Journal of Modern Physics, 2012, 3, 1891-1894
http://dx.doi.org/10.4236/jmp.2012.312238 Published Online December 2012 (http://www.SciRP.org/journal/jmp)
Ab-Initio Structural Study of SrMoO3 Perovskite
Avinash Daga, Smita Sharma
Government Dungar College, Bikaner, India
Email: avinashdaga2009@gmail.com, smita_sharma_bkn@yahoo.com
Received August 10, 2012; revised September 27, 2012; accepted October 10, 2012
ABSTRACT
The equilibrium crystal structure parameter and bulk modulus of the SrMoO3 perovskite has been calculated with
ab-initio method based on density functional theory (DFT) using both local density approximation (LDA) and general-
ized gradient approximation (GGA). The corresponding total free energy along with its various components for SrMoO3
was obtained. The lattice parameter and bulk modulus calculated for SrMoO3 within LDA are 3.99 Å and 143.025 GPa
respectively whereas within GGA are 4.04 Å and 146.14 GPa respectively, both agree well with the available experi-
mental data. The total energy calculated within LDA and GGA is almost the same however lower results are obtained
for GGA. All calculations have been carried out using ABINIT computer code.
Keywords: Perovskite; DFT; LDA; GGA; ABINIT
1. Introduction
Crystal and electronic structures of solid materials are
crucial to understand and improve properties. Many
technological applications, including catalysis, micro-
electronics, substrates for growth of high Tc supercon-
ductors, etc., are based on thin films of ABO3 perovskite.
Until recently, 4d transition metal oxides (TMO) have
attracted less attention because they had the crystalline
defects and having more extended d-orbitals compared to
3d TMO. Yet, numerous studies have shown that this is
no longer the case for some promising TMO such as
molybdates. These oxides are observed to exhibit un-
conventional superconductivity [1] and research is going
on to understand the mechanism [2] of this new quantum
order due to strong electron-electron interaction. Pseudo
gap formation [3], metal-insulator transitions [4,5] and
high-voltage applications are the other significant prop-
erties which draw a considerable attention to 4d TMO.
Some related data were reported by daga et al. [6].
The density functional theory (DFT) is a good ap-
proach for the description of ground state properties of
metals, semiconductors, and insulators. The success of
this technique is that it has allowed us to better under-
stand materials and processes. It has deepened the inter-
pretation of experimental findings. We have used AB-
INIT computer code in order to perform first-principles
DFT calculations. This code is open source ab initio elec-
tronic structure calculation software and has been under
continuous development.
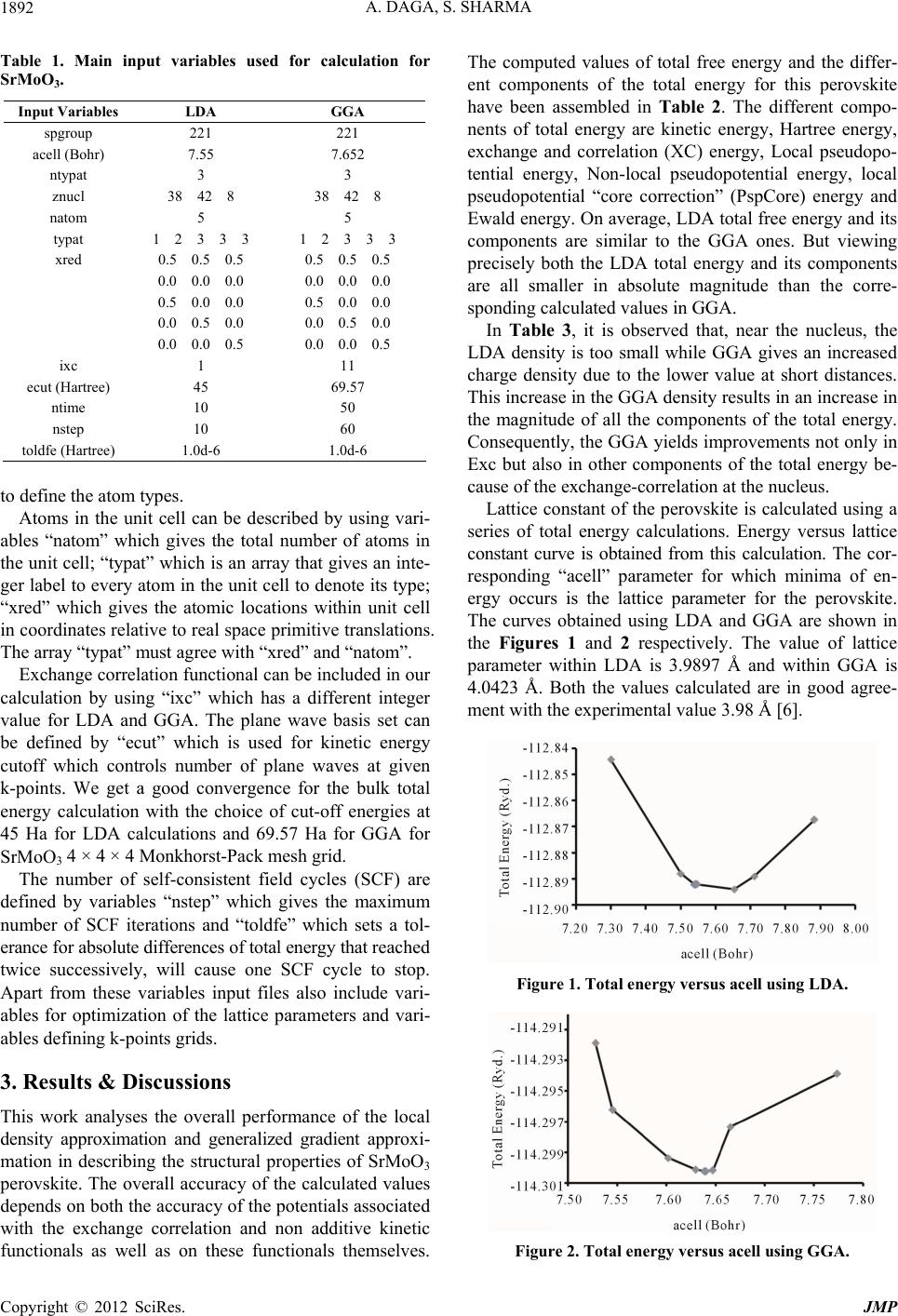
In the present study we calculated lattice constant,
bulk modulus and total energy along with its components
for SrMoO3 perovskite. The chosen perovskite has sim-
ple, cubic Pm3m symmetry.
2. Calculation Method
There are some approximations existing which permit the
calculation of certain physical quantities quite accurately.
In physics the most widely used and the simplest ap-
proximation is the local-density approximation (LDA). It
is based upon exact exchange energy for a uniform elec-
tron gas. The unknown part is exchange-correlation part
of the total-energy functional and it must be approxi-
mated. The functional depends only on the density at the
coordinate. Generalized gradient approximations (GGA)
are still local but also take into account the gradient of
the density at the same coordinate.
In this work, LDA and GGA density functionals are
considered for computation of lattice parameter, bulk
modulus and the total energy. The calculations are carried
out using numerical implementation of the formulation of
DFT; the ABINIT code, based on the DFT using plane
waves and pseudopotentials. Plane waves are used as a
basis set for the electronic wave functions. Many input
variables are needed to run the ABINIT code for the cal-
culations. The main input variables have been listed in
Table 1. “Spgroup” defines space group number of the
perovskite in the input file. “acell” parameter gives the
length scales by which dimensionless primitive transla-
tions are to be multiplied and is specified in Bohr atomic
units. We include input variables “ntypat” and “znucl”,
where “ntypat” gives the number of types of atoms and
“znucl” gives nuclear charge Z of the elements in order
C
opyright © 2012 SciRes. JMP