Paper Menu >>
Journal Menu >>
 Journal of Analytical Sciences, Methods and Instrumentation, 2012, 2, 194-202 http://dx.doi.org/10.4236/jasmi.2012.24030 Published Online December 2012 (http://www.SciRP.org/journal/jasmi) Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate Samy Emara1*, Walaa Zarad1, Maha Kamal2, Ramzia EL-Bagary3 1Pharmaceutical Chemistry Department, Faculty of Pharmacy, Misr International University, Cairo, Egypt; 2Pharmaceutical Analyti- cal Chemistry Department, Faculty of Pharmacy, Modern Sciences and Arts University, 6th of October City Egypt; 3Pharmaceutical Chemistry Department, Faculty of Pharmacy, Cairo University, Giza, Egypt. Email: *emara_miu@yahoo.com Received September 28th, 2012; revised November 9th, 2012; accepted November 18th, 2012 ABSTRACT A simple, sensitive and precise green high-performance liquid chromatographic method including on-line pre-column oxidation combined by column switching with a short Hypersil ODS analytical column (100 mm × 4.0 mm i.d.) for enrichment and separation was developed and validated to determine low levels of methotrexate (MTX). The method was based on oxidative cleavage of MTX into highly fluorescence products, 2,4-diaminopteridine-6-carboxaldehyde and the corresponding 2,4-diaminopteridine-6-carboxylic acid, during the flow of phosphate buffer (0.04 M, pH 3.4) containing the analyte through the packed reactor of cerium (IV) trihydroxyhydroperoxide (CTH) at a flow-rate of 0.2 mL/min and 40˚C. The fluorescent products were enriched on the head of ODS analytical column for the final separa- tion. The separation was performed at room temperature using an environmentally friendly mobile phase consisting of ethanol and phosphate buffer (0.04 M, pH 3.4) in the ratio of 10:90 (v/v). The eluent was monitored at emission and excitation wavelengths of 463 and 367 nm, respectively. The method was successfully applied, without any interference from the excipients, for the determination of drug in tablets and vials with a detection limit of 0.06 ng/mL from 500 L of sample MTX. Keywords: Cerium (IV) Trihydroxyhydroperoxide; HPLC; Methotrexate; ODS Analytical Column; On-Line Pre-Column Oxidative Cleavage 1. Introduction Methotrexate (MTX, 2,4-diamino-4-deoxy-N10-methyl- pteroglutamic acid) is one of the most widely used anti- cancer drugs and acts as an antimetabolite of folic acid. In high doses it is used in treatment of some solid tumors and leukemia. It is also used in a number of autoimmune diseases [1]. Many methods have been developed for the determi- nation of MTX, including spectrofluorimetry [2-4], im- munoassay [5], capillary electrophoresis [6,7] and high- performance liquid chromatography (HPLC) with UV detection [8-13]. Due to its higher sensitivity and selec- tivity, the measurement of low MTX level was more pre- cise in the HPLC with fluorescence detection [14-25]. MTX does not show native fluorescence but it can be derivatized into strongly fluorescent product after variety of successful analytical schemes. Several methodologies have been developed for the oxidation of MTX to a highly fluorescent product, 2,4-diaminopteridine-6-carboxalde- hyde and the corresponding 2,4-diaminopteridine-6-car- boxylic acid. These methods were based on photo-oxida- tive irradiation at 254 nm [14,19,20,23,24], electroche- mical oxidation [16,17], oxidation with permanganate [4,18,25] and hydrogen peroxide [22-24]. Also, flow injection analysis (FIA) of MTX in pharmaceutical for- mulations has been reported [26,27]. These methods are based on the oxidation of the MTX into highly fluores- cent product 2,4-diaminopteridine-6-carboxylic acid by on-line electrochemical oxidation and acidic potassium permanganate; respectively. A sequential injection ana- lysis has also been reported for the determination of MTX using amperometric biosensor detector [28]. Cerium (IV) trihydroxyhydroperoxide (CTH) has been introduced as a packed reactor in a flowing system for conversion of *Corresponding author. Copyright © 2012 SciRes. JASMI 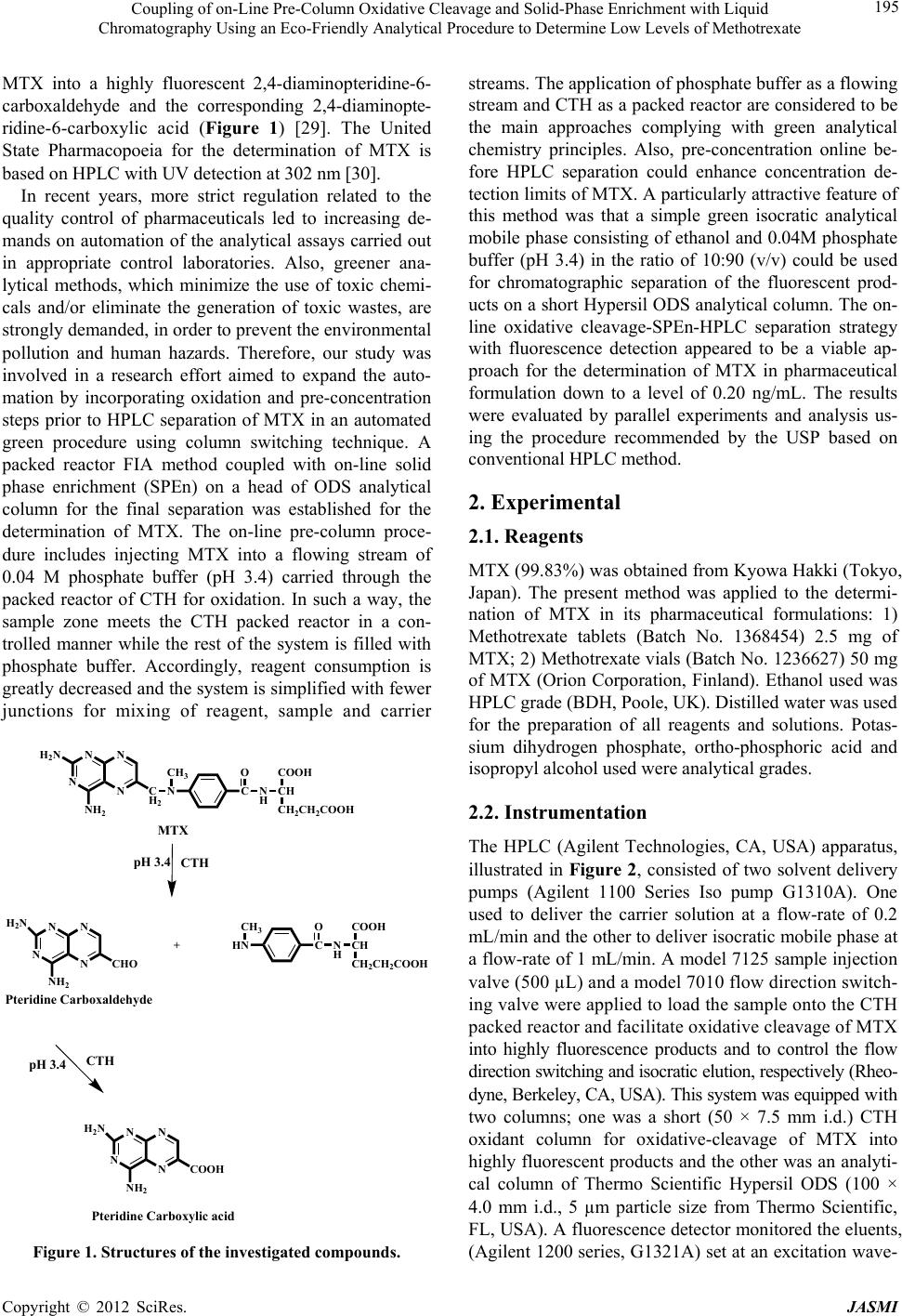 Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate 195 MTX into a highly fluorescent 2,4-diaminopteridine-6- carboxaldehyde and the corresponding 2,4-diaminopte- ridine-6-carboxylic acid (Figure 1) [29]. The United State Pharmacopoeia for the determination of MTX is based on HPLC with UV detection at 302 nm [30]. In recent years, more strict regulation related to the quality control of pharmaceuticals led to increasing de- mands on automation of the analytical assays carried out in appropriate control laboratories. Also, greener ana- lytical methods, which minimize the use of toxic chemi- cals and/or eliminate the generation of toxic wastes, are strongly demanded, in order to prevent the environmental pollution and human hazards. Therefore, our study was involved in a research effort aimed to expand the auto- mation by incorporating oxidation and pre-concentration steps prior to HPLC separation of MTX in an automated green procedure using column switching technique. A packed reactor FIA method coupled with on-line solid phase enrichment (SPEn) on a head of ODS analytical column for the final separation was established for the determination of MTX. The on-line pre-column proce- dure includes injecting MTX into a flowing stream of 0.04 M phosphate buffer (pH 3.4) carried through the packed reactor of CTH for oxidation. In such a way, the sample zone meets the CTH packed reactor in a con- trolled manner while the rest of the system is filled with phosphate buffer. Accordingly, reagent consumption is greatly decreased and the system is simplified with fewer junctions for mixing of reagent, sample and carrier N N N N C H 2 NH 2 H 2 N N C O N HCH COOH CH 2 CH 2 COOH pH 3.4CTH N N N N CHN O N HCH COOH CH 2 CH 2 COOH + NH 2 H 2 N CHO Pteridine Carboxaldehyde CTH pH 3.4 N N N N NH 2 H 2 N COOH Pteridine Carboxylic acid MTX CH 3 CH 3 Figure 1. Structures of the investigated compounds. streams. The application of phosphate buffer as a flowing stream and CTH as a packed reactor are considered to be the main approaches complying with green analytical chemistry principles. Also, pre-concentration online be- fore HPLC separation could enhance concentration de- tection limits of MTX. A particularly attractive feature of this method was that a simple green isocratic analytical mobile phase consisting of ethanol and 0.04M phosphate buffer (pH 3.4) in the ratio of 10:90 (v/v) could be used for chromatographic separation of the fluorescent prod- ucts on a short Hypersil ODS analytical column. The on- line oxidative cleavage-SPEn-HPLC separation strategy with fluorescence detection appeared to be a viable ap- proach for the determination of MTX in pharmaceutical formulation down to a level of 0.20 ng/mL. The results were evaluated by parallel experiments and analysis us- ing the procedure recommended by the USP based on conventional HPLC method. 2. Experimental 2.1. Reagents MTX (99.83%) was obtained from Kyowa Hakki (Tokyo, Japan). The present method was applied to the determi- nation of MTX in its pharmaceutical formulations: 1) Methotrexate tablets (Batch No. 1368454) 2.5 mg of MTX; 2) Methotrexate vials (Batch No. 1236627) 50 mg of MTX (Orion Corporation, Finland). Ethanol used was HPLC grade (BDH, Poole, UK). Distilled water was used for the preparation of all reagents and solutions. Potas- sium dihydrogen phosphate, ortho-phosphoric acid and isopropyl alcohol used were analytical grades. 2.2. Instrumentation The HPLC (Agilent Technologies, CA, USA) apparatus, illustrated in Figure 2, consisted of two solvent delivery pumps (Agilent 1100 Series Iso pump G1310A). One used to deliver the carrier solution at a flow-rate of 0.2 mL/min and the other to deliver isocratic mobile phase at a flow-rate of 1 mL/min. A model 7125 sample injection valve (500 µL) and a model 7010 flow direction switch- ing valve were applied to load the sample onto the CTH packed reactor and facilitate oxidative cleavage of MTX into highly fluorescence products and to control the flow direction switching and isocratic elution, respectively (Rheo- dyne, Berkeley, CA, USA). This system was equipped with two columns; one was a short (50 × 7.5 mm i.d.) CTH oxidant column for oxidative-cleavage of MTX into highly fluorescent products and the other was an analyti- cal column of Thermo Scientific Hypersil ODS (100 × 4.0 mm i.d., 5 µm particle size from Thermo Scientific, FL, USA). A fluorescence detector monitored the eluents, (Agilent 1200 series, G1321A) set at an excitation wave- Copyright © 2012 SciRes. JASMI 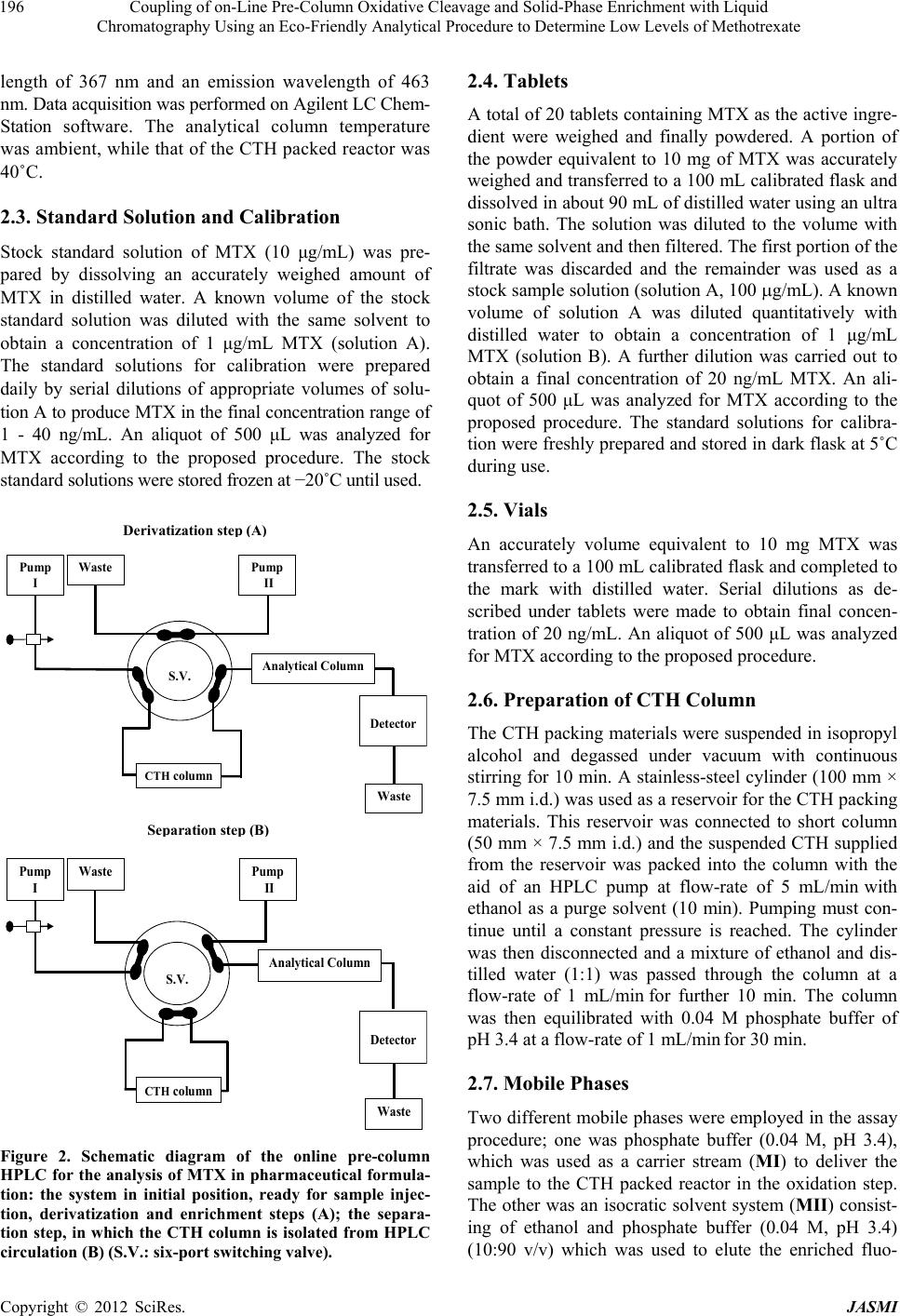 Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate 196 length of 367 nm and an emission wavelength of 463 nm. Data acquisition was performed on Agilent LC Chem- Station software. The analytical column temperature was ambient, while that of the CTH packed reactor was 40˚C. 2.3. Standard Solution and Calibration Stock standard solution of MTX (10 μg/mL) was pre- pared by dissolving an accurately weighed amount of MTX in distilled water. A known volume of the stock standard solution was diluted with the same solvent to obtain a concentration of 1 μg/mL MTX (solution A). The standard solutions for calibration were prepared daily by serial dilutions of appropriate volumes of solu- tion A to produce MTX in the final concentration range of 1 - 40 ng/mL. An aliquot of 500 μL was analyzed for MTX according to the proposed procedure. The stock standard solutions were stored frozen at −20˚C until used. S.V. Analytical Column Detector Waste Pump I CTH column Waste Pump II S.V. Waste Pump I CTH column Waste Analytica l Column Detector Pump II Se p aration ste p ( B ) Derivatization ste p ( A ) Figure 2. Schematic diagram of the online pre-column HPLC for the analysis of MTX in pharmaceutical formula- tion: the system in initial position, ready for sample injec- tion, derivatization and enrichment steps (A); the separa- tion step, in which the CTH column is isolated from HPLC circulation (B) (S.V.: six-port switching valve). 2.4. Tablets A total of 20 tablets containing MTX as the active ingre- dient were weighed and finally powdered. A portion of the powder equivalent to 10 mg of MTX was accurately weighed and transferred to a 100 mL calibrated flask and dissolved in about 90 mL of distilled water using an ultra sonic bath. The solution was diluted to the volume with the same solvent and then filtered. The first portion of the filtrate was discarded and the remainder was used as a stock sample solution (solution A, 100 g/mL). A known volume of solution A was diluted quantitatively with distilled water to obtain a concentration of 1 μg/mL MTX (solution B). A further dilution was carried out to obtain a final concentration of 20 ng/mL MTX. An ali- quot of 500 μL was analyzed for MTX according to the proposed procedure. The standard solutions for calibra- tion were freshly prepared and stored in dark flask at 5˚C during use. 2.5. Vials An accurately volume equivalent to 10 mg MTX was transferred to a 100 mL calibrated flask and completed to the mark with distilled water. Serial dilutions as de- scribed under tablets were made to obtain final concen- tration of 20 ng/mL. An aliquot of 500 μL was analyzed for MTX according to the proposed procedure. 2.6. Preparation of CTH Column The CTH packing materials were suspended in isopropyl alcohol and degassed under vacuum with continuous stirring for 10 min. A stainless-steel cylinder (100 mm × 7.5 mm i.d.) was used as a reservoir for the CTH packing materials. This reservoir was connected to short column (50 mm × 7.5 mm i.d.) and the suspended CTH supplied from the reservoir was packed into the column with the aid of an HPLC pump at flow-rate of 5 mL/min with ethanol as a purge solvent (10 min). Pumping must con- tinue until a constant pressure is reached. The cylinder was then disconnected and a mixture of ethanol and dis- tilled water (1:1) was passed through the column at a flow-rate of 1 mL/min for further 10 min. The column was then equilibrated with 0.04 M phosphate buffer of pH 3.4 at a flow-rate of 1 mL/min for 30 min. 2.7. Mobile Phases Two different mobile phases were employed in the assay procedure; one was phosphate buffer (0.04 M, pH 3.4), which was used as a carrier stream (MI) to deliver the sample to the CTH packed reactor in the oxidation step. The other was an isocratic solvent system (MII) consist- ing of ethanol and phosphate buffer (0.04 M, pH 3.4) (10:90 v/v) which was used to elute the enriched fluo- Copyright © 2012 SciRes. JASMI 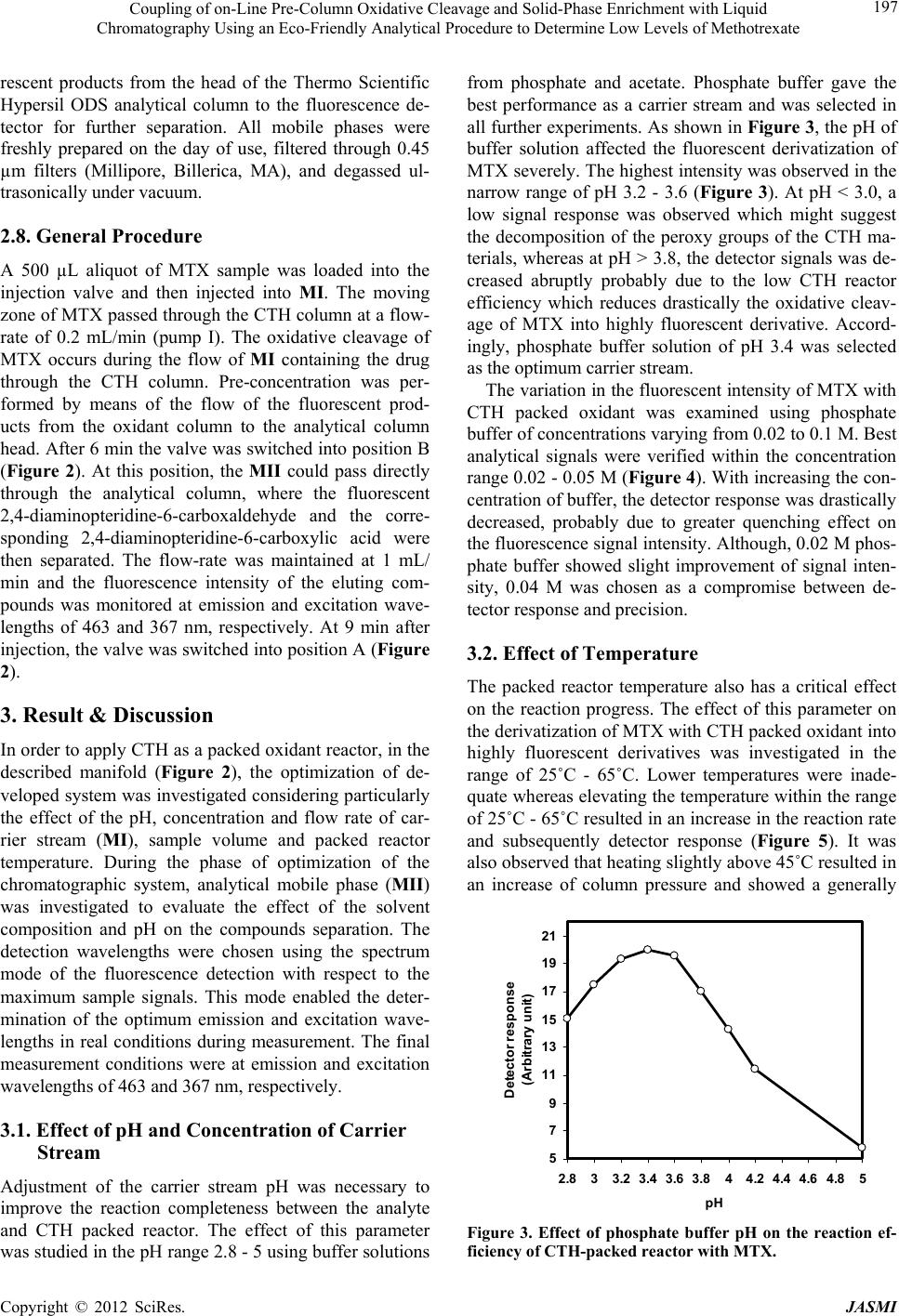 Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate 197 rescent products from the head of the Thermo Scientific Hypersil ODS analytical column to the fluorescence de- tector for further separation. All mobile phases were freshly prepared on the day of use, filtered through 0.45 µm filters (Millipore, Billerica, MA), and degassed ul- trasonically under vacuum. 2.8. General Procedure A 500 µL aliquot of MTX sample was loaded into the injection valve and then injected into MI. The moving zone of MTX passed through the CTH column at a flow- rate of 0.2 mL/min (pump I). The oxidative cleavage of MTX occurs during the flow of MI containing the drug through the CTH column. Pre-concentration was per- formed by means of the flow of the fluorescent prod- ucts from the oxidant column to the analytical column head. After 6 min the valve was switched into position B (Figure 2). At this position, the MII could pass directly through the analytical column, where the fluorescent 2,4-diaminopteridine-6-carboxaldehyde and the corre- sponding 2,4-diaminopteridine-6-carboxylic acid were then separated. The flow-rate was maintained at 1 mL/ min and the fluorescence intensity of the eluting com- pounds was monitored at emission and excitation wave- lengths of 463 and 367 nm, respectively. At 9 min after injection, the valve was switched into position A (Figure 2). 3. Result & Discussion In order to apply CTH as a packed oxidant reactor, in the described manifold (Figure 2), the optimization of de- veloped system was investigated considering particularly the effect of the pH, concentration and flow rate of car- rier stream (MI), sample volume and packed reactor temperature. During the phase of optimization of the chromatographic system, analytical mobile phase (MII) was investigated to evaluate the effect of the solvent composition and pH on the compounds separation. The detection wavelengths were chosen using the spectrum mode of the fluorescence detection with respect to the maximum sample signals. This mode enabled the deter- mination of the optimum emission and excitation wave- lengths in real conditions during measurement. The final measurement conditions were at emission and excitation wavelengths of 463 and 367 nm, respectively. 3.1. Effect of pH and Concentration of Carrier Stream Adjustment of the carrier stream pH was necessary to improve the reaction completeness between the analyte and CTH packed reactor. The effect of this parameter was studied in the pH range 2.8 - 5 using buffer solutions from phosphate and acetate. Phosphate buffer gave the best performance as a carrier stream and was selected in all further experiments. As shown in Figure 3, the pH of buffer solution affected the fluorescent derivatization of MTX severely. The highest intensity was observed in the narrow range of pH 3.2 - 3.6 (Figure 3). At pH < 3.0, a low signal response was observed which might suggest the decomposition of the peroxy groups of the CTH ma- terials, whereas at pH > 3.8, the detector signals was de- creased abruptly probably due to the low CTH reactor efficiency which reduces drastically the oxidative cleav- age of MTX into highly fluorescent derivative. Accord- ingly, phosphate buffer solution of pH 3.4 was selected as the optimum carrier stream. The variation in the fluorescent intensity of MTX with CTH packed oxidant was examined using phosphate buffer of concentrations varying from 0.02 to 0.1 M. Best analytical signals were verified within the concentration range 0.02 - 0.05 M (Figure 4). With increasing the con- centration of buffer, the detector response was drastically decreased, probably due to greater quenching effect on the fluorescence signal intensity. Although, 0.02 M phos- phate buffer showed slight improvement of signal inten- sity, 0.04 M was chosen as a compromise between de- tector response and precision. 3.2. Effect of Temperature The packed reactor temperature also has a critical effect on the reaction progress. The effect of this parameter on the derivatization of MTX with CTH packed oxidant into highly fluorescent derivatives was investigated in the range of 25˚C - 65˚C. Lower temperatures were inade- quate whereas elevating the temperature within the range of 25˚C - 65˚C resulted in an increase in the reaction rate and subsequently detector response (Figure 5). It was also observed that heating slightly above 45˚C resulted in an increase of column pressure and showed a generally 5 7 9 11 13 15 17 19 21 2.833.23.43.63.844.24.44.6 4.85 pH Detect or r espons e (Arbitrary unit) Figure 3. Effect of phosphate buffer pH on the reaction ef- ficiency of CTH-packed reactor with MTX. Copyright © 2012 SciRes. JASMI 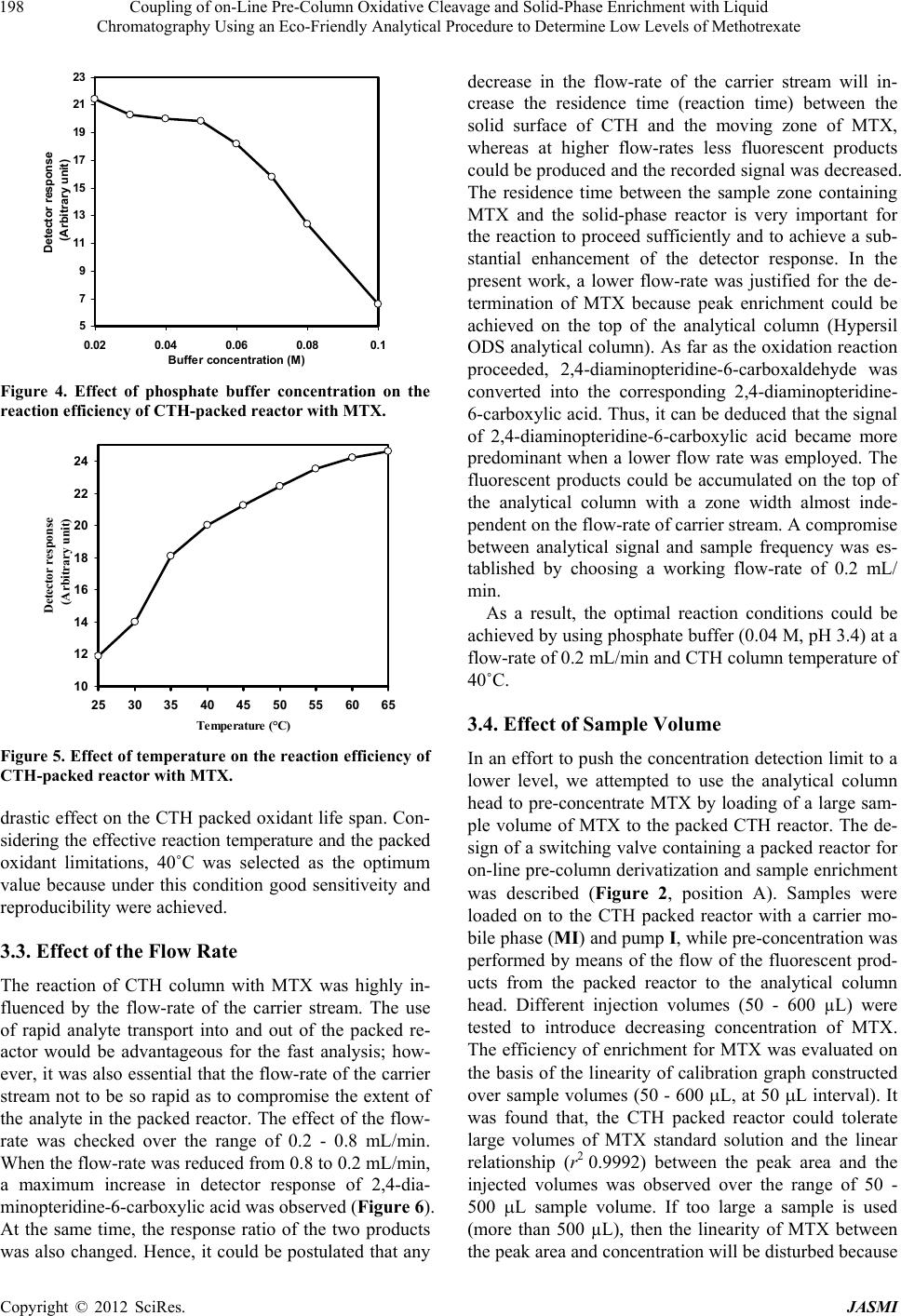 Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate 198 5 7 9 11 13 15 17 19 21 23 0.02 0.04 0.06 0.080.1 Buffer concentration (M) Detector response (Arbitrary unit) Figure 4. Effect of phosphate buffer concentration on the reaction efficiency of CTH-packed reactor with MTX. 10 12 14 16 18 20 22 24 2530 3540 4550 5560 65 Temperature (°C) Detector response (Arbitrary unit) Figure 5. Effect of temperature on the reaction efficiency of CTH-packed reactor with MTX. drastic effect on the CTH packed oxidant life span. Con- sidering the effective reaction temperature and the packed oxidant limitations, 40˚C was selected as the optimum value because under this condition good sensitiveity and reproducibility were achieved. 3.3. Effect of the Flow Rate The reaction of CTH column with MTX was highly in- fluenced by the flow-rate of the carrier stream. The use of rapid analyte transport into and out of the packed re- actor would be advantageous for the fast analysis; how- ever, it was also essential that the flow-rate of the carrier stream not to be so rapid as to compromise the extent of the analyte in the packed reactor. The effect of the flow- rate was checked over the range of 0.2 - 0.8 mL/min. When the flow-rate was reduced from 0.8 to 0.2 mL/min, a maximum increase in detector response of 2,4-dia- minopteridine-6-carboxylic acid was observed (Figure 6). At the same time, the response ratio of the two products was also changed. Hence, it could be postulated that any decrease in the flow-rate of the carrier stream will in- crease the residence time (reaction time) between the solid surface of CTH and the moving zone of MTX, whereas at higher flow-rates less fluorescent products could be produced and the recorded signal was decreased. The residence time between the sample zone containing MTX and the solid-phase reactor is very important for the reaction to proceed sufficiently and to achieve a sub- stantial enhancement of the detector response. In the present work, a lower flow-rate was justified for the de- termination of MTX because peak enrichment could be achieved on the top of the analytical column (Hypersil ODS analytical column). As far as the oxidation reaction proceeded, 2,4-diaminopteridine-6-carboxaldehyde was converted into the corresponding 2,4-diaminopteridine- 6-carboxylic acid. Thus, it can be deduced that the signal of 2,4-diaminopteridine-6-carboxylic acid became more predominant when a lower flow rate was employed. The fluorescent products could be accumulated on the top of the analytical column with a zone width almost inde- pendent on the flow-rate of carrier stream. A compromise between analytical signal and sample frequency was es- tablished by choosing a working flow-rate of 0.2 mL/ min. As a result, the optimal reaction conditions could be achieved by using phosphate buffer (0.04 M, pH 3.4) at a flow-rate of 0.2 mL/min and CTH column temperature of 40˚C. 3.4. Effect of Sample Volume In an effort to push the concentration detection limit to a lower level, we attempted to use the analytical column head to pre-concentrate MTX by loading of a large sam- ple volume of MTX to the packed CTH reactor. The de- sign of a switching valve containing a packed reactor for on-line pre-column derivatization and sample enrichment was described (Figure 2, position A). Samples were loaded on to the CTH packed reactor with a carrier mo- bile phase (MI) and pump I, while pre-concentration was performed by means of the flow of the fluorescent prod- ucts from the packed reactor to the analytical column head. Different injection volumes (50 - 600 µL) were tested to introduce decreasing concentration of MTX. The efficiency of enrichment for MTX was evaluated on the basis of the linearity of calibration graph constructed over sample volumes (50 - 600 L, at 50 L interval). It was found that, the CTH packed reactor could tolerate large volumes of MTX standard solution and the linear relationship (r2 0.9992) between the peak area and the injected volumes was observed over the range of 50 - 500 L sample volume. If too large a sample is used (more than 500 µL), then the linearity of MTX between the peak area and concentration will be disturbed because Copyright © 2012 SciRes. JASMI  Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate Copyright © 2012 SciRes. JASMI 199 D etector res p onse I II 0 3 d 0 3 II I g 0 3 II a I I II 0 3 b I II 0 3 c I II 0 3 e II I 0 3 f Figure 6. Chromatograms obtained after oxidative cleavage of MTX with CTH and fluorimetric detection (20 ng/mL). Peaks: I: 2,4-diaminopteridine-6-carboxylic acid; II: 2,4-diaminopteridine-6-carboxaldehyde. Flow rates: 0.8 mL/min (a); 0.7 mL/ min (b); 0.6 mL/min (c); 0.5 mL/min (d); 0.4 mL/min (e); 0.3 mL/min (f); 0.2 mL/min (g). a long residence time was found necessary to get repro- ducible results upon using large volume of MTX samples. Accordingly, sample volume of 500 µL MTX sample was selected as a compromise between the sensitivity and accuracy. The pre-concentration was effective for the proposed method, achieving detection limit of 0.06 ng/ mL of MTX for a sample volume of 500 µL with fluo- rescence detection. 3.5. Optimization of the Chromatographic System Optimization studies for the oxidative cleavage of MTX with CTH were carried out in the presence of phosphate buffer (0.04 M, pH 3.4) aqueous carrier stream (MI). Also, HPLC experimental set-up required an optimum mobile phase (MII) composition to provide the necessary chromatographic separation of the compounds on the analytical column. It was noted that the CTH reactor needed a long equilibration time with MI after passing of any ethanol containing mobile phase (MII) through it. Thus, the system manifold was set up with column- switching technique and two separate pumps to deliver MI and MII independently in order to eliminate such a long equilibration time (Figure 2). The main objectives of the chromatographic step were to pre-concentrate and separate the highly fluorescent derivatives, 2,4-diaminopteridine-6-carboxaldehyde and the corresponding 2,4-diaminopteridine-6-carboxylic acid. The chromatographic separation was achieved on a short Thermo Scientific Hypersil ODS analytical column (100 mm × 4.0 mm, 5m) using mixture of phosphate buffer (0.04 M, pH 3.4)-ethanol as MII. The optimization pro- cedure was continued by changing the pH of MII as well as changing the ratios of phosphate buffer and ethanol. Variation in the pH of mobile phase in the range of 2.0 - 6 weakly affected retention behavior of the fluorescent products. While variation of ethanol concentration strongly affected retention behaviors and band broadening. A green mobile phase of ethanol and phosphate buffer (0.04 M, pH 3.4) (10:90 v/v) was found to give acceptable separation at a flow rate of 1.0 mL/min and 25˚C column temperature (Figure 6). 3.6. Method Validation 3.6.1. Linearity The linearity was evaluated and established by triplicate analysis of the standard solutions of MTX. The obtained peak areas were plotted against the corresponding con- centrations to generate calibration curve. Good linearity was evident (r2 = 0.9997) over the examined concentra- tion range 1 - 40 ng/mL. The equation for the best-fit straight line was determined by the linear regression analysis as Y = a + bC, where Y is the peak area and C denotes the concentration in ng/mL of MTX. Character- istic parameters of the linear calibration curve are shown in Table 1. 3.6.2. Limit of Detection and Quantification The limit of detection (LOD), defined as the lowest con- centration of MTX that can be clearly detected above the base line signal, is estimated as three-times the signal- to-noise ratio. The LOD was determined (n = 3) by injec- tion of MTX in decreasing concentrations. The LOD was found to be 0.06 ng/mL (Table 1). The LOQ is often defined as 10 times the signal-to-noise ratio. The LOQ was determined (n = 3) by injection of MTX in decreas- ing concentrations. The precision was calculated for each concentration. Then, the LOQ was calculated as the con-  Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate 200 centration, where the precision was less than or equal to 15% and was found to be 0.20 ng/mL (Table 1). 3.6.3. Precision and Accuracy The relative standard deviation (RSD %) and the relative error (RE %) of the mean measured concentration were served as measures of accuracy and precision for valida- tion of the assay procedure. The intra- and inter-day as- say precision and accuracy for MTX are summarized in Table 2. Within the examined range, the intra-day re- producibility and accuracy of the assay were excellent, with RSD % being in the range of 0.23 - 0.28 and with RE % ranged from −0.33 to −0.49. The inter-day RSD% were 0.26 - 0.32 and the mean RE % ranged from −0.36 to −0.56. Repeatability and reproducibility of MTX sam- ples with high and low concentration levels were below 0.60%, indicating a reliable measurement using the pro- posed method (Table 2). RE was evaluated by back- calculation and expressed as the percent deviation be- tween concentration added and concentration found ac- cording to the following: concentration foundconcentrationadded RE 100 concentration added Table 1. Characteristic parameters for the regression equa- tions of the proposed methoda. Parameters MTX Calibration range (ng/mL) 1-40 Detection limit (ng/mL) 0.06 Quantitation limit (ng/mL) 0.20 Slope (b) 1.4465 Standard error of the slope 0.0104 Intercept (a) 0.2421 Standard error of the intercept 0.2149 Correlation coefficient (r2) 0.9997 aY = a + bC, where C is the concentration of MTX in ng/mL and Y is the peak area. Table 2. Precision and accuracy validation of MTX. Concentration (ng/mL) Recovery % ± RSD Mean RE (%) Intra-assaya 5 99.54 ± 0.24 −0.46 10 99.65 ± 0.27 −0.35 20 99.67 ± 0.23 −0.33 40 99.51 ± 0.28 −0.49 Inter-assaya 5 99.48 ± 0.26 −0.52 10 99.62 ± 0.31 −0.38 20 99.64 ± 0.26 −0.36 40 99.44 ± 0.32 −0.56 a Average of five determinations. 3.6.4. Interference Studies In order to examine the selectivity of the proposed method, the effect of common excipients normally used in pharmaceutical formulations was studied. Solutions containing MTX (20 ng/mL) in the presence of more than 100 folds of common additives such as maize starch, calcium hydrogen phosphate, magnesium stearate, man- nitol, red ferric oxide, lactose, methylhydroxypropylcel- lulose, sodium stearyl fumarate, microcrystalline cellu- lose, hydrophobic colloidal silica and sucrose were pre- pared. The undissolved materials were filtered off before injection. No significant changes were observed on the results and recoveries in the range of 99.42% - 99.75% were obtained in all cases. 3.7. Drug Content Analysis The proposed on-line pre-column oxidative cleavage method was applied for the determination of MTX in its commercial formulations together with the official USP method [30]. As indicated, the assay results obtained by the proposed method were in accordance with those ob- tained by the official method. The accuracy and precision of the developed method were further judged by applying t- and F-test at 95% confidence level. The experimental t- and F-values did not exceed the theoretical values, which support the similar accuracy and precision of the proposed and official USP methods (Table 3). The accu- racy and precision of the developed method were further judged by applying t- and F-test at 95% confidence level. The experimental t- and F-values did not exceed the theoretical values, which support the similar accuracy and precision of the proposed and official USA methods (Table 3). Table 3. Determination of MTX in commercial formula- tions by the proposed and official methods. Recovery (%)a ± RSD Commercial formulationProposed method Official method Tablets (20 ng/mL) 99.84 ± 0.34 99.86 ± 0.30 Student’s t-test 0.09 (2.30)b F-test 1.22 (6.38)b Vials (20 ng/mL) 99.85 ± 0.27 99.87 ± 0.29 0.13 (2.30)b 1.14 (6.38)b Recovery (%)c ± RSD Tablets 99.48 ± 0.35 Vials 99.50 ± 0.32 aAverage of five determinations; bTheoretical values at p = 0.05; cFor stan- dard addition of 50% of the nominal content. Copyright © 2012 SciRes. JASMI  Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate 201 Mean value are very close to the theoretical concentra- tions, showing method % recoveries from 99.48 to 99.50 and RSD from 0.32 to 0.35. These results indicate that the effects of the common additives and ingredients of the pharmaceutical formulations do not interfere with the determination of MTX. 3.8. Robustness To determine robustness of the proposed method, ex- perimental conditions such as flow-rate, pH and concen- tration of the buffer solution used as a carrier stream, packed reactor temperature and organic content of the mobile phase were purposely altered and the detector responses were evaluated. Variation of each parameter by ±2% did not have a significant effect on the detector response. 4. Conclusion In this study, a green on-line pre-column oxidation-SPEn- HPLC separation strategy using a packed oxidant reactor of CTH and a short Hypersil ODS analytical column (100 mm × 4.0 mm i.d.) has been developed to determine low levels of MTX. The method was a powerful analytic- cal technique that had excellent sensitivity, sufficient accuracy and required relatively simple and inexpensive instrumentation. These advantages made the proposed method an attractive procedure for the routine quality control and dosage form analysis of MTX at very low concentration level, down to 0.20 ng/mL. The applicabil- ity of this method was evaluated by the determination of MTX in pharmaceutical formulations. Common excipi- ents used as additives in pharmaceutical preparations did not interfere. Furthermore, the proposed method is worthy contributed to the existing environmental friendly analytical chemistry due to reducing or elimination of the use and generation of hazardous substances. REFERENCES [1] S. Shen, T. O’Brien, L. M. Yap, H. M. Prince and C. J. McCormack, “The Use of Methotrexate in Dermatology: A Review,” Australasian Journal of Dermatology, Vol. 53, No. 1, 2102, pp. 1-18. [2] M. Bărcă, M. Ilie, D. L. Baconi, A. M. Ciobanu, D. Bălă- lău and G. T. Burcea, “Spectrofluorimetric Methotrexate Assay in Human Plasma,” Farmacia, Vol. 58, No. 1, 2010, pp. 95-101. [3] S. M. Sabry, M. Abdel-Hady, M. Elsayed, O. T. Fahmy and H. M. Mahr, “Study of Stability of Methotrexate in Acidic Solution. Spectrofluorimetric Determination of Me- thotrexate in Pharmaceutical Preparations through Acid- Catalyzed Degradation Reaction,” Journal of Pharma- ceutical and Biomedical Analysis, Vol. 32, No. 3, 2003, pp. 409-423. doi:10.1016/S0731-7085(03)00239-5 [4] A. Espinosa-Mansilla, I. Durán Merás, A. Zamora Madera, L. Pedano and C. Ferreyra, “Kinetic Fluorimetric Deter- mination of the Antineoplastic Methotrexate (MTX) in Human Serum,” Journal of Pharmaceutical and Biomedi- cal Analysis, Vol. 29, No. 5, 2002, pp. 851-858. doi:10.1016/S0731-7085(02)00212-1 [5] H. Hayashi, C. Fujimaki, S. Tsuboi, T. Matsuyama, T. Daimon and K. Itoh, “Application of Fluorescence Polari- zation Immunoassay for Determination of Methotrexate- Polyglutamates in Rheumatoid Arthritis Patients,” The Tohoku Journal of Experimental Medicine, Vol. 215, No. 1, 2008, pp. 95-101. doi:10.1620/tjem.215.95 [6] H. L. Cheng, S. S. Chiou, Y. M. Liao, C. Y. Lu, Y. L. Chen and S. M. Wu, “Analysis of Methotrexate and Its Eight Metabolites in Cerebrospinal Fluid by Solid-Phase Extraction and Triple-Stacking Capillary Electrophore- sis,” Analytical and Bioanalytical Chemistry, Vol. 398, No. 5, 2010, pp. 2183-2190. doi:10.1007/s00216-010-4152-3 [7] H. L. Cheng, Y. M. Liao, S. S. Chiou and S. M. Wu, “On- Line Stacking Capillary Electrophoresis for Analysis of Methotrexate and Its Eight Metabolites in Whole Blood,” Electrophoresis, Vol. 29, No. 17, 2008, pp. 3665-3673. doi:10.1002/elps.200800029 [8] S. Fang, C. P. Lollo, C. Derunes and M. J. LaBarre, “De- velopment and Validation of a Liquid Chromatography Method for Simultaneous Determination of Three Proc- ess-Related Impurities: Yeastolates, Triton X-100 and Methotrexate,” Journal of Chromatography B, Vol. 879, No. 30, 2011, pp. 3612-3619. doi:10.1016/j.jchromb.2011.10.003 [9] E. Klapkova, J. Kukacka, K. Kotaska, I. Suchanska, R. Urinovska and R. Prusa, “The Influence of 7-OH Meth- otrexate Metabolite on Clinical Relevance of Methotrexate Determination,” Clinical Laboratory, Vol. 57, No. 7-8, 2011, pp. 599-606. [10] K. Michail and M. S. Moneeb, “Determination of Meth- otrexate and Indomethacin in Urine Using SPE-LC-DAD after Derivatization,” Journal of Pharmaceutical and Bio- medical Analysis, Vol. 55, No. 2, 2011, pp. 317-324. doi:10.1016/j.jpba.2011.01.032 [11] L. Hu, Y. Liu and S. Cheng, “Simultaneous Determina- tion of Six Analytes by HPLC-UV for High Throughput Analysis in Permeability Assessment,” Journal of Chro- matographic Science, Vol. 49, No. 2, 2011, pp. 124-128. doi:10.1093/chrsci/49.2.124 [12] T. Sartori, F. S.Murakami, A. P. Cruz and A. M. de Cam- pos, “Development and Validation of a Fast RP-HPLC Method for Determination of Methotrexate Entrapment Efficiency in Polymeric Nanocapsules,” Journal of Chro- matographic Science, Vol. 46, No. 6, 2008, pp. 505-509. [13] D. A. el-Hady, N. A. el-Maali, R. Gotti, C. Bertucci, F. Mancini and V. Andrisano, “Methotrexate Determination in Pharmaceuticals by Enantioselective HPLC,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 37, No. 5, 2005, pp. 919-925. doi:10.1016/j.jpba.2004.07.046 [14] M. Uchiyama, T. Matsumoto, T. Matsumoto, S. Jimi, Y. Copyright © 2012 SciRes. JASMI  Coupling of on-Line Pre-Column Oxidative Cleavage and Solid-Phase Enrichment with Liquid Chromatography Using an Eco-Friendly Analytical Procedure to Determine Low Levels of Methotrexate Copyright © 2012 SciRes. JASMI 202 Takamatsu, K. Tamura and S. Hara, “Simple and Sensi- tive HPLC Method for the Fluorometric Determination of Methotrexate and Its Major Metabolites in Human Plasma by Post-Column Photochemical Reaction,” Biomedical Chro- matography, Vol. 26, No. 1, 2012, pp. 76-80. doi:10.1002/bmc.1628 [15] L. van Haandel and J. F. Stobaugh, “Bioanalytical Method Development for a Generation 5 Polyamidoamine Folic Acid Methotrexate Conjugated Nanoparticle,” Analytical and Bioanalytical Chemistry, Vol. 397, No. 5, 2010, pp. 1841-1852. doi:10.1007/s00216-010-3716-6 [16] S. Chen and Z. Zhang, “Molecularly Imprinted Solid- Phase Extraction Combined with Electrochemical Oxida- tion Fluorimetry for the Determination of Methotrexate in Human Serum and Urine,” Spectrochimica Acta Part A, Vol. 70, No. 1, 2008, pp. 36-41. doi:10.1016/j.saa.2007.07.009 [17] H. Li, W. Luo, Q. Zeng, Z. Lin, H. Luo and Y. Zhang, “Method for the Determination of Blood Methotrexate by High Performance Liquid Chromatography with Online Post-Column Electrochemical Oxidation and Fluorescence Detection,” Journal of Chromatography B, Vol. 845, No. 1, 2007, pp. 164-168. doi:10.1016/j.jchromb.2006.07.026 [18] I. Durán Merás, A. Espinosa Mansilla and M. J. Rodríguez Gómez, “Determination of Methotrexate, Several Pteridi- nes, and Creatinine in Human Urine, Previous Oxidation with Potassium Permanganate, Using HPLC with Photo- metric and Fluorimetric Serial Detection,” Analytical Bio- chemistry, Vol. 346, No. 2, 2005, pp. 201-209. doi:10.1016/j.ab.2005.07.038 [19] E. A. McCrudden and S. E. Tett, “Improved High-Per- formance Liquid Chromatography Determination of Meth- otrexate and Its Major Metabolite in Plasma Using a Poly (Styrene-Divinylbenzene) Column,” Journal of Chromatog- raphy B, Vol. 721, No. 1, 1999, pp. 87-92. doi:10.1016/S0378-4347(98)00439-3 [20] Z. Yu, D. Westerlund and K. S. Boose, “Determination of Methotrexate and Its Metabolite 7-Hydroxymethotrexate by Direct Injection of Human Plasma into a Column- Switching Liquid Chromatographic System Using Post- Column Photochemical Reaction with Fluorimetric De- tection,” Journal of Chromatography B, Vol. 689, No. 2, 1997, pp. 379-386. doi:10.1016/S0378-4347(96)00357-X [21] T. Suzuki, H. Hashimoto and N. Ichinose, “Microanalysis of Methotrexate by High-Performance Liquid Chroma- tography Using a Fluorescence Detector,” Fresenius Jour- nal of Analytical Chemistry, Vol. 351, No. 8, 1995, pp. 806-807. doi:10.1007/BF00323644 [22] H. Kubo, Y. Umiguchi, M. Fukumoto and T. Kinoshita, “Fluorimetric Determination of Methotrexate in Serum by High-Performance Liquid Chromatography Using in-Line Oxidation with Hydrogen Peroxide,” Analytical Sciences, Vol. 8, No. 6, 1992, pp. 789-792. doi:10.2116/analsci.8.789 [23] J. Šalamoun, M. Smrž, F. Kiss and A. Šalamounová, “Column Liquid Chromatography of Methotrexate and Its Metabolites Using a Post-Column Photochemical Reactor and Fluorescence Detection,” Journal of Chromatogra- phy B, Vol. 419, 1987, pp. 213-223. doi:10.1016/0378-4347(87)80279-7 [24] J. Šalamoun and J. František, “Determination of Meth- otrexate and Its Metabolites 7-Hydroxymethotrexate and 2,4-Diamino-N10-methylpteroic Acid in Biological Fluids by Liquid Chromatography with Fluorimetric Detection,” Journal of Chromatography B, Vol. 378, No. 1, 1986, pp. 173-181. [25] J. A. Nelson, B. A. Harris, W. J. Decker and D. Farquhar, “Analysis of Methotrexate in Human Plasma by High- Performance Liquid Chromatography with Fluorescence Detection,” Cancer Research, Vol. 37, No. 11, 1977, pp. 3970-3973. [26] S. Chen, Z. Zhang, D. He, Y. Hu, H. Zheng and C. He, “Flow-Injection-Electrochemical Oxidation Fluorimetry for Determination of Methotrexate,” Luminescence, Vol. 22, No. 4, 2007, pp. 338-342. doi:10.1002/bio.968 [27] S. Emara, “Determination of Methotrexate in Pharmaceu- tical Formulations by Flow Injection Analysis Exploiting the Reaction with Potassium Permanganate,” II Farmaco, Vol. 59, No. 10, 2004, pp. 827-833. doi:10.1016/j.farmac.2004.06.005 [28] R. I. Stefan, R. G. Bokretsion, J. F. van Staden and H. Y. Aboul-Enein, “Simultaneous Determination of L- and D- Methotrexate Using a Sequential Injection Analysis/Am- perometric Biosensors System,” Biosensors and Bioelec- tronics, Vol. 19, No. 3, 2003, pp. 261-267. doi:10.1016/S0956-5663(03)00210-0 [29] S. Emara, S. Razee, A.-N. El-Shorbagy and T. Masujima, “Flow Injection Method for the Determination of Meth- otrexate with a Column-Packed Oxidizing Agent,” Ana- lyst, Vol. 121, No. 2, 1996, pp. 183-188. doi:10.1039/an9962100183 [30] United States Pharmacopeial Convention, “The United States Pharmacopeia and National Formulary (USP 28-NF 23),” The Official Compendia of Standards, Asian Edi- tion, The United States Pharmacopeial Convention Inc., Rockville, 2005. |