14-prostaglandin J(2),” Neurotoxicology, Vol. 29, No. 6, 2008, pp. 1054-1061. doi:10.1016/j.neuro.2008.08.003
- H. S. Kuehn, M. Y. Jung, M. A. Beaven, D. D. Metcalfe and A. M. Gilfillan, “Distinct PGE(2)-Responder and Non-Responder Phenotypes in Human Mast Cell Populations: ‘All or Nothing’ Enhancement of Antigen-Dependent Mediator Release,” Immunology Letters, Vol. 141, No. 1, 2011, pp. 45-54. doi:10.1016/j.imlet.2011.07.002
- M. Prijatelj, T. Celhar and I. Mlinaric-Rascan, “Prostaglandin EP4 Receptor Enhances BCR-Induced Apoptosis of Immature B Cells,” Prostaglandins & Other Lipid Mediators, Vol. 95, No. 1-4, 2011, pp. 19-26. doi:10.1016/j.prostaglandins.2011.04.001
- [61] R. E. Carroll, et al., “Phase IIa Clinical Trial of Curcumin for the Prevention of Colorectal Neoplasia,” Cancer Prevention Research (Philadelphia, Pa.), Vol. 4, 2011, pp. 354-364. doi:10.1158/1940-6207.CAPR-10-0098
- [62] R. Rao, et al., “Prostaglandin E2-EP4 Receptor Promotes Endothelial Cell Migration via ERK Activation and Angiogenesis in Vivo,” The Journal of Biological Chemistry, Vol. 282, 2007, pp. 16959-16968. doi:10.1074/jbc.M701214200
- [63] V. R. Holla, H. Wu, Q. Shi, D. G. Menter and R. N. Dubois, “The Nuclear Orphan Receptor NR4A2 Modulates Fatty Acid Oxidation Pathways in Colorectal Cancer,” The Journal of Biological Chemistry, Vol. 286, 2011, pp. 30003-30009. doi:10.1074/jbc.M110.184697
- [64] H. Shigeishi, et al., “PGE Targets Squamous Cell Carcinoma Cell with the Activated Epidermal Growth Factor Receptor Family for Survival against 5-Fluorouracil through NR4A2 Induction,” Cancer Letters, Vol. 307, No. 2, 2011, pp. 227-236. doi:10.1016/j.canlet.2011.04.008
- [65] K. Brecht, et al., “Macrophages Programmed by Apoptotic Cells Promote Angiogenesis via Prostaglandin E2,” The FASEB Journal, Vol. 25, No. 7, 2011, pp. 2408- 2417.
- [66] D. Chen, S. Reierstad, F. Fang and S. E. Bulun, “JunD and JunB Integrate Prostaglandin E2 Activation of Breast Cancer-Associated Proximal Aromatase Promoters,” Molecular Endocrinology (Baltimore, Md.), Vol. 25, No. 5, 2011, pp. 767-775. doi:10.1210/me.2010-0368
- [67] V. D. Haakensen, et al., “Serum Estradiol Levels Associated with Specific Gene Expression Patterns in Normal Breast Tissue and in Breast Carcinomas,” BMC Cancer, Vol. 11, No. 1, 2011, p. 332. doi:10.1186/1471-2407-11-332
- [68] X. Ma, N. Kundu, P. D. Collin, O. Goloubeva and A. M. “Fulton, Frondoside A Inhibits Breast Cancer Metastasis and Antagonizes Prostaglandin E Receptors EP4 and EP2,” Breast Cancer Research and Treatment, Vol. 132, No. 3, 2011, pp. 1001-1008.
- [69] A. D. Kim, et al., “Ginseng Saponin Metabolite Induces Apoptosis in MCF-7 Breast Cancer Cells through the Modulation of AMP-Activated Protein Kinase,” Environmental Toxicology and Pharmacology, Vol. 30, 2010, pp. 134-140. doi:10.1016/j.etap.2010.04.008
- [70] S. P. Grekova, A. Angelova, L. Daeffler and Z. Raykov, “Pancreatic Cancer Cell Lines Can Induce Prostaglandin e2 Production from Human Blood Mononuclear Cells,” Journal of Oncology, Vol. 2011, 2011, p. 741868. doi:10.1155/2011/741868
- [71] Y. Zheng, J. D. Ritzenthaler, X. Sun, J. Roman and S. Han, “Prostaglandin E2 Stimulates Human Lung Carcinoma Cell Growth through Induction of Integrin-Linked Kinase: The Involvement of EP4 and Sp1,” Cancer Research, Vol. 69, 2009, pp. 896-904. doi:10.1158/0008-5472.CAN-08-2677
- [72] L. Liu, et al., “Activation of Telomerase by Seminal Plasma in Malignant and Normal Cervical Epithelial Cells,” The Journal of Pathology, Vol. 225, No. 2, 2011, pp. 203-211. doi:10.1002/path.2914
- [73] I. Surh, et al., “The EP1 Receptor for Prostaglandin E2 Promotes the Development and Progression of Malignant Murine Skin Tumors,” Molecular Carcinogenesis, Vol. 50, No. 6, 2011, pp. 458-468. doi:10.1002/mc.20730
- [74] M. C. Kuppner, Y. Sawamura, M. F. Hamou and N. de Tribolet, “Influence of PGE2- and cAMP-Modulating Agents on Human Glioblastoma Cell Killing by Interleukin-2-Activated Lymphocytes,” Journal of Neurosurgery, Vol. 72, No. 4, 1990, pp. 619-625. doi:10.3171/jns.1990.72.4.0619
- [75] Y. Akasaki, et al., “Induction of a CD4+ T Regulatory Type 1 Response by Cyclooxygenase-2-Overexpressing Glioma,” The Journal of Immunology, Vol. 173, No. 7, 2004, pp. 4352-4359.
- [76] Y. Nakano, E. Kuroda, T. Kito, A. Yokota and U. Yamashita, “Induction of Macrophagic Prostaglandin E2 Synthesis by Glioma Cells,” Journal of Neurosurgery, Vol. 104, No. 4, 2006, pp. 574-582. doi:10.3171/jns.2006.104.4.574
- [77] M. Fujita, et al., “COX-2 Blockade Suppresses Gliomagenesis by Inhibiting Myeloid-Derived Suppressor Cells,” Cancer Research, Vol. 71, 2011, pp. 2664-2674. doi:10.1158/0008-5472.CAN-10-3055
- [78] N. S. Zeineddine, M. D. Avina, C. C. Williams, H. T. Wepsic and M. R. Jadus, “Macrophages that Kill Glioma Cells Expressing the Membrane Form of Macrophage Colony Stimulating Factor Are Resistant to Prostaglandin E2 and Interleukin-10,” Immunology Letters, Vol. 70, No. 1, 1999, pp. 63-68. doi:10.1016/S0165-2478(99)00129-7
- [79] T. Payner, et al., “Microsomal Prostaglandin E Synthase-1 Regulates Human Glioma Cell Growth via Prostaglandin E(2)-Dependent Activation of Type II Protein Kinase A,” Molecular Cancer Therapeutics, Vol. 5, 2006, pp. 1817-1826. doi:10.1158/1535-7163.MCT-05-0548
- [80] P. Bidwell, K. Joh, H. A. Leaver and M. T. Rizzo, “Prostaglandin E2 Activates cAMP Response Element-Binding Protein in Glioma Cells via a Signaling Pathway Involving PKA-Dependent Inhibition of ERK,” Prostaglandins & Other Lipid Mediators, Vol. 91, No. 1-2, 2010, pp. 18- 29. doi:10.1016/j.prostaglandins.2009.12.002
- [81] W. T. Chiu, et al., “Contribution of Reactive Oxygen Species to Migration/Invasion of Human Glioblastoma Cells U87 via ERK-Dependent COX-2/PGE(2) Activation,” Neurobiology of Disease, Vol. 37, No. 1, 2010, pp. 118-129. doi:10.1016/j.nbd.2009.09.015
- [82] L. Lalier, et al., “Increase in PGE2 Biosynthesis Induces a Bax Dependent Apoptosis Correlated to Patients’ Survival in Glioblastoma Multiforme,” Oncogene, Vol. 26, 2007, pp. 4999-5009. doi:10.1038/sj.onc.1210303
- [83] N. Baryawno, et al., “Tumor-Growth-Promoting Cyclooxygenase-2 Prostaglandin E2 Pathway Provides Medulloblastoma Therapeutic Targets,” Journal of Neuro-Oncology, Vol. 10, No. 5, 2008, pp. 661-674. doi:10.1215/15228517-2008-035
- [84] F. S. Hodi, et al., “Improved Survival with Ipilimumab in Patients with Metastatic Melanoma,” The New England Journal of Medicine, Vol. 363, 2010, pp. 711-723. doi:10.1056/NEJMoa1003466
- [85] N. Nagata, Y. Kusakari, Y. Fukunishi, T. Inoue and Y. Urade, “Catalytic Mechanism of the Primary Human Prostaglandin F2alpha Synthase, Aldo-Keto Reductase 1B1- Prostaglandin D2 Synthase Activity in the Absence of NADP(H),” The FEBS Journal, Vol. 278, No. 8, 2011, pp. 1288-1298. doi:10.1111/j.1742-4658.2011.08049.x
- [86] C. G. Park, et al., “Effects of Prostaglandin F2alpha on Small Intestinal Interstitial Cells of Cajal,” World Journal of Gastroenterology, Vol. 17, No. 9, 2011, pp. 1143-1151. doi:10.3748/wjg.v17.i9.1143
- [87] G. Pugliesi, et al., “Effects of Inhibition of Prostaglandin F(2alpha) Biosynthesis during Preluteolysis and Luteolysis in Heifers,” Theriogenology, Vol. 76, No. 4, 2011, pp. 640-651. doi:10.1016/j.theriogenology.2011.03.017
- [88] G. Hollo, et al., “Long-Term Outcomes of Prostaglandin Analog versus Timolol Maleate in Ocular Hypertensive or Primary Open-Angle Glaucoma Patients in Europe,” Journal of Ocular Pharmacology and Therapeutics, Vol. 27, No. 5, 2011, pp. 493-498.
- [89] B. Bobrowska, A. Tokarz, S. Bialek and M. Seweryn, “Effect of Dietary Supplementation on the Prognostic Value of Urinary and Serum 8-Isoprostaglandin F2alpha in Chemically-Induced Mammary Carcinogenesis in the Rat,” Lipids in Health and Disease, Vol. 10, 2011, p. 40. doi:10.1186/1476-511X-10-40
- [90] A. Dogukan, et al., “A Tomato Lycopene Complex Protects the Kidney from Cisplatin-Induced Injury via Affecting Oxidative Stress as well as Bax, Bcl-2, and HSPs Expression,” Nutrition and Cancer, Vol. 63, No. 3, 2011, pp. 427-434. doi:10.1080/01635581.2011.535958
- [91] Y. Kibar, et al., “The Effect of Intravesical Acetylsalicylic Acid Instillation on Tissue Prostaglandin Levels after Partial Bladder Outlet Obstruction in Rabbits,” Neurourology and Urodynamics, Vol. 30, No. 8, 2011, pp. 1646-1651. doi:10.1002/nau.21153
- [92] N. Miwa, H. Sugino, R. Ueno and O. Hayaishi, “Prostaglandin Induces Ca2+ Influx and Cyclic GMP Formation in Mouse Neuroblastoma X Rat Glioma Hybrid NG108- 15 Cells in Culture,” Journal of Neurochemistry, Vol. 50, No. 5, 1988, pp. 1418-1424.
- [93] S. Zhao, X. Jiang, D. Xue and D. Chen, “Glioma Prostaglandin Levels Correlate with Brain Edema,” Journal of Tongji Medical University, Vol. 18, No. 2, 1998, pp. 115- 118.
- [94] W. Xu, C. L. Chou, D. D. Israel, A. J. Hutchinson and J. W. Regan, “PGF(2alpha) Stimulates FP Prostanoid Receptor Mediated Crosstalk between Ras/Raf Signaling and Tcf Transcriptional Activation,” Biochemical and Biophysical Research Communications, Vol. 381, No. 4, 2009, pp. 625-629.
- [95] B. Zhang, P. C. Tsang, J. L. Pate and M. A. Moses, “A Role for Cysteine-Rich 61 in the Angiogenic Switch during the Estrous Cycle in Cows: Regulation by Prostaglandin f2alpha,” Biology of Reproduction, Vol. 85, No. 2, 2011, pp. 261-268.
- [96] H. J. Kang, et al., “Activation of Peroxisome Proliferators Activated Receptor {Delta} (PPAR{Delta}) Promotes Blastocyst Hatching in Mice,” Molecular Human Reproduction, Vol. 17, No. 10, 2011, pp. 653-660. doi:10.1093/molehr/gar030
- [97] R. L. Keith, et al., “Oral Iloprost Improves Endobronchial Dysplasia in Former Smokers,” Cancer Prevention Research (Phila), Vol. 4, 2011, pp. 793-802. doi:10.1158/1940-6207.CAPR-11-0057
- [98] M. C. Cathcart, et al., “Prostacyclin Synthase Expression and Epigenetic Regulation in Nonsmall Cell Lung Cancer,” Cancer, Vol. 117, No. 22, 2011, pp. 5121-5132. doi:10.1002/cncr.26168
- [99] Y. Pehlivan, et al., “Possible Anti-Metastatic Effect of Iloprost in a Patient with Systemic Sclerosis with Lung Cancer: A Case Study,” Rheumatology International, Vol. 32, No. 5, 2011, pp. 1437-1441.
- [100] R. Ortmann, “Effect of PGI2 and Stable Endoperoxide Analogues on Cyclic Nucleotide Levels in Clonal Cell Lines of CNS Origin,” FEBS Letter, Vol. 90, No. 2, 1978, pp. 348-352. doi:10.1016/0014-5793(78)80402-5
- [101] N. Jaiswal, D. I. Diz, E. A. Tallant, M. C. Khosla and C. M. Ferrario, “The Nonpeptide Angiotensin II Antagonist DuP 753 is a Potent Stimulus for Prostacyclin Synthesis,” American Journal of Hypertension, Vol. 4, No. 3, 1991, pp. 228-233. doi:10.1093/ajh/4.3.228
- [102] A. Cavalié, S. E. Philipp and H. Meves, “ONO-54918-07, a Stable Prostacyclin Analogue, Mimics the Effect of Prostaglandin PGE1 on NG108-15 Cells,” Naunyn-Schmiedeberg’s Archives of Pharmacology, Vol. 376, No. 3, 2007, pp. 165-173. doi:10.1093/ajh/4.3.228
- [103] E. J. Adie, I. Mullaney, F. R. McKenzie and G. Milligan, “Concurrent Down-Regulation of IP Prostanoid Receptors and the Alpha-Subunit of the Stimulatory GuanineNucleotide-Binding Protein (Gs) during Prolonged Exposure of Neuroblastoma x Glioma Cells to Prostanoid Agonists. Quantification and Functional Implications,” Biochemical Journal, Vol. 285, Pt. 2, 1992, pp. 529-536.
- [104] K. M. Chu, K. B. Chow, Y. H. Wong and H. Wise, “Prostacyclin Receptor-Mediated Activation of Extracellular Signal-Regulated Kinases 1 and 2,” Cell Signal, Vol. 16, No. 4, 2004, pp. 477-486. doi:10.1016/j.cellsig.2003.09.004
- [105] Y. M. Seok, H. J. Cho, B. Y. Cha, J. T. Woo and I. K. Kim, “Honokiol Attenuates Vascular Contraction through the Inhibition of the RhoA/Rho-Kinase Signalling Pathway in Rat Aortic Rings,” The Journal of Pharmacy and Pharmacology, Vol. 63, No. 9, 2011, pp. 1244-1251. doi:10.1111/j.2042-7158.2011.01332.x
- [106] M. C. Cathcart, et al., “Examination of Thromboxane Synthase as a Prognostic Factor and Therapeutic Target in Non-Small Cell Lung Cancer,” Molecular Cancer, Vol. 10, 2011, p. 25. doi:10.1186/1476-4598-10-25
- [107] M. G. Castelli, et al., “Prostaglandin and Thromboxane Synthesis by Human Intracranial Tumors,” Cancer Research, Vol. 49, No. 6, 1989, pp. 1505-1508.
- [108] Y. Obara, H. Kurose and N. Nakahata, “Thromboxane A2 Promotes Interleukin-6 Biosynthesis Mediated by an Activation of Cyclic AMP-Response Element-Binding Protein in 1321N1 Human Astrocytoma Cells,” Molecular Pharmacology, Vol. 68, No. 3, 2005, pp. 670-679.
- [109] M. Saito, H. Tanaka, M. Sasaki, H. Kurose and N. Nakahata, “Involvement of Aquaporin in Thromboxane A2 Receptor-Mediated, G 12/13/RhoA/NHE-Sensitive Cell Swelling in 1321N1 Human Astrocytoma Cells,” Cell Signal, Vol. 22, No. 1, 2010, pp. 41-46. doi:10.1016/j.cellsig.2009.09.006
- [110] E. McCoy and H. Sontheimer, “Expression and Function of Water Channels (Aquaporins) in Migrating Malignant Astrocytes,” Glia, Vol. 55, No. 10, 2007, pp. 1034-1043. doi:10.1002/glia.20524
- [111] A. K. Schauff, et al., “Inhibition of Invasion-Associated Thromboxane Synthase Sensitizes Experimental Gliomas to Gamma-Radiation,” Journal of Neuro-Oncology, Vol. 91, No. 3, 2009, pp. 241-249. doi:10.1007/s11060-008-9708-0
- [112] N. O. Schmidt, et al., “Inhibition of Thromboxane Synthase Activity Improves Glioblastoma Response to Alkylation Chemotherapy,” Translational Oncology, Vol. 3, No. 1, 2010, pp. 43-49.
- [113] T. Inazumi, et al., “Prostaglandin E2-EP4 Signaling Suppresses Adipocyte Differentiation in Mouse Embryonic Fibroblasts via an Autocrine Mechanism,” Journal of Lipid Research, Vol. 52, No. 8, 2011, pp. 1500-1508. doi:10.1194/jlr.M013615
- [114] J. Tuettenberg, et al., “Continuous Low-Dose Chemotherapy plus Inhibition of Cyclooxygenase-2 as an Antiangiogenic Therapy of Glioblastoma Multiforme,” Journal of Cancer Research and Clinical Oncology, Vol. 131, No. 1, 2005, pp. 31-40. doi:10.1007/s00432-004-0620-5
- [115] N. Guo, C. J. Baglole, C. W. O’Loughlin, S. E. Feldon and R. P. Phipps, “Mast Cell-Derived Prostaglandin D2 Controls Hyaluronan Synthesis in human Orbital Fibroblasts via DP1 Activation: Implications for Thyroid Eye Disease,” The Journal of Biological Chemistry, Vol. 285, No. 21, 2010, pp. 15794-15804. doi:10.1074/jbc.M109.074534
- [116] U. H. Gandhi, et al., “Selenoprotein-Dependent Up-Regulation of Hematopoietic Prostaglandin D2 Synthase in Macrophages Is mediated through the Activation of Peroxisome Proliferator-activated Receptor (PPAR) {Gamma},” The Journal of Biological Chemistry, Vol. 286, No. 31, 2011, pp. 27471-27482. doi:10.1074/jbc.M111.260547
- [117] D. Kajiwara, et al., “Role of Hematopoietic Prostaglandin D Synthase in Biphasic Nasal Obstruction in Guinea Pig Model of Experimental Allergic Rhinitis,” European Journal of Pharmacology, Vol. 667, No. 1-3, 2011, pp. 389-395. doi:10.1016/j.ejphar.2011.05.041
- [118] T. Nabe, et al., “Inhibition of Hematopoietic Prostaglandin D Synthase Improves Allergic Nasal Blockage in Guinea Pigs,” Prostaglandins & Other Lipid Mediators, Vol. 95, No. 1-4, 2011, pp. 27-34. doi:10.1016/j.prostaglandins.2011.05.001
- [119] Y. H. Choi, et al., “The ERK MAPK/RSK1 Cascade Phosphorylates CREB to Induce MUC5B Gene Expression via DP1 Receptor Signaling,” The Journal of Biological Chemistry, 2011.
- [120] Z. H. Zhang, et al., “EP3 Receptors Mediate PGE2-Induced Hypothalamic Paraventricular Nucleus Excitation and Sympathetic Activation. American Journal of physiology,” Heart and Circulatory Physiology, 2011.
- [121] C. Suzuki, C. Miyamoto, T. Furuyashiki, S. Narumiya and K. Ohinata, “Central PGE(2) Exhibits AnxiolyticLike Activity via EP(1) and EP(4) Receptors in a Manner Dependent on Serotonin 5-HT(1A), Dopamine D(1) and GABA(A) Receptors,” FEBS letters, Vol. 585, No. 14, 2011, pp. 2357-2362. doi:10.1016/j.febslet.2011.06.004
- [122] B. Cha, J. W. Lim, K. H. Kim and H. Kim, “15-DeoxyD12,14-Prostaglandin J2 Suppresses RANTES Expression by Inhibiting NADPH Oxidase Activation in Helicobacter Pylori-Infected Gastric Epithelial Cells,” Journal of Physiology and Pharmacology, Vol. 62, No. 2, 2011, pp. 167-174.
- [123] J. Kim, et al., “Beraprost Enhances the APC Function of B Cells by Upregulating CD86 Expression Levels,” Journal of Immunology (Baltimore, Md.: 1950), Vol. 186, No. 7, 2011, pp. 3866-3873.
- [124] G. Prasanna, et al., “Effect of PF-04217329 a Prodrug of a Selective Prostaglandin EP(2) Agonist on Intraocular Pressure in Preclinical Models of Glaucoma,” Experimental Eye Research, Vol. 93, No. 3, 2011, pp. 256-264. doi:10.1016/j.exer.2011.02.015
- [125] Y. Ikeda-Matsuo, H. Tanji, S. Narumiya and Y. Sasaki, “Inhibition of Prostaglandin E(2) EP3 Receptors Improves Stroke Injury via Anti-Inflammatory and AntiApoptotic Mechanisms,” Journal of Neuroimmunology, Vol. 238, No. 1-2, 2011, pp. 34-43. doi:10.1016/j.jneuroim.2011.06.014
- [126] T. Ueno and K. Fujimori, “Novel Suppression Mechanism Operating in Early Phase of Adipogenesis by Positive Feedback Loop for Enhancement of Cyclooxygenase-2 Expression through Prostaglandin F(2alpha) Receptor Mediated Activation of MEK/ERK-CREB Cascade,” The FEBS Journal, Vol. 278, No. 16, 2011, pp. 2901-2912. doi:10.1111/j.1742-4658.2011.08213.x
- [127] R. Yamagishi, M. Aihara and M. Araie, “Neuroprotective Effects of Prostaglandin Analogues on Retinal Ganglion Cell Death Independent of Intraocular Pressure Reduction,” Experimental Eye Research, Vol. 93, No. 3, 2011, pp. 265-270. doi:10.1016/j.exer.2011.06.022
- [128] S. S. Barbieri, et al., “Tobacco Smoke Regulates the Expression and Activity of Microsomal Prostaglandin E Synthase-1: Role of Prostacyclin and NADPH-Oxidase,” The FASEB Journal, Vol. 25, No. 10, 2011, pp. 3731- 3740. doi:10.1096/fj.11-181776
- [129] E. C. Turner, E. P. Mulvaney, H. M. Reid and B. T. Kinsella, “Interaction of the Human Prostacyclin Receptor with the PDZ Adapter Protein PDZK1: Role in Endothelial Cell Migration and Angiogenesis,” Molecular Biology of the Cell, Vol. 22, No. 15, 2011, pp. 2664-2679. doi:10.1091/mbc.E11-04-0374
- [130] C. Tamburrelli, et al., “Epoprostenol Inhibits Human Platelet-Leukocyte Mixed Conjugate and Platelet Microparticle Formation in Whole Blood,” Thrombosis Research, Vol. 128, No. 5, 2011, pp. 446-451. doi:10.1016/j.thromres.2011.05.010
- [131] A. Pavelescu and R. Naeije, “Effects of Epoprostenol and Sildenafil on Right Ventricular Function in Hypoxic Volunteers: A Tissue Doppler Imaging Study,” European Journal of Applied Physiology, Vol. 112, No. 4, 2011, pp. 1285-1294. doi:10.1007/s00421-011-2085-y
- [132] N. Boulos, et al., “Notch3 is Essential for Regulation of the Renal Vascular Tone,” Hypertension, Vol. 57, No. 6, 2011, pp. 1176-1182. doi:10.1161/HYPERTENSIONAHA.111.170746
- [133] N. Ghonem, et al., “Treprostinil, a Prostacyclin Analog, Ameliorates Ischemia-Reperfusion Injury in Rat Orthotopic Liver Transplantation,” American Journal of Transplantation, Vol. 11, No. 11, 2011, pp. 2508-2516. doi:10.1111/j.1600-6143.2011.03568.x
- [134] T. H. Lin, et al., “Upregulation of Heme Oxygenase-1 Inhibits the Maturation and Mineralization of Osteoblasts,” Journal of Cellular Physiology, Vol. 222, No. 3, 2010, pp. 757-768.
- [135] P. Raman, B. L. Kaplan, J. T. Thompson, J. P. Vanden Heuvel and N. E. Kaminski, “15-Deoxy-delta12,14-prostaglandin J2-Glycerol Ester, a Putative Metabolite of 2-Arachidonyl Glycerol, Activates Peroxisome Proliferator Activated Receptor Gamma,” Molecular Pharmacology, Vol. 80, No. 1, 2011, pp. 201-209. doi:10.1124/mol.110.070441
- [136] H. Li, Y. Zhao, S. Zhou and C. K. Heng, “Serum Amyloid A Activates Peroxisome Proliferator-Activated Receptor Gamma through Extracellularly Regulated Kinase 1/2 and COX-2 Expression in Hepatocytes,” Biochemistry, Vol. 49, No. 44, 2010, pp. 9508-9517. doi:10.1021/bi100645m
- [137] Y. Yokoyama, et al., “15-Deoxy-Delta12,14-prostaglandin J2 Inhibits Angiotensin II-Induced Fibronectin Expression via Hepatocyte Growth Factor Induction in Human Peritoneal Mesothelial Cells,” Therapeutic Apheresis and Dialysis, Vol. 14, No. 1, 2010, pp. 43-51. doi:10.1111/j.1744-9987.2009.00702.x
- [138] K. Bach-Ngohou, et al., “Enteric Glia Modulate Epithelial Cell Proliferation and Differentiation through 15- Deoxy-12,14-Prostaglandin J2,” The Journal of Physiology, Vol. 588, Pt. 14, 2010, pp. 2533-2544. doi:10.1113/jphysiol.2010.188409
- [139] M. A. Syed, et al., “Expression of TREM-1 is Inhibited by PGD2 and PGJ2 in Macrophages,” Experimental Cell Research, Vol. 316, No. 19, 2010, pp. 3140-3149. doi:10.1016/j.yexcr.2010.08.009
- [140] T. Katura, T. Moriya and N. Nakahata, “15-Deoxy-delta 12,14-prostaglandin J2 Biphasically Regulates the Proliferation of Mouse Hippocampal Neural Progenitor Cells by Modulating the Redox State,” Molecular Pharmacology, Vol. 77, No. 4, 2010, pp. 601-611. doi:10.1124/mol.109.061010
- [141] C. Cherdon, et al., “BM-573 Inhibits the Development of Early Atherosclerotic Lesions in Apo E Deficient Mice by Blocking TP Receptors and Thromboxane Synthase,” Prostaglandins & Other Lipid Mediators, Vol. 94, No. 3-4, 2011, pp. 124-132. doi:10.1016/j.prostaglandins.2011.03.001
- [142] A. Drouin, et al., “Up-Regulation of Thromboxane A(2) impairs Cerebrovascular eNOS Function in Aging Atherosclerotic Mice,” Pflugers Archiv: European Journal of Physiology, Vol. 462, No. 3, 2011, pp. 371-383.
- [143] A. Koumura, et al., “Fasudil and Ozagrel in Combination Show Neuroprotective Effects on Cerebral Infarction after Murine Middle Cerebral Artery Occlusion,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 338, No. 1, 2011, pp. 337-344. doi:10.1124/jpet.110.177675
- [144] J. M. Hernandez and L. J. Janssen, “TP-Receptor Activation Amplifies airway Stretch-Activated Contractions Assessed in Perfused Intact Bovine Bronchial Segments,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 339, No. 1, 2011, pp. 248-256. doi:10.1124/jpet.111.182246
- [145] F. Y. Senouvo, et al., “Improved Bioavailability of Epoxy-Eicosatrienoic Acids Reduces TP-Receptor-AgonistInduced Tension in Human Bronchi,” American journal of physiology,” Lung Cellular and Molecular Physiology, Vol. 30, No. 5, 2011, pp. L675-L682. doi:10.1152/ajplung.00427.2010
- [146] P. Toth, B. Rozsa, Z. Springo, T. Doczi and A. Koller, “Isolated Human and Rat Cerebral Arteries Constrict to Increases in Flow: Role of 20-HETE and TP Receptors,” Journal of Cerebral Blood Flow and Metabolism, Vol. 31, No. 10, 2011, pp. 2096-2105. doi:10.1038/jcbfm.2011.74
- [147] P. F. Lesault, et al., “Daily Administration of the TP Receptor Antagonist Terutroban Improved Endothelial Function in High-Cardiovascular-Risk Patients with Atherosclerosis,” British Journal of Clinical Pharmacology, Vol. 71, No. 6, 2011, pp. 844-851. doi:10.1111/j.1365-2125.2010.03858.x
NOTES
*Corresponding author.