T. YAMAGUCHI, K. SAITOH
194
The reason why the difference at the center occurs is
that the apex of pyramidal structure is the same size but
is not scaled in total size. However, it is believed that
hydrostatic stress at the surface of pyramidal structure
and the Ge-Si interface is constant without dependence
on size of pyramidal structure.
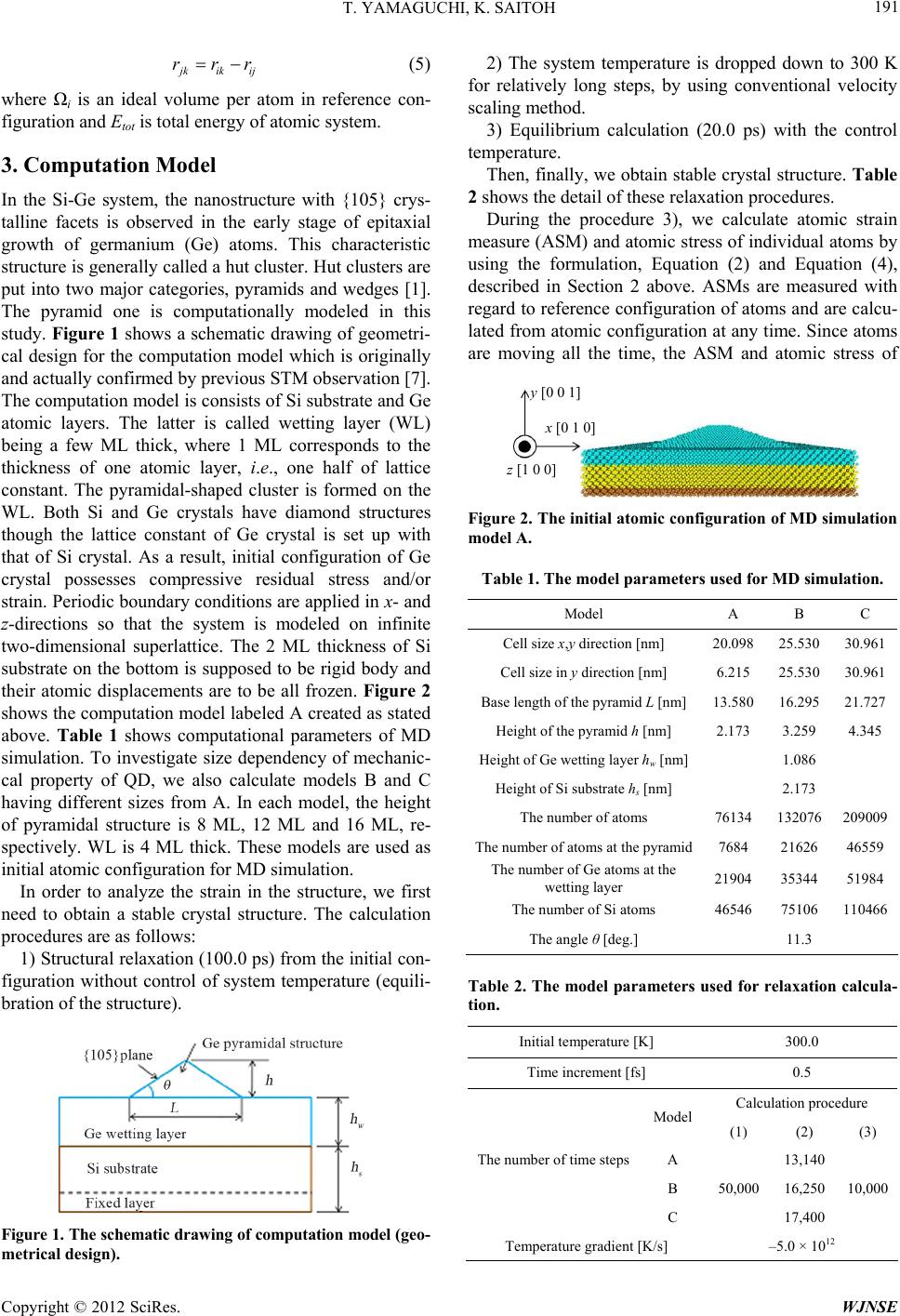
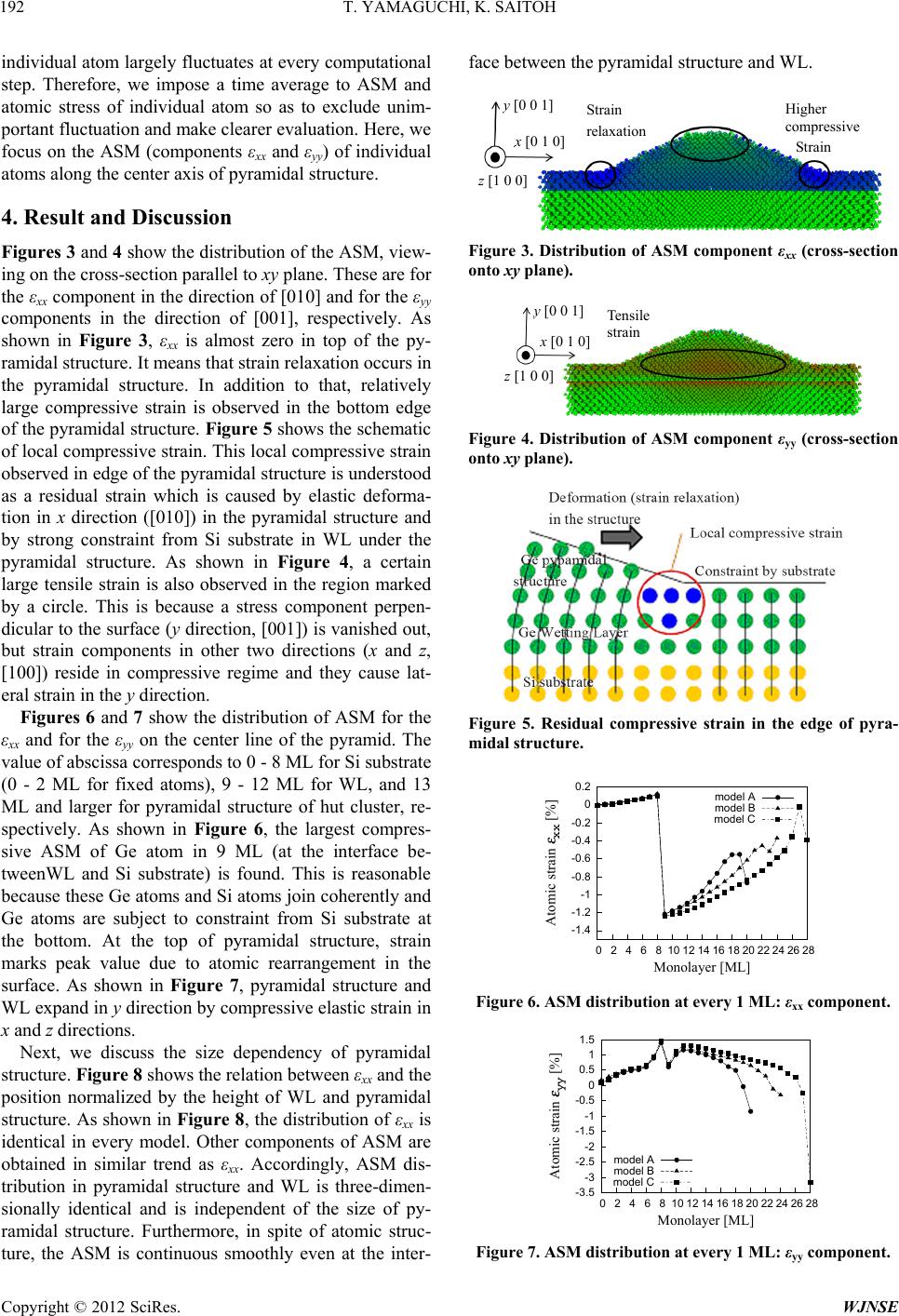
Figures 11 and 12 show the relationship between hy-
drostatic stress σm and atomic volumetric strain εV. Here,
atomic volumetric strain is calculated from components
of ASM, Figure 11 shows plots for atoms on the center
line of the pyramid, just as Figures 6 and 7 above. Sev-
eral plots of positive σm (>0) are found at the surface re-
gion, and their distribution seems at random. Figure 12
is a magnified figure of Figure 11 for its densely plotted
area. The relationship between hydrostatic stress and
atomic volumetric strain is basically linear for all models,
except for the surface of pyramidal structure and Ge-Si
interface. This means that there is a reasonable correla-
tion between atomic stress proposed in the present study
and atomic strain measure, ASM.
5. Conclusions
We perform molecular dynamics simulation for investi-
gating mechanical characteristic in an uncapped pyrami-
dal structure in the Ge/Si(001) system with lattice mis-
match. We estimate the strain by mean of atomic strain
measure (ASM) which is formulated on the Green’s defi-
-6
-4
-2
0
2
4
6
8
-35-30-25-20-15-10-5 0
σ
m
[GPa]
ε
V
[%]
model A
model B
model C
Figure 11. The relationship between hydrostatic stress σm
and atomic volumetric strain εV.
-5
-4
-3
-2
-1
0
-2.5-2-1.5-1-0.5 0 0.
σ
m
[GPa]
ε
V
[%]
model A
model B
model C
Figure 12. The correlation between hydrostatic stress σm
V
nition of continuum strain and
and atomic volumetric strain ε.
is expressed with atomic
ss in atomic scale can be
qu
strain in x and z directions is
ob
tu
ion between atomic
st
ed by “Strategic Project to
[1] L. V. Arapkintomic Structure of
positions, and atomic stress for triplet interaction. Fol-
lowing results are obtained.
1) Elastic strain and stre
alitatively calculated.
2) Local compressive
served in edge of the pyramidal structure. This local
compressive strain is understood as a residual strain
which is caused by elastic deformation in x and z direc-
tions in the pyramidal structure and by strong constraint
from Si substrate in WL under the pyramidal structure.
3) Hydrostatic stress at the surface of pyramidal struc-
re and the Ge-Si interface is constant without depend-
ence on size of pyramidal structure.
4) There is a reasonable correlat
ress proposed in the present study and atomic strain
measure, ASM, except for the surface of pyramidal struc-
ture and Ge-Si interface.
6. Acknowledgements
This study is partly support
Support the Formation of Research Bases at Private
Universities: Matching Fund Subsidy from MEXT (Min-
istry of Education Culture, Sports, Science and Technol-
ogy) (2012)”.
REFERENCES
a and V. A. Yuryev, “A
Ge Quantum Dots on the Si(001) Surface,” JETP Letters,
Vol. 91, No. 6, 2010, pp. 281-285.
doi:10.1134/S0021364010060056
[2] A. A. Williams, et al., “Strain Relaxation during the Ini-
tial Stages of Growth in Ge/Si(001),” Physical Review B,
Vol. 43, No. 6, 1991, pp. 5001-5011.
doi:10.1103/PhysRevB.43.5001
[3] A. I. Nikiforov, V. A. Cherepanov, O. P. Pchelyakov, A.
V. Dvurechenskii and A. I. Yakimov, “In situ RHEED
Control of Self-Organized Ge Quantum Dots,” Thin Solid
Films, Vol. 380, No. 1-2, 2000, pp. 158-163.
doi:10.1016/S0040-6090(00)01493-0
[4] P. Raiteri, F. Valentinotti and L. Miglio, “Stress, Strain
and Elastic Energy at Nanometric Ge Dots on Si(001),”
Applied Surface Science, Vol. 188, No. 1-2, 2002, pp. 4-8.
doi:10.1016/S0169-4332(01)00702-4
[5] W. Yu and A. Madhukar, “Molecular Dynamics Study of
Coherent Island Energetics, Stresses, and Strains in Highly
Strained Epitaxy,” Physical Review Letters, Vol. 79, No.
5, 1997, pp. 905-908. doi:10.1103/PhysRevLett.79.905
[6] Y. Kikuchi, H. Sugai and K. Shintani, “Strain Profiles in
Pyramidal Quantum Dots by Means of Atomistic Simula-
tion,” Journal of Applied Physics, Vol. 89, No. 2, 2001,
pp. 1191-1196. doi:10.1063/1.1335822
[7] Y.-W. Mo, D.E. Savage, B. S. Swarzentruber and M. G.
Lagally, “Kinetic Pathway in Stranski-Krastanov Growth
of Ge on Si(001),” Physical Review Letters, Vol. 65, No.
Copyright © 2012 SciRes. WJNSE