International Journal of Clinical Medicine
Vol.2 No.4(2011), Article ID:7569,14 pages DOI:10.4236/ijcm.2011.24086
Mitocans as Novel Agents for Anticancer Therapy: An Overview
![]()
1Department of Pharmacology, Principal K. M. Kundnani College of Pharmacy, Mumbai, India; 2Department of Pharmaceutics, Principal K. M. Kundnani College of Pharmacy, Mumbai, India; 3Karlsruhe Institute of Technology, Institute of toxicology and Genetics, Karlsruhe, Germany.
Email: *mail to:vspanda@rediffmail.com
Received March 10th, 2011; revised July 15th, 2011; accepted July 31th, 2011.
Keywords: Mitocans, Mitochondria, Cancer, Apoptosis, Membrane Permeabilization, Energy Metabolism
ABSTRACT
Many conventional anticancer drugs have an associated lack of safety by their toxicity. Relatively faster mutations in tumor cells pose a significant obstacle in treatment of cancer. Recently, “Mitocans” have emerged as a novel class of anticancer agents selectively targeting tumor cells and thus, are much less toxic than conventional anticancer chemotherapeutic agents. Mitocans are drugs that act directly on mitochondria within the cell, thus causing changes in energy metabolism of the cell. Amongst these mitocans, a-Tocopheryl succinate or vitamin E analogs are studied very well by researchers. This review discusses mitochondrial drug targeting strategies and a variety of novel mitochondrial drug targets of mitocans, such as electron transport chain, mitochondrial permeability transition, Bcl-2 family proteins and mitochondrial DNA. The purpose of this review is to focus on the various classes of mitocans, the mechanisms by which these drugs specifically act on tumor cells and their applications in cancer chemotherapeutics.
1. Introduction
Despite a steady development in cancer chemotherapeutics in recent years, this millennium has been a witness to various types of cancers becoming a major cause for mortality. Various chemotherapeutic or chemopreventive drugs that target cell microtubules and cytoskeleton components have been used for cancer treatment over the years [1,2]. The treatment of cancer to date remains a significant challenge, particularly due to continuous mutations, which make the tumor cells resistant to established chemotherapeutic molecules or drugs. Hence, for resolving the current problem, newer approaches are to be sought and put into practice. As mitochondria are the “power house” of the cell and are essential for its survival, they are novel targets showing considerable promise for intervention and treatment of cancer [3-5]. Anticancer agents specifically targeting cancer cell mitochondria are referred to as “mitocans”. Mitocans act by destabilizing mitochondria and unleashing their apoptotic potential, leading to death of tumor cells or reducing their growth drastically [6,7]. They destabilize mitochondria and cause the cytosolic release of modulators of apoptosis [8]. Some drugs have the potential to affect mitochondrial function directly while others may have different primary drug targets at other cellular locations and alter mitochondrial function as a side effect [3,8]. Thus, mitochondria become a major target in cancer cells.
The purpose of this review is to present an update on the action of various therapeutically active substances affecting mitochondria in tumor cells. This review describes various drug molecules acting on the mitochondria (mitocans), different classes of mitocans and their mechanisms of action. We also discuss about the scope of these drugs with examples like betulinic acid and α-TOS, which are more researched drugs in this field.
2. Mitochondria: The Energy House of a Cell
Mitochondria play a central role in energy metabolism (energy generation process) within the cell [9]. Impaired respiratory chain functioning can occur due to mitochondrial DNA mutations, leading to decreased ATP production [10], free radical generation [11] and alterations in cellular calcium handling [12]. This altogether initiates peroxidation of biomolecules like mitochondrial DNA, proteins, lipids and alters the mitochondrial permeability by opening transition pores, leading to apoptotic cell death[13,14]. Electrons are transported through the mitochondrial respiratory complexes during oxidative phosphorylation and this establishes a proton gradient across the mitochondrial membrane for ATP production. Production of superoxide radicals is one of the important biochemical events associated with energy metabolism. When an electron escapes from the electron transport chain in mitochondria especially at complex I or III, it may react with molecular oxygen to form the superoxide radical [15,16]. During the cellular metabolism, superoxide radicals are constantly generated and may be converted into hydrogen peroxide and/or other reactive oxygen species (ROS), which are highly reactive and toxic to the cells when accumulated to higher levels. These species need to be removed from cells using a antioxidant system which includes metabolic enzymes such as superoxide dismutase (SOD), peroxidases and other redox molecules [17]. Maintenance of appropriate levels of intracellular ROS is vital for redox balance and signalling cellular proliferation under physiological conditions [18]. However, overproduction of ROS or a decreased ability of the cell to remove ROS can result in increased levels of ROS, causing cell death due to lipid peroxidation [19], oxidative DNA changes and protein and enzyme inactivation. This cell damaging effect may also kill the cancer cells if ROS are inductively accumulated in malignant cells which are metabolically active and produce higher ROS. Malignant cells are already under oxidative stress and are more susceptible to further stress by exogenous ROS generating compounds [20-23]. It is well known that when oxidative stress due to ROS reaches a threshold, cancer cell apoptosis occurs as shown in case of 2-methylestradiol [24,25]. Once apoptosis signalling pathways are initiated, cytochrome C and apoptotic factors are released into cytoplasm. This is associated with changes in mitochondrial ultrastructure, membrane permeability and transmembrane potential [17].
3. Mitochondria as Pharmacological Target
Mitochondria provide a novel targeting site which can selectively kill cancer cells without affecting normal cells, unlike other anticancer agents [26]. For this reason, many drugs that target mitochondria are currently in clinical trials for treating cancer. There are several reports suggesting the possible role of mitochondria in various complex processes such as apoptosis and cardioprotection [3, 7,8,27]. Mitochondrial dysfunctions lead to various neurodegenerative disorders like Alzheimer’s disease [28-31], Parkinson’s disease [32-34], Huntington’s disease [35,36] and Amyotrophic lateral sclerosis [37-39], the so-called “mitochondrial diseases”. Thus, identification of mitochondria as a primary or secondary target of a drug may help us to understand better its mechanism of action and open new perspectives for its application in chemotherapy [40]. Various therapeutically active drugs like Paclitaxel (anticancer) [41-44], Cyclosporin A & Rapamycin (immunosuppressant) [45-47], antiviral nucleotide analogues [48-50], some sulfonylureas [51-53] and anesthetics [54,55] are known to act on the mitochondria. These drugs selectively disrupt energy metabolism in cancer cells leading to ROS generation and cell death. Because of their mitochondrial action and anticancer property, these drugs have been termed “mitocans”.
4. Cell Biology of Apoptosis
Apoptosis is a process that develops in 3 phases [6] as follows;
1) Initiation phase, which varies depending on the apoptosis-inducing agent and the biochemical pathway activated by it;
2) Commitment phase, which is common to different types of apoptosis, during which the cell “decides” to commit suicide;
3) Common degradation phase, which is characterized by activation of catabolic hydrolases (caspases and nucleases).
Before cells lyse, permeabilization of both, inner as well as the outer mitochondrial membrane takes place with dissipation of inner transmembrane potential (∆ψm) and/or release of apoptogenic proteins, such as cytochrome c and apoptosis-inducing factor (AIF) via the outer membrane [56-59]. This suggests that caspase activation may not be always required for apoptotic cell death to occur. Rather, cell death is intimately associated with the permeabilization of mitochondrial membranes [60]. A number of triggers such as Bax [61], Bak [62], c-Myc [62], PML [63], Fas-associated death domain [64], glucocorticoid receptor occupancy [58], tumor necrosis factor [65], growth factor withdrawal [66], CXCR4 (CXC chemokine receptor 4 also called fusin), cross-linking [67], and chemotherapeutic agents, such as etoposide [58], camptothecin [68], or cisplatin [69] induce cell death without activation of caspases and nucleases.
With the use of cell-free systems where subcellular fractions (e.g., mitochondria, nuclei, and cytosol) are mixed together, Costantini et al. have demonstrated that apoptosis of mammalian cells develops in several steps in vitro [4]. Pro-apoptotic second messengers like ceramide, Ca2+ and nitric oxide, whose nature depends on the apoptosis-inducing agent, accumulate in the cytosol during initiation phase. These agents then induce mitochondrial membrane permeabilization, allowing cells to enter the commitment phase. The apoptotic changes in the mitochondria include ∆ψm loss, transient swelling of the mitochondrial matrix, mechanical rupture of outer membrane and/or its nonspecific permeabilization by giant protein-permeant pores, and release of soluble intermembrane proteins (SIMPs) like cytochrome c and adenylate kinase 2, through the outer membrane [59]. Once the mitochondrial membrane integrity is lost, a collapse in bioenergetic status, redox equilibrium and ion homeostasis takes place leading to cell death. The activation of proteases (caspases) and nucleases by SIMPs is necessary for acquisition of apoptotic morphology [59], the degradation step, to the point of no return of apoptotic process [70]. Different SIMPs provide a molecular link between mitochondrial membrane permeabilization and activation of catabolic hydrolases: cytochrome c (a heme protein that participates in caspase activation) [71], certain procaspases (in particular, procaspases 2 and 9, which, in some cell types, are selectively enriched in mitochondria) [72] and AIF [59,70]. AIF is a nuclearencoded intermembrane flavoprotein that translocates to nucleus where it induces caspase-independent peripheral chromatin condensation and degradation of DNA into 50 Kbp fragments [70].
5. Mechanism of Action of Mitocans
The tumor cells are under increased intrinsic oxidative stress and are prone to apoptosis induced by the generated free radicals (Figure 1). Huang et al. reported a novel strategy to enhance superoxide generation and block mitochondrial electron transport chain in leukemic cells which in turn will increase the apoptosis induced by anticancer drugs [24]. This mechanism has already been proven using a specific mitochondrial electron transport chain complex I inhibitor, i.e. rotenone [73]. Partial inhibition of mitochondrial respiration enhances electron leakage from the transport chain, leading to an increased generation of superoxide radicals and thus leukemic cells become more sensitive to anticancer agents (i.e. those agents which act by free radical generation). The antileukemic agent As2O3 also inhibits mitochondrial respiratory function and increases generation of free radicals in cultured as well as primary leukemic cells [74]. Thus As2O3 can be used in combination with other anticancer agents which act by increasing free radical generation [17,75,76].
6. Molecular Mechanisms of Mitochondrial Membrane Permeabilization
The mechanism of mitochondrial membrane permeabilization is not completely understood. Some investigators have proposed that pro-apoptotic members of the Bcl-2 family are inserted in the outer membrane [77] where they oligomerize and form cytochrome c permeant pores in an autonomous fashion, not requiring the interaction with other mitochondrial membrane proteins [77,78]. However, Bax (pro-apoptotic Bcl-2 protein)-induced membrane permeabilization is inhibited by cyclosporin A (CsA) and bongkrekic acid (BA), two inhibitors of formation of the permeability transition pore, suggesting that sessile mitochondrial proteins (targets of CsA and BA) are involved in this process [78]. The permeability transition pore has a polyprotein structure that is formed at the contact sites between the inner and outer membranes [79,80]. One of the key proteins of the permeability transition pore complex (PTPC) is adenine nucleotide
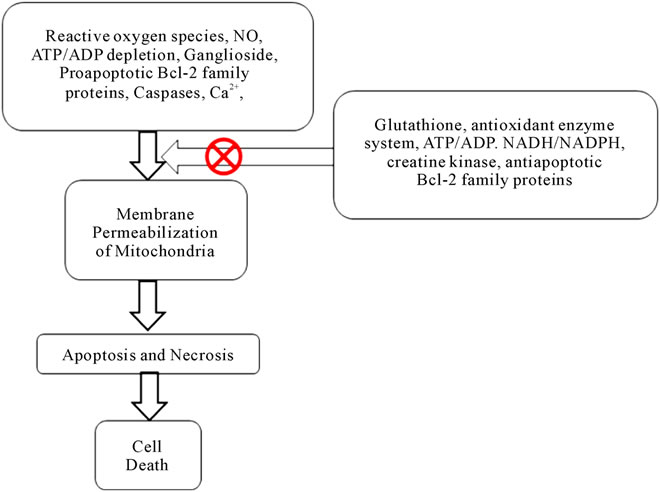
Figure 1. Mechanism of cell death through mitochondrial dysfunction.
translocator (ANT). ANT, the target of BA, is the most abundant inner membrane protein [78]. ANT normally functions as a specific carrier protein for the exchange of adenosine triphosphate (ATP) and adenosine diphosphate (ADP), but it can become a nonspecific pore (Figures 2 and 3) [4].
The adenine nucleotide translocator (ANT) plays a dual role in the process of pore formation on the mitochondrial membrane as follows:
It functions as a highly specific transporter by exchanging adenosine diphosphate (ADP) and adenosine triphosphate (ATP) on the inner mitochondrial membrane.
The ANT can also become a pore, acting at several levels of conductance, specificity, and reversibility. Bcl-2 inhibits pore formation whereas Bax, atractyloside, palmitate, Ca2+, thiol oxidants, and viral protein R (Vpr) from human immunodeficiency virus-1 favor pore formation.
Due to the presence of a pore protein, termed porin or voltage-dependent anion channel (VDAC), the outer mitochondrial membrane is permeable to polar molecules of up to five kDA and is impermeable to cytochrome c. However, VDAC can allow for leaking of cytochrome c by interacting with Bcl-2 and Bax, thus facilitating pore formation.
ANT, thus has two functions—that of a vital ATP/ ADP carrier and that of a lethal pore (Figure 2) [81]. ANT interacts with another equally abundant protein of outer membrane, VDAC, as well as a soluble protein of the mitochondrial matrix, cyclophilin D, the target of CsA [80]. ANT and/or VDAC physically interact with Bcl-2 and Bax [78,79], the peripheral benzodiazepine receptor (PBR), as well as with several proteins involved
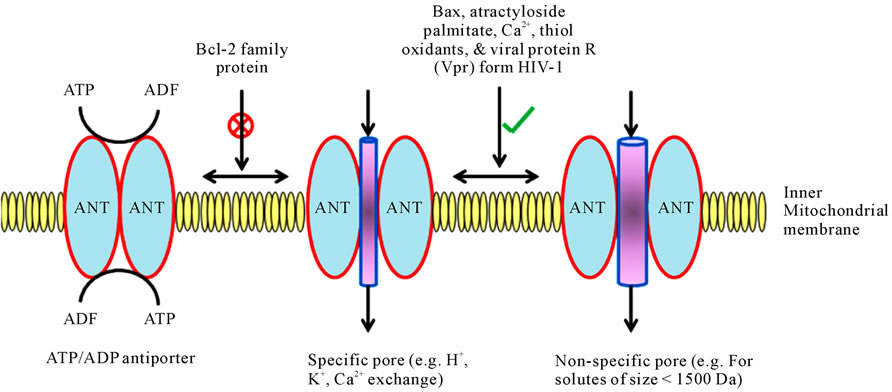
Figure 2. Role of ANT (adenine nucleotide translocator) in specific or nonspecific pore formation on the inner mitochondrial membrane.
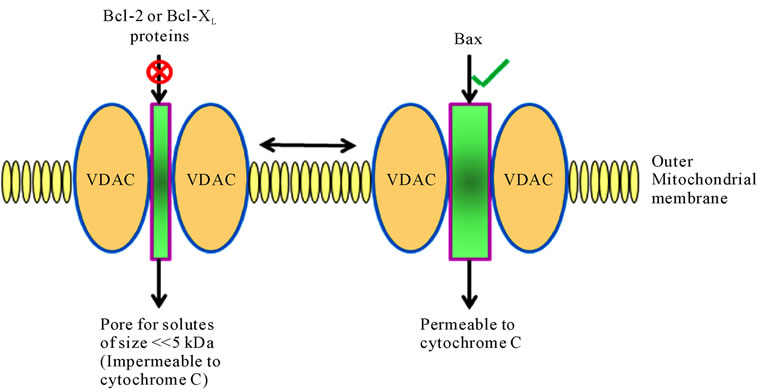
Figure 3. Function of VDAC (voltage dependent anion channel) in pore formation on the outer mitochondrial membrane.
in the regulation of energy metabolism (e.g., hexokinase II and creatine kinase) [82]. It appears that the PTPC simultaneously controls the permeability of the outer membrane (pore forming protein: VDAC and/or Bax) [83] and that of the inner membrane (pore-forming protein: ANT and Bax) (Figure 3) and participates in energy metabolism (via the kinases and ANT). Because of the presence of multiple proteins, each of which influences pore opening, the PTPC senses a large number of metabolic conditions: redox couples (e.g., reduced versus oxidized glutathione, nicotinamide adenine dinucleotide [NAD+] versus nicotinamide adenine dinucleotide phosphate [NADPH], local ATP/ADP concentrations, different metabolites (e.g., glucose and creatine), ions (Ca2+ and Mg2+), the pH, and the ∆ψm. All of these factors determine the probability of membrane permeabilization by the PTPC [57]. Recombinant Bcl-2 as well as Bcl-xL (both of which are anti-apoptotic) have a direct inhibitory effect on the PTPC [79], as well as on ion channel formation by purified ANT [84] and VDAC [83]. In contrast, Bax (which is pro-apoptotic) aids ANT [79,84] or VDAC [83] to create large channels. The permeabilization of inner and/or outer mitochondrial membranes causes oxidation of reduced NADPH and glutathione, the depletion of ATP, the dissipation of ∆ψm and release of Ca2+ from the matrix leading to altered homeostasis of intracellular ions. All of these changes themselves increase the probability of PTPC opening [57]. This has two important implications: firstly, opening of PTPC in a self amplification loop co-ordinates lethal response among mitochondria within the same cells and secondly, the final result of PTPC opening does not depend on initiating a stimulus, be it a specific pro-apoptotic signal transduction cascade or nonspecific damage at the energy or redox levels [4,57,85].
PTPC (and its components) may not be the only mechanism by which mitochondrial membranes are permeabilized. Pro-apoptotic members of the Bcl-2 family, such as Bax or Bid, may cause outer-membrane permeabilization without inducing an immediate dissipation [86]. Apoptosis without a complete ∆ψm loss has also been reported to occur in some cell lines, such as human promyelocytic leukemia cells (HL60) [87]. It is possible that Bax and other pro-apoptotic members of Bcl-2 family act independently by forming giant channels and/or acting on mitochondrial structures other than PTPC [83,88,89].
7. Pro-Apoptotic Signal-Transducing Molecules Acting on Mitochondria
7.1. Indirect Apoptotic Signals
Conventional anticancer agents, such as etoposide, doxorubicin, cisplatin, or paclitaxel (Taxol), trigger apoptosis by Inducing: 1) p53 expression [90,91]; 2) ceramide/GD3 pathway [92,93]; 3) CD95/CD95L ligand system [67,94]
Affecting Bcl-2 like proteins [83,95]
Compromising redox or energy balance [22]
Thus, these agents elicit mitochondrial permeabilization in an indirect fashion by induction of endogenous effectors that are involved in physiologic control of apoptosis. Following are important endogenous effectors that exert a direct effect on mitochondria and its PTPC.
7.2. Direct Apoptotic SignalsRedox Metabolism
De novo production of ROS resulting from over-expression of pro-apoptotic anti-oncogene p53 [90] or from treatment of cells with a second messenger such as ceramide [96] may cause changes in cellular redox potentials, depletion of reduced glutathione, or NADPH, all of which may induce or facilitate PTPC opening [57]. Peroxynitrite (formed by reaction of nitric oxide with superoxide anion) is also a potent PTPC trigger [97]. The mitochondrial mega-channel possesses several redoxsensitive sites, one of which is modulated by NADPH whereas a second is affected by mitochondrial matrix glutathione levels, is most likely located on ANT [98], and thiol oxidation will convert ANT to become a large nonspecific pore [99].
7.3. Energy Metabolism
ADP and ATP are physiologic ligands of ANT and they function as endogenous inhibitors of PTPC [57,100] and/ or pore formation induced by atractyloside [101], ROS, or thiol cross-linkers [84,93]. Their depletion, therefore, might facilitate PTPC opening. Matrix alkalinization and/or ∆ψm reduction also trigger PTPC opening [80]. Thus, uncoupling or inhibition of respiratory chain (which leads to a decrease in ∆ψm) may be expected to favour mitochondrial membrane permeabilization [102].
7.4. Lipid Messengers
Ceramide is generated in cells exposed to several apoptosis-inducing stimuli, including signalling via the Fas/ Apo-1/CD95 receptor or tumor necrosis factor (TNF) receptor, nonspecific stress, or cytotoxic drugs [93]. When added to cells, ceramide induces mitochondrial membrane permeabilization [94,103] but not the PTPC opening [103]. To induce apoptosis, ceramide must be converted into ganglioside GD3 in the Golgi apparatus. GD3 then translocates to mitochondria and causes PTPC opening [94].
7.5. Cytosolic Calcium
Ca2+ ions are among the most efficient triggers of the PTPC. At supraphysiologic doses (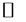 10 mM), Ca2+ suffices to induce permeability transition whereas at lower doses, it facilitates induction of permeability transition (PT) by other stimuli [57,104].
10 mM), Ca2+ suffices to induce permeability transition whereas at lower doses, it facilitates induction of permeability transition (PT) by other stimuli [57,104].
7.6. Pro-Apoptotic Members of Bcl-2 Family
On induction of apoptosis, several pro-apoptotic Bcl-2 family members can translocate from the cytosol (Bax, Bid, and Bad) or from the microtubules (Bim) to mitochondria, where they can incorporate into the outer membrane and undergo a conformational change [77, 105]. The mechanism\of Bax translocation to mitochondria involves activation by JNK through phosphorylation of 14-3-3, a cytoplasmic anchor of Bax [106]. However, Bid translocation likely involves its cleavage by caspase 8 [107], and Bad translocation may involve its dephosphorylation, causing its release from cytosolic 14-3-3 protein [108]. Pro-apoptotic signalling may also lead to the inactivation of anti-apoptotic members of Bcl-2 family. Inactivation of Bcl-2 is achieved by chemotherapeutic agents (such as paclitaxel), which act on microtubule assembly and cause its hyperphosphorylation and, simultaneously, favour opening of PT pore [109]. In addition, Bcl-2 can be cleaved by caspase 3 in a reaction that yields a pro-apoptotic product [110].
7.7. Caspases
Ligation of some receptors can lead to a rapid proteolytic activation of caspases within seconds or minutes. Caspases can act on members of the Bcl-2 family (e.g., caspase 8 cleaves Bid, caspase 1 cleaves Bcl-xL, and caspase 3 cleaves Bcl-2), thereby activating pro-apoptotic members of the Bcl-2 family (Bid) or inactivating anti-apoptotic members (Bcl-2 and Bcl-xL) [60,65,66,72, 87,110].
8. Chemotherapeutic Agents Acting Directly on Mitochondria
Mitocans affect mitochondria-associated activities such as hexokinase inhibition, activation of mitochondrial permeability transition pore (MPTP), inhibition of Bcl-2 anti-apoptotic proteins, and blocking the electron transport/respiratory chain [3,4,6,7,17,89,111-114]. The classification of mitocans is shown in Table 1 [5], and examples of some of the prominent members of these groups are discussed below.
The first class of Mitocans is hexokinase inhibitors which selectively induce apoptosis in cancer cells that metabolize anaerobically [115]. The glucose metabolites, 2-deoxyglucose (2DG), oxamate, and 3-bromopyruvate are hexokinase-inhibiting mitocans [116]. In tumor cells, hexokinase isoforms HK-I and HK-II bind to voltage-dependent anion channel (VDAC) on the outer mitochondrial membrane to inhibit cytochrome c release, and protect against apoptotic cell death [117]. Use of hexokinase inhibitory drugs should enhance the efficacy of conventional cancer chemotherapeutics and radiation regimens that focus on aerobic cancer cells. In addition, hexokinase inhibitors may be used in conjunction with anti-angiogenic agents to limit oxygen supply to tumor cells [5,117-121].
The second class of mitocans includes novel small molecule inhibitors of Bcl-2 and Bcl-xL anti-apoptotic proteins that are overexpressed in cancer cells [122]. The BH4 (Bcl-2 homology 4) helix-containing anti-apoptotic proteins Bcl-2 and Bcl-xL both share a hydrophobic groove on their surface whose function is to bind the BH3 amphipathic helix of pro-apoptotic family members, thereby preventing apoptosis. This hydrophobic groove also binds a range of small molecules, including the natural compound gossypol, that result in blocking of BH3 binding [123]. Thus, small molecules blocking Bcl-2 and Bcl-xL will enable BH3 family of pro-apoptotic inducers to then freely bind to their relevant targets and induce apoptosis. The BH3 mimetics are only recently entering clinical trial in patients with cancer [113,124- 126].
Mitocans from the arsenite class of compounds (trivalent inorganic salt formed by arsenic trioxide) have been

Table 1. Classification of mitocans.
used medically for many years to treat cancers and are effective against hematological and other malignancies [74,75,127]. Hence, arsenic oxides and their derivatives have become established as effective treatments for acute promyelocytic leukemia and are in trials for other hematological cancers, including myelodysplastic syndromes, multiple myeloma, and chronic myelogenous leukemia [128]. The arsenite compounds are likely to modulate critical cysteine residues in the ANT channel, thereby inhibiting its activity [129,130].
Another group of mitocans is represented by the ANT channel-inhibiting drugs such as lonidamine. Lonidamine (an indazole carboxylate derivative) induces activation of MPTP and causes mitochondrial membrane permeabilization by binding and affecting ANT in mitochondrial inner membrane of tumor cells [129,131]. Although lonidamine is a potent anti-proliferative anticancer drug, a recent clinical trial have shown it to have little or no additional benefit over conventional therapies and, as a result, has not been pursued further as a broad-spectrum anticancer therapy [132].
CD437 (6[3-adamantyl-4-hydroxyphenyl]-2-naphthalene carboxylic acid) is a new synthetic retinoid acid receptor g (RARg) agonist inducing apoptosis of human breast, lung, cervical, and ovarian carcinomas, melanoma, prostate cancer, neuroblastoma, and acute promyelocytic leukemia (APL). Several mechanisms of induction of cell death process have been reported; activation of AP-1 complex; increase of p53, p21, and Bax; decrease of Bcl-xL; cell-cycle arrest; and activation of caspase 3 and caspase 7 [132,133]. Moreover, in intact cells CD437- dependent caspase activation is preceded by release of cytochrome C from mitochondria. CD437 also causes membrane permeabilization and this effect is prevented by PTPC inhibitors CsA and BA. Since CD437-mediated cell killing is suppressed by CsA and BA, it appears plausible that CD437 exerts its cytotoxic effects via PTPC [133-136].
Attempts have been made to use cationic lipophilic toxins like MKT-077, as anti-cancer agents. MKT-077, a cationic rhodacyanine dye, is selectively toxic to carcinoma cells in vitro and in vivo [137], perhaps owing to higher ∆ψm in carcinoma cells versus normal cells. Two mechanisms accounting for mitochondrial toxicity of MKT-077 have been proposed [138,139], namely, a selective MKT-077-driven depletion of mitochondrial DNA in carcinoma cells but not in normal epithelial cells and inhibition of mitochondrial respiration with a decrease in the activities of succinate–cytochrome c reductase and cytochrome oxidase. All of these effects are enhanced by photoactivation. Because of its selectivity toward tumor cells, MKT-077 is currently being evaluated in phase I clinical trials [137,140]. Another cationic lipophilic dye, chloromethyl-X-rosamine, also acts as a photosensitizer [141].
Betulinic acid, a pentacyclic triterpene, is a novel experimental anticancer drug. It possesses anti-tumor activity in vitro and in vivo in melanoma, neuroectodermal tumors, and glioma cell lines. Fulda et al. have shown that betulinic acid induces apoptosis via direct mitochondrial alterations [142]. In betulinic acid-induced apoptosis, perturbation of mitochondrial function, including loss of mitochondrial permeability transition, precedes other key features of apoptosis such as activation of caspase cascade and nuclear fragmentation via the liberation of soluble factors, such as cytochrome C or AIF [142]. Bcl-2 and Bcl-xL block all mitochondrial and cellular manifestations of apoptosis induced by betulinic acid, as does bongkrekic acid, an inhibitor of PTPC [143, 144].
The redox-silent analogs of vitamin E (VE) are a new group of mitocans which unlike antioxidant VE, selectively induce apoptosis in malignant cells via mitochondria-dependent apoptotic signalling [120]. Alpha tocopheryl succinate (α-TOS), a prominent example of this class is postulated to act by two mechanisms: the major one, in which it inhibits oxidative respiration at the level of complex II (CII), and an auxiliary role, which involves its binding to Bcl-2 and Bcl-xL to allow Bax to form mitochondrial membrane channels [92]. Thus, upon inhibiting the activity of CII, α-TOS impairs transfer of electrons along the redox chain, which leads to generation of ROS, such as the superoxide anion radicals. In the cytosol, ROS oxidize cysteine residues on Bax monomers to form disulfide bridges between monomers of Bax, causing a conformational change and dimerisation that is followed by Bax mobilisation to the mitochondrial outer membrane to form (mega) channels. In the mitochondria, ROS trigger the oxidase activity of cytochrome C that leads to oxidative modification of cardiolipin (CL) with ensuing release of cytochrome C from its binding to the mitochondrial inner membrane phospholipid, followed by cytochrome C extrusion via the Bax channel to cytosol [92,145]. α-TOS may also complex with BH3 binding hydrophobic groove of Bcl-2 and Bcl-xL, preventing capture of pro-apoptotic Bax homologue Bak by anti-apoptotic Bcl-2 and Bcl-xL. As a result, there is increase in Bak, which is required to form channels in mitochondrial outer membrane, leading to induction of apoptosis especially in prostate cancer cells [125]. In particular, hemisuccinate and two proximal isoprenyl units of side chain were shown to play a critical role in ligand anchoring and Bcl-2 protein-ligand complex formation. From the relationship of α-TOS with UbQ, it is speculated that Bcl-2 and Bcl-xL bind both ubiquinone and quinine related structures. In fact, many small chemical molecules that have been found recently to bind these proteins are likely to mimic ubiquinones [5]. For example, antimycin A, a well-described inhibitor of quinonebinding site on cytochrome bc1 in mitochondrial respiratory chain, has also been shown to compete for binding with BH3 for hydrophobic groove of either Bcl-2 or BclxL [146]. Furthermore, a 2-methoxy antimycin A derivative with no inhibitory effects on the respiratory chain retains selectivity for Bcl-xL. Hence, a role for quinones such as UbQ in binding anti-apoptotic Bcl-2-related family members, affecting their ability to form dimer with BH3 pro-apoptotic family members, seems very likely [5].
The mitochondrially targeted form of α-TOS called MitoVES is a recent development in the VE class of mitocans. Mitochondrially targeted vitamin E succinate (MitoVES) is modified so that it is preferentially localized into an ideal position across the interface of the mitochondrial inner membrane and matrix, greatly enhancing its pro-apoptotic and anti-cancer activity. Using genetically manipulated cells, MitoVES causes apoptosis and generation of reactive oxygen species (ROS) in CII-proficient malignant cells but not their CII-dysfunctional counterparts. The agent has no effect either on the enzymatic activity of CI or on electron transfer from CI to CIII [147].
Mastoparan, a peptide isolated from wasp venom, is the first peptide known to induce mitochondrial membrane permeabilization and apoptosis via a CsA-inhibitable mechanism [148]. This peptide has an α-helical structure and possesses some positive charges that are distributed on one side of the helix. A similar peptide (KLAKLAKKLAKLAK or (KLAKLAK)2 (K = lysine, L = alanine, and A = leucine) has been found recently to disrupt mitochondrial membranes when it is added to purified mitochondria, although the mechanisms of this effect have not been elucidated [149]. The pro-apoptotic 96 amino acid protein viral protein R (Vpr) from human immunodeficiency virus-1 contains an a-helix with several cationic charges that concentrate on same side of helix [150]. Vpr induces apoptosis via a direct effect on the mitochondrial PTPC, causes dissipation of Δψm as well as the mitochondrial release of apoptogenic proteins, such as cytochrome C or AIF.
9. Conclusions
The search for a selective and efficient anticancer agent for treating neoplastic diseases has yet to deliver a universally suitable compound. Recently, a novel target for potential anticancer agents, the mitochondrion, was discovered which shows a considerable promise for future clinical applications. Anticancer agents specifically targeting cancer cell mitochondria are the “mitocans”, which achieve cancer cell death by mitochondrial mediated apoptosis. But, further clinical studies are required for mitocans to be used as established anticancer therapies or in conjunction with existing chemotherapies. As mitocans have shown limited side effects on normal “healthy” cells in vivo, they offer great potential in cancer therapy.
REFERENCES
- S. D. John, A. Hadfield, N. Hirst and A. T. McGown, “Tubulin and Microtubules as Targets for Anticancer Drugs,” Progress in Cell Cycle Research, Vol. 5, 2003, pp. 309-325.
- M. C. S. Stig Linder, “Lysosomes and Endoplasmic Reticulum: Targets for Improved, Selective Anticancer Therapy,” Drug Resistance Updates, Vol. 8, No. 4, 2005, pp. 199-204. doi:10.1016/j.drup.2005.06.004
- A. Szewczyk and L. Wojtczak, “Mitochondria as a Pharmacological Target,” Pharmacological Reviews, Vol. 54, No. 1, 2002, pp. 101-127. doi:10.1124/pr.54.1.101
- P. Costantini, E. Jacotot, D. Decaudin and G. Kroemer, “Mitochondrion as a Novel Target of Anticancer Chemotherapy,” Journal of National Cancer Institute, Vol. 92, No. 13, 2000, pp. 1042-1053. doi:10.1093/jnci/92.13.1042
- J. Neuzil, M. Tomasetti, Y. Zhao, L.-F. Dong, M. Birringer, X.-F. Wang, P. Low, K. Wu, B. A. Salvatore and S. J. Ralph, “Vitamin E Analogs, a Novel Group of “Mitocans,” as Anticancer Agents: The Importance of Being Redox-Silent,” Molecular Pharmacology, Vol. 71, No. 5, 2007, pp. 1185-1199. doi:10.1124/mol.106.030122
- P. P. G. Kroemer, N. Zamzami, J. L. Vayssiere and B. Mignotte, “The Biochemistry of Programmed Cell Death,” FASEB Journal, Vol. 9, 1995, pp. 1277-1287.
- J. Neuzil, J. C. Dyason, R. Freeman, L.-F. Dong, L. Prochazka, X.-F. Wang, I. Scheffler and S. J. Ralph, “Mitocans as Anti-Cancer Agents Targeting Mitochondria: Lessons from Studies with Vitamin E Analogues, Inhibitors of Complex II,” Journal of Bioenergetics and Biomembranes, Vol. 39, No. 1, 2007, pp. 65-72. doi:10.1007/s10863-006-9060-z
- D. R. Green and J. C. Reed, “Mitochondria and Apoptosis,” Science, Vol. 281, No. 5381, 1998, pp. 1309-1312. doi:10.1126/science.281.5381.1309
- S. Passarella and A. Atlante, “Teaching the Role of Mitochondrial Transport in Energy Metabolism,” Biochemistry and Molecular Biology Education, Vol. 35, No. 2, 2007, pp. 125-132. doi:10.1002/bmb.31
- R. Shepherd, N. Checcarelli, A. Naini, D. De Vivo, S. DiMauro and C. Sue, “Measurement of ATP Production in Mitochondrial Disorders,” Journal of Inherited Metabolic Disease, Vol. 29, No. 1, 2006, pp. 86-91. doi:10.1007/s10545-006-0148-8
- H. P. Indo, M. Davidson, H.-C. Yen, S. Suenaga, K. Tomita, T. Nishii, M. Higuchi, Y. Koga, T. Ozawa and H. J. Majima, “Evidence of ROS Generation by Mitochondria in Cells with Impaired Electron Transport Chain and Mitochondrial DNA Damage,” Mitochondrion, Vol. 7, No. 1-2, 2007, pp. 106-118. doi:10.1016/j.mito.2006.11.026
- Y. Chen, W. H. Yuen, J. Fu, G. Huang, A. J. Melendez, F. B. M. Ibrahim, H. Lu and X. Cao, “The Mitochondrial Respiratory Chain Controls Intracellular Calcium Signaling and NFAT Activity Essential for Heart Formation in Xenopus Laevis,” Molecular and Cellular Biology, Vol. 27, No. 18, 2007, pp. 6420-6432. doi:10.1128/MCB.01946-06
- N. Gattermann, “Mitochondrial DNA Mutations in the Hematopoietic System,” Leukemia, Vol. 18, 2003, pp. 18- 22. doi:10.1038/sj.leu.2403209
- A. M. Porcelli, A. Angelin, A. Ghelli, E. Mariani, A. Martinuzzi, V. Carelli, V. Petronilli, P. Bernardi and M. Rugolo, “Respiratory Complex I Dysfunction Due to Mitochondrial DNA Mutations Shifts the Voltage Threshold for Opening of the Permeability Transition Pore toward Resting Levels,” Journal of Biological Chemistry, Vol. 284, 2009, pp. 2045-2052. doi:10.1074/jbc.M807321200
- H. Saybaşili, M. Yüksel, G. Haklar and A. S. Yalçin, “Effect of Mitochondrial Electron Transport Chain Inhibitors on Superoxide Radical Generation in Rat Hippocampal and Striatal Slices,” Antioxidants and Redox Signaling, Vol. 3, 2001, pp. 1099-1104. doi:10.1089/152308601317203602
- K. Staniek, L. Gille, A. V. Kozlov and H. Nohl, “Mitochondrial Superoxide Radical Formation is Controlled by Electron Bifurcation to the High and Low Potential Pathways,” Free Radical Research, Vol. 36, No. 4, 2002, pp. 381-387. doi:10.1080/10715760290021225
- H. Pelicano, L. Feng, Y. Zhou, J. S. Carew, E. O. Hileman, W. Plunkett, M. J. Keating and P. Huang, “Inhibition of Mitochondrial Respiration a Novel Strategy to Enhance Drug-Induced Apoptosis in Human Leukemia Cells by a Reactive Oxygen Species-Mediated Mechanism,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 37832- 37839. doi:10.1074/jbc.M301546200
- M. J. F. G. A. Murrell and L. Bromley, “Modulation of fibroblast Proliferation by Oxygen Free Radicals,” Biochemical Journal, Vol. 265, 1990, pp. 659-665.
- J. Emerit, J. M. Klein, A. Coutellier and F. Congy, “Free Radicals and Lipid Peroxidation in Cell Biology: Physiopathologic Prospects,” Pathologie-Biologie, Vol. 39, No. 316, 1991.
- T. P. Szatrowskiand and C. F. Nathan, “Production of Large Amounts of Hydrogen Peroxide by Human Tumor Cells,” Cancer Research, Vol. 51, 1991, pp. 794-798.
- S. Toyokunia, K. Okamoto, J. Yodoi and H. Hiai, “Persistent Oxidative Stress in Cancer,” FEBS Letters, Vol. 358, No. 1, 1995, pp. 1-3. doi:10.1016/0014-5793(94)01368-B
- S. Toyokuni, “Oxidative Stress and Cancer: The Role of Redox Regulation,” Biotherapy, Vol. 11, No. 2-3, 1998, pp. 47-154. doi:10.1023/A:1007934229968
- E. A. Hileman, G. Achanta and P. Huang, “Superoxide Dismutase: An Emerging Target for Cancer Therapeutics,” Expert Opinion on Therapeutic Targets, Vol. 5, No. 6, 2001, pp. 697-710. doi:10.1517/14728222.5.6.697
- P. Huang, L. Feng, E. A. Oldham, M. J. Keating and W. Plunkett, “Superoxide Dismutase as a Target for the Selective Killing of Cancer Cells,” Nature, Vol. 407, 2000, pp. 390-395. doi:10.1038/35030140
- J. G. Ğb, D. Nowis, M. Skrzycki, H. Czeczot, A. Barańczyk-Kuźma, G. M. Wilczyński, M. Makowski, P. Mróz, K. Kozar, R. Kamiński, A. Jalili, M. Kopeć, T. Grzela and M. Jakóbisiak, “Antitumor Effects of Photodynamic Therapy Are Potentiated by 2-Methoxyestradiol A Superoxide Dismutase Inhibitor,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 407-414.
- S. J. Ralph, P. Low, L. Dong, A. Lawen and J. Neuzil, “Mitocans: Mitochondrial Targeted Anti-Cancer Drugs as improved Therapies and Related Patent Documents,” Recent Patents on Anti-Cancer Drug Discovery, Vol. 1, No. 3, 2006, pp. 327-346. doi:10.2174/157489206778776952
- R. J. Green, “Mitochondrial Targeting as a Novel Paradigm of Cancer Therapy: The Emergence of Mitocans,” Science, Vol. 281, 1998, pp. 1309-1312.
- S. M. Cardoso, I. Santana, R. H. Swerdlow and C. R. Oliveira, “Mitochondria Dysfunction of Alzheimer’s Disease Cybrids Enhances Abeta Toxicity,” Journal of Neurochemistry, Vol. 89, No. 6, 2004, pp. 1417-1426. doi:10.1111/j.1471-4159.2004.02438.x
- M.T. Lin and M. F. Beal, “Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases,” Nature, Vol. 443, 2006, pp. 787-795. doi:10.1038/nature05292
- X. Chen, D. Stern and S. Du Yan, “Mitochondrial Dysfunction and Alzheimers Disease,” Current Alzheimer Research, Vol. 3, No. 5, 2006, pp. 515-520. doi:10.2174/156720506779025215
- R. Castellani, K. Hirai, G. Aliev, K. L. Drew, A. Nunomura, A. Takeda, A. D. Cash, M. E. Obrenovich, G. Perry and M. A. Smith, “Role of Mitochondrial Dysfunction in Alzheimer’s Disease,” Journal of Neuroscience Research, Vol. 70, No. 3, pp. 357-360. doi:10.1002/jnr.10389
- C. P. Ramsey and B. I. Giasson, “Role of Mitochondrial Dysfunction in Parkinson’s Disease: Implications for Treatment,” Drugs Aging, Vol. 24, No. 2, 2007, pp. 95- 105. doi:10.2165/00002512-200724020-00002
- S. S. Ghosh, R. H. Swerdlow, S. W. Miller, B. Sheeman, W. D. Parker Jr. and R. E. Davis, “Use of Cytoplasmic Hybrid Cell Lines for Elucidating the Role of Mitochondrial Dysfunction in Alzheimer’s Disease and Parkinson’s Disease,” Annals of the New York Academy of Sciences, Vol. 893, 1999, pp. 176-191. doi:10.1111/j.1749-6632.1999.tb07825.x
- J. T. Greenamyre, G. MacKenzie, T. I. Peng and S. E. Stephans, “Mitochondrial Dysfunction in Parkinson’s Disease,” Biochemical Society Symposia, Vol. 66, 1999, pp. 85-97.
- J. M. A. Oliveira, M. B. Jekabsons, S. Chen, A. Lin, A. C. Rego, J. Gonçalves, L. M. Ellerby and D. G. Nicholls, “Mitochondrial Dysfunction in Huntington’s Disease: The Bioenergetics of Isolated and in Situ Mitochondria from Transgenic Mice,” Journal of Neurochemistry, Vol. 101, No. 1, 2007, pp. 241-249. doi:10.1111/j.1471-4159.2006.04361.x
- R. A. Quintanillaand and G. V. W. Johnson, “Role of Mitochondrial Dysfunction in the Pathogenesis of Huntington’s Disease,” Brain Research Bulletin, Vol. 80, 2009, pp. 242-247.
- P. Shi, J. Gal, D. M. Kwinter, X. Liu and H. Zhu, “Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis,” Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease, Vol. 1802, No. 1, 2010, pp. 45-51. doi:10.1016/j.bbadis.2009.08.012
- F. M. Menzies, M. R. Cookson, R. W. Taylor, D. M. Turnbull, Z. M. A. Chrzanowska-Lightowlers, L. Dong, D. A. Figlewicz and P. J. Shaw, “Mitochondrial Dysfunction in a Cell Culture Model of Familial Amyotrophic Lateral Sclerosis,” Brain, Vol. 125, No. 7, 2002, pp. 1522- 1533. doi:10.1093/brain/awf167
- I. Hervias, M. F. Beal and G. Manfredi, “Mitochondrial Dysfunction and Amyotrophic Lateral Sclerosis,” Muscle & Nerve, Vol. 33, No. 5, 2006, pp. 598-608. doi:10.1002/mus.20489
- E. Seppet, M. Gruno, A. Peetsalu, Z. Gizatullina, H.P. Nguyen, S. Vielhaber, M.H.P. Wussling, S. Trumbeckaite, O. Arandarcikaite, D. Jerzembeck, M. Sonnabend, K. Jegorov, S. Zierz, F. Striggow and F. N. Gellerich, “Mitochondria and Energetic Depression in Cell Pathophysiology,” International Journal of Molecular Sciences, Vol. 10, No. 5, 2009, pp. 2252-2303. doi:10.3390/ijms10052252
- H. J. Ahn, Y. S. Kim, J.-U. Kim, S. M. Han, J. W. Shin and H. O. Yang, “Mechanism of Taxol-Induced Apoptosis in Human SKOV3 Ovarian Carcinoma Cells,” Journal of Cellular Biochemistry, Vol. 91, 2004, pp. 1043-1052. doi:10.1002/jcb.20006
- N. Andre, D. Braguer, G. Brasseur, A. Goncalves, D. Lemesle-Meunier, S. Guise, M. A. Jordan and C. Briand, “Paclitaxel Induces Release of Cytochrome c from Mitochondria Isolated from Human Neuroblastoma Cells,” Cancer Research, Vol. 60, 2000, pp. 5349-5353.
- J. F. Kidd, M. F. Pilkington, M. J. Schell, K. E. Fogarty, J. N. Skepper, C. W. Taylor and P. Thorn, “Paclitaxel Affects Cytosolic Calcium Signals by Opening the Mitochondrial Permeability Transition Pore,” The Journal of Biological Chemistry, Vol. 277, 2002, pp. 6504-6510. doi:10.1074/jbc.M106802200
- K. Ozgurand and L. Anthony, “Alteration of the Mitochondrial Apoptotic Pathway Is Key to Acquired Paclitaxel Resistance and Can be Reversed by ABT-737,” Cancer Res, Vol. 68, 2008, pp. 7985-7994. doi:10.1158/0008-5472.CAN-08-1418
- M. Crompton and L. Andreeva, “On the Interactions of Ca2+ and Cyclosporin A With a Mitochondrial Inner Membrane Pore: A Study Using Cobaltammine Complex Inhibitors of the Ca2+ Uniporter,” Biochemical Journal, Vol. 302, 1994, pp. 181-185.
- D. W. Jung, P. C. Bradshaw and D. R. Pfeiffer, “Properties of a Cyclosporin-Insensitive Permeability Transition Pore in Yeast Mitochondria,” The Journal of Biological Chemistry, Vol. 272, 1997, pp. 21104-21112. doi:10.1074/jbc.272.34.21104
- K. M. Broekemeier and D. R. Pfeiffer, “Inhibition of the Mitochondrial Permeability Transition by Cyclosporin A during long Time Frame Experiments: Relationship between Pore Opening and the Activity of Mitochondrial Phospholipases,” Biochemistry, Vol. 34, No. 50, 1995, pp. 16440- 16449. doi:10.1021/bi00050a027
- R. Scatena, P. Bottoni, G. Botta, G. E. Martorana and B. Giardina, “The Role of Mitochondria in Pharmacotoxicology: A Reevaluation of an Old, Newly Emerging Topic,” American Journal of Cell Physiology, Vol. 293, No. 1, 2007, pp. C12-21. doi:10.1152/ajpcell.00314.2006
- A. A. Johnson, A. S. Ray, J. Hanes, Z. Suo, J. M. Colacino, K. S. Anderson and K. A. Johnson, “Toxicity of Antiviral Nucleoside Analogs and the Human Mitochondrial DNA Polymerase,” The Journal of Biological Chemistry, Vol. 276, 2001, pp. 40847-40857. doi:10.1074/jbc.M106743200
- L. F. Pereira, M. B. M. Oliveira and E. G. S. Carnieri, “Mitochondrial Sensitivity to AZT,” Cell Biochemistry and Function, Vol. 16, No. 3, 1998, pp. 173-181. doi:10.1002/(SICI)1099-0844(199809)16:3<173::AID-CBF783>3.0.CO;2-4
- T. Sato, H. Nishida, M. Miyazaki and H. Nakaya, “Effects of Sulfonylureas on Mitochondrial ATP-Sensitive Channels in Cardiac Myocytes: Implications for Sulfonylurea Controversy,” Diabetes/Metabolism Research and Reviews, Vol. 22, No. 5, 2006, 341-347. doi:10.1002/dmrr.621
- A. Szewczyk, “Intracellular Targets for Antidiabetic Sulfonylureas and Potassium Channel Openers,” Biochemical Pharmacology, Vol. 54, No. 9, 1997, pp. 961-965. doi:10.1016/S0006-2952(97)00136-6
- P. A. Smith, P. Proks and A. Moorhouse, “Direct Effects of Tolbutamide on Mitochondrial Function, Intracellular Ca2+ and Exocytosis in Pancreatic β-Cells,” Pflügers Archiv European Journal of Physiology, Vol. 437, 1999, pp. 577-588.
- M. Zaugg, E. Lucchinetti, D. R. Spahn, T. Pasch, C. Garcia and M. C. Schaub, “Differential Effects of Anesthetics on Mitochondrial K(ATP) Channel Activity and Cardiomyocyte Protection,” Anesthesiology, Vol. 97, No. 1, 2002, pp. 15-23. doi:10.1097/00000542-200207000-00004
- Y. Zhang, Y. Dong, X. Wu, Y. Lu, Z. Xu, A. Knapp, Y. Yue, T. Xu and Z. Xie, “The Mitochondrial Pathway of Anesthetic Isoflurane-Induced Apoptosis,” The Journal of Biological Chemistry, Vol. 285, 2010, pp. 4025-4037. doi:10.1074/jbc.M109.065664
- M. G. D. Spierings, M. Saleh, C. Bender, J. Chipuk, U. Maurer and D. R. Green, “Connected to Death: The (Unexpurgated) Mitochondrial Pathway of Apoptosis,” Science, Vol. 310, No. 5745, 2005, p. 2. doi:10.1126/science.1117105
- M. Zoratti and I. Szabò, “The Mitochondrial Permeability Transition,” Biochimica et Biophysica Acta, Vol. 1241, 1995, pp. 139-176.
- T. Hirsch, P. Marchetti, S. A. Susin, B. Dallaporta, N. Zamzami, I. Marzo, M. Geuskens and G. Kroemer, “The Apoptosis-Necrosis Paradox. Apoptogenic Proteases Activated after Mitochondrial Permeability Transition Determine the Mode of Cell Death,” Oncogene, Vol. 15, No. 13, 1997, pp. 1573-1581. doi:10.1038/sj.onc.1201324
- N. Zamzami, S. A. Susin, P. Marchetti, T. Hirsch, I. Gómez-Monterrey, M. Castedo and G. Kroemer, “Mitochondrial Control of Nuclear Apoptosis,” Journal of Experimental Medicine, Vol. 183, No. 4, 1996, pp. 1533- 1544. doi:10.1084/jem.183.4.1533
- [61] D. Green and G. Kroemer, “The Central Executioners of Apoptosis: Caspases or Mitochondria?” Trends in Cell Biology, Vol. 8, No. 7, 1998, pp. 267-271. doi:10.1016/S0962-8924(98)01273-2
- [62] J. G. Pastorino, S.-T. Chen, M. Tafani, J. W. Snyder and J. L. Farber, “The Overexpression of Bax Produces Cell Death upon Induction of the Mitochondrial Permeability Transition,” Journal of Biological Chemistry, Vol. 273, 1998, pp. 7770-7775. doi:10.1074/jbc.273.13.7770
- [63] N. J. McCarthy, M. K. B. Whyte, C. S. Gilbert and G. I. Evan, “Inhibition of Ced-3/ICE Related Proteases Does not Prevent Cell Death Induced by Oncogenes, DNA Damage, or the Bcl-2 Homologue Bak,” Journal of Cell Biology, Vol. 136, No. 1, 1997, pp. 215-227. doi:10.1083/jcb.136.1.215
- [64] F. Quignon, F. D. Bels, M. Koken, J. Feunteun, J.-C. Ameisen and H. D. Thé, “PML Induces a Novel Caspase-Independent Death Process,” Nature Genetics, Vol. 20, 1998, pp. 259-265. doi:10.1038/3068
- [65] A. Kawahara, Y. Ohsawa, H. Matsumura, Y. Uchiyama and S. Nagata, “Caspase Independent Cell Killing by Fas-Associated Protein with Death Domain,” Journal of Cell Biology, Vol. 143, No. 5, 1998, pp. 1353-1360. doi:10.1083/jcb.143.5.1353
- [66] D. Vercammen, R. Beyaert, G. Denecker, V. Goossens, G. V. Loo, W. Declercq, J. Grooten, W. Fiers and P. Vandenabeele, “Inhibition of Caspases Increases the Sensitivity of L929 Cells to Necrosis Mediated by Tumor Necrosis Factor,” Journal of Experimental Medicine, Vol. 187, No. 9, 1998, pp. 1477-1485. doi:10.1084/jem.187.9.1477
- [67] H. K. Bojes, X. Feng, J. P. Kehrer and G. M. Cohen, “Apoptosis in Hematopoietic Cells (FL5.12) Caused by Interleukin-3 Withdrawal: Relationship to Caspase Activity and the Loss of Glutathione,” Cell Death and Differentiation, Vol. 6, No. 1, 1999, pp. 61-70. doi:10.1038/sj.cdd.4400452
- [68] C. Berndt, B. Moepps, S. Angermueller, P. Gierschik and P.H. Krammer, “CXCR4 and CD4 Mediate a Rapid CD95-Independent Cell Death in CD4(+) T Cells,” Proceedings of The National Academy of Science USA, Vol. 95, 1998, pp. 12556-12561.
- [69] D. E. Wood and E. W. Newcomb, “Caspase-Dependent Activation of Calpain during Drug-Induced Apoptosis,” Journal of Biological Chemistry, Vol. 274, 1999, pp. 8309-8315. doi:10.1074/jbc.274.12.8309
- [70] K. M. Henkels and J. J. Turchi, “Cisplatin-Induced Apoptosis Proceeds by Caspase-3-Dependent and -Independent Pathways in Cisplatin-Resistant and -Sensitive Human Ovarian Cancer Cell Lines,” Cancer Research, Vol. 59, 1999, pp. 3077-3083.
- [71] S. A. Susin, H. K. Lorenzo, N. Zamzami, I. Marzo, B. E. Snow, G. M. Brothers, J. Mangion, E. Jacotot, P. Costantini, M. Loeffler, N. Larochette, D. R. Goodlett, R. Aebersold, D. P. Siderovski, J. M. Penninger and G. Kroemer, “Molecular Characterization of Mitochondrial Apoptosis-Inducing Factor,” Nature, Vol. 397, 1999, pp. 441- 446.
- [72] X. Liu, C. N. Kim, J. Yang, R. Jemmerson and X. Wang, “Induction of Apoptotic Program in Cell-Free Extracts: Requirement for dATP and Cytochrome c,” Cell, Vol. 86, 1996, pp. 147-157.
- [73] S. A. Susin, H. K. Lorenzo, N. Zamzami, I. Marzo, C. Brenner, N. Larochette, M.-C. Prévost, P. M. Alzari and G. Kroemer, “Mitochondrial Release of Caspase-2 and -9 during the Apoptotic Process,” Journal of Experimental Medicine, Vol. 189, No. 2, 1999, pp. 381-394. doi:10.1084/jem.189.2.381
- [74] Y. Chen, E. McMillan-Ward, J. Kong, S. J. Israels and S. B. Gibson, “Mitochondrial Electron-Transport-Chain Inhibitors of Complexes I and II Induce Autophagic Cell Death Mediated by Reactive Oxygen Species,” Journal of Cell Science, Vol. 120, 2007, pp. 4155-4166. doi:10.1242/jcs.011163
- [75] H.-S. Huang, Z.-M. Liu, L. Ding, W.-C. Chang, P.-Y. Hsu, S.-H. Wang, C.-C. Chi and C.-H. Chuang, “Opposite Effect of ERK1/2 and JNK on p53-Independent p21 WAF1/CIP1 Activation Involved in the Arsenic Trioxide-Induced Human Epidermoid Carcinoma A431 Cellular Cytotoxicity,” Journal of Biomedical Science, Vol. 13, No. 1, 2006, pp. 113-125. doi:10.1007/s11373-005-9040-z
- [76] P. Kumar, Q. Gao, Y. Ning, Z. Wang, P. H. Krebsbach and P. J. Polverini, “Arsenic Trioxide Enhances the Therapeutic Efficacy of Radiation Treatment of Oral Squamous Carcinoma While Protecting Bone,” Molecular Cancer Therapeutics, Vol. 7, No. 7, 2008, pp. 2060- 2069. doi:10.1158/1535-7163.MCT-08-0287
- [77] C. Yedjou, L. Thuisseu, C. Tchounwou, M. Gomes, C. Howard and P. Tchounwou, “Ascorbic Acid Potentiation of Arsenic Trioxide Anticancer Activity Against Acute Promyelocytic Leukemia,” Archives of Drug Information, Vol. 2, 2009, pp. 59-65.
- [78] K. G. Wolter, Y.-T. Hsu, C. L. Smith, A. Nechushtan, X.-G. Xi and R. J. Youle, “Movement of Bax from the Cytosol to Mitochondria during Apoptosis,” Journal of Cell Biology, Vol. 139, No. 5, 1997, pp. 1281-1292. doi:10.1083/jcb.139.5.1281
- [79] M. Narita, S. Shimizu, T. Ito, T. Chittenden, R. J. Lutz, H. Matsuda and Y. Tsujimoto, “Bax Interacts with the Permeability Transition Pore to Induce Permeability Transition and Cytochrome c Release in Isolated Mitochondria,” Proceedings of The National Academy of Science USA, Vol. 95, No. 25, 1998, pp. 14681-14686. doi:10.1073/pnas.95.25.14681
- [80] I. Marzo, C. Brenner, N. Zamzami, J. M. Jürgensmeier, S. A. Susin, H. L. A. Vieira, M.-C. Prévost, Z. Xie, S. Matsuyama, J. C. Reed and G. Kroemer, “Bax and Adenine Nucleotide Translocator Cooperate in the Mitochondrial Control of Apoptosis,” Science, Vol. 281, 1998, pp. 2027- 2031.
- [81] M. Crompton, S. Virji and J. M. Ward, “Cyclophilin-D Binds Strongly to Complexes of the Voltage-Dependent Anion Channel and the Adenine Nucleotide Translocase to Form the Permeability Transition Pore,” European Journal of Biochemistry, Vol. 258, No. 2, 1998, pp. 729- 735. doi:10.1046/j.1432-1327.1998.2580729.x
- [82] N. Brustovetsky and M. Klingenberg, “Mitochondrial ADP/ATP Carrier Can be Reversibly Converted into a Large Channel by Ca2+,” Biochemistry, Vol. 35, 1996, pp. 8483-8488.
- [83] G. Beutner, A. Rück, B. Riede, W. Welte and D. Brdiczka, “Complexes between Kinases, Mitochondrial Porin and Adenylate Translocator in Rat Brain Resemble the Permeability Transition Pore,” FEBS Letters, Vol. 396, No. 2-3, 1996, pp. 189-195. doi:10.1016/0014-5793(96)01092-7
- [84] S. Shimizu, M. Narita and Y. Tsujimoto, “Bcl-2 Family Proteins Regulate the Release of Apoptogenic Cytochrome c by the Mitochondrial Channel VDAC,” Nature, Vol. 399, 1999, pp. 483-487.
- [85] C. Brenner, H. Cadiou, H. L. Vieira, N. Zamzami, I. Marzo, Z. Xie, B. Leber, D. Andrews, H. Duclohier, J. C. Reed and G. Kroemer, “Bcl-2 and Bax Regulate the Channel Activity of the Mitochondrial Adenine Nucleotide Translocator,” Oncogene, Vol. 19, 2000, pp. 329- 336.
- [86] J. E. Belizário, J. Alves, J. M. Occhiucci, M. Garay-Malpartida and A. Sesso, “A Mechanistic View of Mitochondrial Death Decision Pores,” Brazilian Journal of Medical and Biological Research, Vol. 40, 207, pp. 1011- 1024.
- [87] S. Desagher, A. Osen-Sand, A. Nichols, R. Eskes, S. Montessuit, S. Lauper, K. Maundrell, B. Antonsson and J.-C. Martinou, “Bid-Induced Conformational Change of Bax Is Responsible for Mitochondrial Cytochrome c Release during Apoptosis,” Journal of Cell Biology, Vol. 144, No. 5, 1999, pp. 5891-5901. doi:10.1083/jcb.144.5.891
- [88] E. Bossy-Wetzel, D. D. Newmeyer and D. R. Green, “Mitochondrial Cytochrome c Release in Apoptosis Occurs Upstream of DEVD-Specific Caspase Activation and Independently of Mitochondrial Transmembrane Depolarization,” EMBO Journal, Vol. 17, 1998, pp. 37-49. doi:10.1093/emboj/17.1.37
- [89] J. C. Reed, “Bcl-2 Family Proteins,” Oncogene, Vol. 17, 1998, pp. 3225-3236.
- [90] Z. Huang, “Bcl-2 Family Proteins as Targets for Anticancer Drug Design,” Oncogene, Vol. 19, 2000, pp. 6627-6631.
- [91] K. Polyak, Y. Xia, J. L. Zweier, K. W. Kinzler and B. Vogelstein, “A Model for p53-Induced Apoptosis,” Nature, Vol. 389, 1997, pp. 300-305.
- [92] Z. Wang and Y. Sun, “Targeting p53 for Novel Anticancer Therapy,” Translational Oncology, Vol. 3, 2010, pp. 1-12.
- [93] J. Neuzil, X.-F. Wang, L.-F. Dong, P. Low and S. J. Ralph, “Molecular Mechanism of ‘Mitocan’-Induced Apoptosis in Cancer Cells Epitomizes the Multiple Roles of Reactive Oxygen Species and Bcl-2 Family Proteins,” FEBS Letters, Vol. 580, No. 22, 2006, pp. 5125-5129. doi:10.1016/j.febslet.2006.05.072
- [94] R. N. Kolesnick, “Regulation of Ceramide Production and Apoptosis,” Annual Review of Physiology, Vol. 60, 1998, pp. 643-665. doi:10.1146/annurev.physiol.60.1.643
- [95] R. D. Maria, L. Lenti, F. Malisan, F. D’Agostino, B. Tomassini, A. Zeuner, M. R. Rippo and R. Testi, “Requirement for GD3 Ganglioside in CD95- and Ceramide-Induced Apoptosis,” Science, Vol. 277, 1997, pp. 1652-1655.
- [96] H. Basma, H. El-Refaey, M. K. Sgagias, K. H. Cowan, X. Luo and P.-W. Cheng, “BCL-2 Antisense and Cisplatin Combination Treatment of MCF-7 Breast Cancer Cells with or without Functional P53,” Journal of Biomedical Science, Vol. 12, No. 6, 2005, pp. 999-1011. doi:10.1007/s11373-005-9025-y
- [97] A. Quillet-Mary, J.-P. Jaffrézou, V. Mansat, C. Bordier, J. Naval and G. Laurent, “Implication of Mitochondrial Hydrogen Peroxide Generation in Ceramide-induced Apoptosis,” Journal of Biological Chemistry, Vol. 272, 1997, pp. 21388-21395. doi:10.1074/jbc.272.34.21388
- [98] J. L. Scarlett, M. A. Packer, C. M. Porteous and M. P. Murphy, “Alterations to Glutathione and Nicotinamide Nucleotides during the Mitochondrial Permeability Transition Induced by Peroxynitrite 1,” Biochemical Pharmacology, Vol. 52, No. 7, 1996, pp. 1047-1055. doi:10.1016/0006-2952(96)99426-5
- [99] P. Costantini, B.V. Chernyak, V. Petronilli, and P. Bernardi, “Modulation of the Mitochondrial Permeability Transition Pore by Pyridine Nucleotides and Dithiol Oxidation at Two Separate Sites,” Journal of Biological Chemistry, Vol. 271, 1996, pp. 6746-6751. doi:10.1074/jbc.271.12.6746
- [100] P. Costantini, A.-S. Belzacq, H. L. Vieira, N. Larochette, M. A. D. Pablo, N. Zamzami, S. A. Susin, C. Brenner and G. Kroemer, “Oxidation of a Critical Thiol Residue of the Adenine Nucleotide Translocator Enforces Bcl-2-Independent Permeability Transition Pore Opening and Apoptosis,” Oncogene, Vol. 19, 2000, pp. 307-314.
- [101] M. Klingenberg, “The ADP-ATP Translocation in Mitochondria, a Membrane Potential Controlled Transport,” Journal of Membrane Biology, Vol. 56, No. 2, 1980, pp. 97-105. doi:10.1007/BF01875961
- [102] E. Chávez and A. Osornio, “Temperature Dependence of the Atractyloside-Induced Mitochondrial Ca2+ Release,” International Journal of Biochemistry, Vol. 20, No. 7, 1988, pp. 731-736. doi:10.1016/0020-711X(88)90169-3
- [103] G. Kroemer, L. Galluzzi and C. Brenner, “Mitochondrial Membrane Permeabilization in Cell Death,” Physiological Reviews, Vol. 87, 2007, pp. 99-163.
- [104] S. A. Susin, N. Zamzami, M. Castedo, E. Daugas, H.-G. Wang, S. Geley, F. Fassy, J. C. Reed and G. Kroemer, “The Central Executioner of Apoptosis: Multiple Connections between Protease Activation and Mitochondria in Fas/APO-1/CD95- and Ceramide-induced Apoptosis,” Journal of Experimental Medicine, Vol. 86, No. 1, 1997, pp. 25- 37. doi:10.1084/jem.186.1.25
- [105] P. Bernardi and V. Petronilli, “The Permeability Transition Pore as a Mitochondrial Calcium Release Channel: A Critical Appraisal,” Journal of Bioenergetics and Biomembranes, Vol. 28, No. 2, 1996, pp. 131-138. doi:10.1007/BF02110643
- [106] H. Puthalakath, D. C. S. Huang, L. A. O’Reilly, S. M. King and A. Strasser, “The Proapoptotic Activity of the Bcl-2 Family Member Bim Is Regulated by Interaction with the Dynein Motor Complex,” Molecular Cell, Vol. 3, No. 3, 1999, pp. 287-296. doi:10.1016/S1097-2765(00)80456-6
- [107] F. Tsuruta, J. Sunayama, Y. Mori, S. Hattori, S. Shimizu, Y. Tsujimoto, K. Yoshioka, N. Masuyama and Y. Gotoh, “JNK Promotes Bax Translocation to Mitochondria through Phosphorylation of 14-3-3 Proteins,” The EMBO Journal, Vol. 23, 2004, pp. 1889-1899. doi:10.1038/sj.emboj.7600194
- [108] H. Li, H. Zhu, C.-J. Xu and J. Yuan, “Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis,” Cell, Vol. 94, 1998, pp. 491-501.
- [109] J. Zha, H. Harada, E. Yang, J. Jockel and S. J. Korsmeyer, “Serine Phosphorylation of Death Agonist BAD in Response to Survival Factor Results in Binding to 14-3-3 Not BCL-XL,” Cell, Vol. 87, 1996, pp. 619-628.
- [110] Y. V. Evtodienko, V. V. Teplova, S. S. Sidash, F. Ichas, and J.-P. Mazat, “Microtubule-Active Drugs Suppress the Closure of the Permeability Transition Pore in Tumour Mitochondria,” FEBS Letters, Vol. 393, No. 1, 1996, pp. 86-88. doi:10.1016/0014-5793(96)00875-7
- [111] D. G. Kirsch, A. Doseff, B. N. Chau, D.-S. Lim, N. C. D. Souza-Pinto, R. Hansford, M. B. Kastan, Y. A. Lazebnik and J. M. Hardwick, “Caspase-3-Dependent Cleavage of Bcl-2 Promotes Release of Cytochrome c,” Journal of Biological Chemistry, Vol. 274, 1999, pp. 21155-21161. doi:10.1074/jbc.274.30.21155
- [112] J. Armstrong, “Mitochondrial Medicine: Pharmacological Targeting of Mitochondria in Disease,” British Journal of Pharmacology, Vol. 151, No. 8, 2007, pp. 1154-1165. doi:10.1038/sj.bjp.0707288
- [113] A. Lena, M. Rechichi, A. Salvetti, B. Bartoli, D. Vecchio, V. Scarcelli, R. Amoroso, L. Benvenuti, R. Gagliardi, V. Gremigni and L. Rossi, “Drugs Targeting the Mitochondrial Pore Act as Citotoxic and Cytostatic Agents in Temozolomide-Resistant Glioma Cells,” Journal of Translational Medicine, Vol. 7, 2009, pp. 1-13. doi:10.1186/1479-5876-7-13
- [114] V. Labi, M. Erlacher, S. Kiessling and A. Villunger, “BH3-Only Proteins in Cell Death Initiation, Malignant Disease and Anticancer Therapy,” Cell Death and Differentiation, Vol. 13, 2006, pp. 1325-1338. doi:10.1038/sj.cdd.4401940
- [115] S. J. Ralph, “Arsenic-Based Antineoplastic Drugs and Their Mechanisms of Action,” Metal-Based Drugs, Vol. 1, 2008, pp. 13.
- [116] H. Pelicano, D. S. Martin, R.-H. Xu and P. Huang, “Glycolysis Inhibition for Anticancer Treatment,” Oncogene, Vol. 25, 2006, pp. 4633-4646. doi:10.1038/sj.onc.1209597
- [117] Y. H. Ko, B. L. Smith, Y. Wang, M. G. Pomper, D. A. Rini, M. S. Torbenson, J. Hullihen and P. L. Pedersen, “Advanced Cancers: Eradication in All Cases Using 3 Bromopyruvate Therapy to Deplete ATP,” Biochemical and Biophysical Research Communications, Vol. 324, No. 1, 2004, pp. 269-275. doi:10.1016/j.bbrc.2004.09.047
- [118] L. Arzoine, N. Zilberberg, R. Ben-Romano and V. Shoshan-Barmatz, “Voltage-dependent Anion Channel 1-based Peptides Interact with Hexokinase to Prevent Its Anti-apoptotic Activity,” Journal of Biological Chemistry, Vol. 284, 2009, pp. 3946-3955. doi:10.1074/jbc.M803614200
- [119] S. Abu-Hamad, H. Zaid, A. Israelson, E. Nahon and V. Shoshan-Barmatz, “Hexokinase-I Protection against Apoptotic Cell Death Is Mediated via Interaction with the Voltage-Dependent Anion Channel-1 Mapping the Site of Binding,” Journal of Biological Chemistry, Vol. 283, 2008, pp. 13482-13490. doi:10.1074/jbc.M708216200
- [120] F. Chiara, D. Castellaro, O. Marin, V. Petronilli, W.S. Brusilow, M. Juhaszova, S. J. Sollott, M. Forte, P. Bernardi and A. Rasola, “Hexokinase II Detachment from Mitochondria Triggers Apoptosis through the Permeability Transition Pore Independent of Voltage-Dependent Anion Channels,” PLoS ONE, Vol. 3, 2008, p. 1852. doi:10.1371/journal.pone.0001852
- [121] J. Neuzil, T. Marco, M. A. Sleiman, A. Renata, S. B. A. B. Marc and M. W. Fariss, “Vitamin E Analogues: A New Class of Inducers of Apoptosis with Selective AntiCancer Effects,” Current Cancer Drug Targets, Vol. 4, No. 4, 2004, pp. 355-372. doi:10.2174/1568009043332943
- [122] W. Kim, J.-H. Yoon, J.-M. Jeong, G.-J. Cheon, T.-S. Lee, J.-I. Yang, S.-C. Park and H.-S. Lee, “Apoptosis-Inducing Antitumor Efficacy of Hexokinase II Inhibitor in Hepatocellular Carcinoma,” Molecular Cancer Therapeutics, Vol. 6, 2007, pp. 2554-2562. doi:10.1158/1535-7163.MCT-07-0115
- [123] J. O’Neill, M. Manion, P. Schwartz and D. M. Hockenbery, “Promises and Challenges of Targeting Bcl-2 Anti-Apoptotic Proteins for Cancer Therapy,” Biochimica et Biophysica Acta, Vol. 1705, 2004, pp. 43-51.
- [124] W.-S. Yeow, A. Baras, A. Chua, D. M. Nguyen, S. S. Sehgal, D. S. Schrump and D. M. Nguyen, “Gossypol, a Phytochemical with BH3-Mimetic Property, Sensitizes Cultured Thoracic Cancer Cells to Apo2 Ligand/Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand,” The Journal of Thoracic and Cardiovascular Surgery, Vol. 132, No. 6, 2006, pp. 1356-1362. doi:10.1016/j.jtcvs.2006.07.025
- [125] A. Degterev, A. Lugovskoy, M. Cardone, B. Mulley, G. Wagner, T. Mitchison and J. Yuan, “Identification of Small-Molecule Inhibitors of Interaction between the BH3 Domain and Bcl-xL,” Nature Cell Biology, Vol. 3, 2001, pp. 173-182. doi:10.1038/35055085
- [126] C.-W. Shiau, J.-W. Huang, D.-S. Wang, J.-R. Weng, C.-C. Yang, C.-H. Lin, C. Li and C.-S. Chen, “α-Tocopheryl Succinate Induces Apoptosis in Prostate Cancer Cells in Part through Inhibition of Bcl-xL/Bcl-2 Function,” Journal of Biological Chemistry, Vol. 281, 2006, pp. 11819- 11825. doi:10.1074/jbc.M511015200
- [127] K. N. Prasad, B. Kumar, X.-D. Yan, A. J. Hanson and W. C. Cole, “α-Tocopheryl Succinate, the Most Effective Form of Vitamin E for Adjuvant Cancer Treatment: A Review,” Journal of the American College of Nutrition, Vol. 22, 2003, pp. 108-117.
- [128] K. B. Kim, A. Y. Bedikian, L. H. Camacho, N. E. Papadopoulos and C. McCullough, “A Phase II Trial of Arsenic Trioxide in Patients with Metastatic Melanoma,” Cancer, Vol. 104, No. 8, 2005, pp. 1687-1692. doi:10.1002/cncr.21386
- [129] S. Amadori, P. Fenaux, H. Ludwig, M. O’Dwyer and M. Sanz, “Use of Arsenic Trioxide in Haematological Malignancies: Insight into the Clinical Development of a Novel Agent,” Current Medical Research and Opinion, Vol. 21, No. 3, 2005, pp. 403-411. doi:10.1185/030079904X20349
- [130] A.-S. Belzacq, C. E. Hamel, H. L. A. Vieira, I. Cohen, D. Haouzi, D. Métivier, P. Marchetti, C. Brenner and G. Kroemer, “Adenine Nucleotide Translocator Mediates the Mitochondrial Membrane Permeabilization Induced by Lonidamine, Arsenite and CD437,” Oncogene, Vol. 20, No. 52, 2001, pp. 7579-7587. doi:10.1038/sj.onc.1204953
- [131] L. Biasutto, L.-F. Dong, M. Zoratti and J. Neuzil, “Mitochondrially Targeted Anti-Cancer Agents,” Mitochondrion, 2010.
- [132] L. Ravagnan, I. Marzo, P. Costantini, S. A. Susin, N. Zamzami, P. X. Petit, F. Hirsch, M. Goulbern, M.-F. Poupon, L. Miccoli, Z. Xie, J. C. Reed and G. Kroemer, “Lonidamine Triggers Apoptosis via a Direct, Bcl-2-Inhibited Effect on the Mitochondrial Permeability Transition Pore,” Oncogene, Vol. 18, No. 16, 1999, pp. 2537- 2546. doi:10.1038/sj.onc.1202625
- [133] S.-Y. Sun, P. Yue, G. S. Wu, W. S. El-Deiry, B. Shroot, W. K. Hong and R. Lotan, “Mechanisms of Apoptosis Induced by the Synthetic Retinoid CD437 in Human Non-Small Cell Lung Carcinoma Cells,” Oncogene, Vol. 18, No. 14, 1999, pp. 2357-2365. doi:10.1038/sj.onc.1202625
- [134] P. Marchetti, N. Zamzami, B. Joseph, S. SchraenMaschke, C. Méreau-Richard, P. Costantini, D. Métivier, S. A. Susin, G. Kroemer and P. Formstecher, “The Novel Retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-Naphtalene Carboxylic Acid Can Trigger Apoptosis through a Mitochondrial Pathway Independent of the Nucleus,” Cancer Research, Vol. 59, 1999, pp. 6257-6266.
- [135] M. Pfahl and F. J. Piedrafita, “Retinoid Targets for Apoptosis Induction,” Oncogene, Vol. 22, 2003, pp. 9058- 9062. doi:10.1038/sj.onc.1207109
- [136] D. Schadendorf, M. A. Kern, M. Artuc, H. L. Pahl, T. Rosenbach, I. Fichtner, W. Nürnberg, S. Stüting, E. V. Stebut, M. Worm, A. Makki, K. Jurgovsky, G. Kolde and B. M. Henz, “Treatment of Melanoma Cells with the Synthetic Retinoid CD437 Induces Apoptosis via Activation of AP-1 in Vitro, and Causes Growth Inhibition in Xenografts in vivo,” Journal of Cell Biology, Vol. 135, No. 6, 1996, pp. 1889-1898. doi:10.1083/jcb.135.6.1889
- [137] X. Zhao and R. A. Spanjaard, “The Apoptotic Action of the Retinoid CD437/AHPN: Diverse Effects, Common Basis,” Journal of Biomedical Science, Vol. 10, No. 1, 2003, pp. 44-49. doi:10.1007/BF02255996
- [138] J. S. Modica-Napolitano, K. Koya, E. Weisberg, B. T. Brunelli, Y. Li and L. B. Chen, “Selective Damage to Carcinoma Mitochondria by the Rhodacyanine MKT- 077,” Cancer Research, Vol. 56, 1996, pp. 544-550.
- [139] D. J. Propper, J. P. Braybrooke, D. J. Taylor, R. Lodi, P. Styles, J. A. Cramer, W. C. J. Collins, N. C. Levitt, D. C. Talbot, T. S. Ganesan and A. L. Harris, “Phase I Trial of the Selective Mitochondrial Toxin MKT 077 in ChemoResistant Solid Tumours,” Annals of Oncology, Vol. 10, No. 8, 1999, pp. 923-927. doi:10.1023/A:1008336904585
- [140] R. Wadhwa, T. Sugihara, A. Yoshida, H. Nomura, R. R. Reddel, R. Simpson, H. Maruta and S. C. Kaul, “Selective Toxicity of MKT-077 to Cancer Cells Is Mediated by Its Binding to the hsp70 Family Protein mot-2 and Reactivation of p53 Function,” Cancer Research, Vol. 60, 2000, pp. 6818-6821.
- [141] J. S. Modica-Napolitano, B. T. Brunelli, K. Koya and L.B. Chen, “Photoactivation Enhances the Mitochondrial Toxicity of the Cationic Rhodacyanine MKT-077,” Cancer Research, Vol. 58, 1998, pp. 71-75.
- [142] T. Minamikawa, A. Sriratana, D. A. Williams, D. N. Bowser, J. S. Hill and P. Nagley, “Chloromethyl-XRosamine (MitoTracker Red) Photosensitises Mitochondria and Induces Apoptosis in Intact Human Cells,” Journal of Cell Science, Vol. 112, 1999, pp. 2419-2430.
- [143] S. Fulda, C. Scaffidi, S. A. Susin, P. H. Krammer, G. Kroemer, M. E. Peter and K.-M. Debatin, “Activation of Mitochondria and Release of Mitochondrial Apoptogenic Factors by Betulinic Acid,” Journal of Biological Chemistry, Vol. 273, 1998, pp. 33942-33948. doi:10.1074/jbc.273.51.33942
- [144] S. Fulda and K.-M. Debatin, “Sensitization for Anticancer Drug-Induced Apoptosis by Betulinic Acid,” Neoplasia, Vol. 7, 2005, pp. 162-170. doi:10.1593/neo.04442
- [145] S. Fulda and G. Kroemer, “Targeting Mitochondrial Apoptosis by Betulinic Acid in Human Cancers,” Drug Discovery Today, Vol. 14, No. 17-18, 2009, pp. 885-890. doi:10.1016/j.drudis.2009.05.015
- [146] V. E. Kagan, A. Bayir, H. Bayir, D. Stoyanovsky, G. G. Borisenko, Y. Y. Tyurina, P. Wipf, J. Atkinson, J. S. Greenberger, R. S. Chapkin and N. A. Belikova, “Mitochondria-Targeted Disruptors and Inhibitors of Cytochrome C/Cardiolipin Peroxidase Complexes: A New Strategy in Anti-Apoptotic Drug Discovery,” Molecular Nutrition & Food Research, Vol. 53, No. 1, 2009, pp. 104-114. doi:10.1002/mnfr.200700402
- [147] X. Gao, X. Wen, L. Esser, B. Quinn, L. Yu, C.-A. Yu and D. Xia, “Structural Basis for the Quinone Reduction in the bc1 Complex: A Comparative Analysis of Crystal Structures of Mitochondrial Cytochrome bc1 with Bound Substrate and Inhibitors at the Qi Site,” Biochemistry, Vol. 42, No. 30, 2003, pp. 9067-9080. doi:10.1021/bi0341814
- [148] L. F. Dong, V. J. A. Jameson, D. Tilly, J. Cerny, E. Mahdavian, A. Marín-Hernández, L. Hernández-Esquivel, S. Rodríguez-Enríquez, J. Stursa and P. K. Witting, “Mitochondrial Targeting of Vitamin E Succinate Enhances Its Pro-apoptotic and Anti-cancer Activity via Mitochondrial Complex II,” Journal of Biological Chemistry, Vol. 286, p. 3717. doi:10.1074/jbc.M110.186643
- [149] D. R. Pfeiffer, T. I. Gudz, S. A. Novgorodov and W. L. Erdahl, “The Peptide Mastoparan Is a Potent Facilitator of the Mitochondrial Permeability Transition,” Journal of Biological Chemistry, Vol. 270, 1995, pp. 4923-4932. doi:10.1074/jbc.270.9.4923
- [150] H. M. Ellerby, W. Arap, L. M. Ellerby, R. Kain, R. Andrusiak, G. D. Rio, S. Krajewski, C. R. Lombardo, R. Rao, E. Ruoslahti, D. E. Bredesen and R. Pasqualini, “Anti-Cancer Activity of Targeted Pro-Apoptotic Peptides,” Nature Medicine, Vol. 5, 1999, pp. 1032-1038. doi:10.1038/12469
- [151] W. Schuler, K. Wecker, H. D. Rocquigny, Y. Baudat, J. Sire and B. P. Roques, “NMR Structure of the (52-96) C-Terminal Domain of the HIV-1 Regulatory Protein Vpr: Molecular Insights into Its Biological Functions,” Journal of Molecular Biology, Vol. 285, No. 5, 1999, pp. 2105- 2117. doi:10.1006/jmbi.1998.2381