Paper Menu >>
Journal Menu >>
 Open Journal of Blood Diseases, 2012, 2, 71-80 http://dx.doi.org/10.4236/ojbd.2012.24014 Published Online December 2012 (http://www.SciRP.org/journal/ojbd) 71 Incidence of Sickle Cell Anaemia and Thalassaemia in Central India Bhaskar P. Urade Department of Anthropology, University of Pune, Pune, India. Email: druradebp@gmail.com Received June 25th, 2012; revised August 13th, 2012; accepted August 26th, 2012 ABSTRACT Haemoglobinopathies are group of diseases characterized by abnormalities both quantitative and qualitative in the syn- thesis of haemoglobin. Haemoglobinopathies consist of sickle cell anaemia (SCA), thalassaemia (βT) and variant haemoglobins. In India, they are responsible for the largest number of genetic disorders and hence are of great public health hazardous. In India major concerned haemoglobinopathic disorders are sickle cell anaemia and β-thalassaemia. Of the several abnormal haemoglobin molecules, four which are widely prevalent in India include: HbS, HbβT, HbE and HbD. Examination of 6463 individuals showed high incidences for haemoglobin variants, HbS and HbβT in different ethnic groups, the frequency being varies from 0% - 20% and 0% - 9% respectively. The frequency of HbS in Brahmins is 4.17%, in Kalar 5.41%, in Rajput 2.04%, in Muslims 3.73% in Maratha 2.08% in Bania 9.09% while in Teli it is 3.65%. Among the Scheduled castes and Nomadic tribal groups HbS ranges from 1% - 12%; in backward caste catego- ries it varies from 3% - 16%; while in Scheduled tribes it ranges from 0% - 20%. The high magnitude of sickle cell trait has been noticed in the Pardhan (20.31%) followed by the Marar (16.10%), the Dhiwar (11.90%), the Gond (11.89%), the Mahar (11.81%) and the Bania (9.90%). A considerable high frequency (9.27%) of β-thalassaemia has been ob- served among the Sindhi population. Sporadic occurrence of HbβT and HbD among other communities suggested the gradual spread of the genes into the region. The present findings in 11 communities with the thalassaemia syndrome suggest that the β-thalassaemia is accompanied by raised level of HbA2. Unusual greater mean RBC and WBC suggest the high concentration of hypochromic microcytosis in anaemia. The mean MCV and MCH in HbβT and HbD are much lower than the normal ranges compared to HbS. The mean MCHC is much lower in HbβT, HbDD and HbS than the normal range. The cumulative gene frequency of haemoglobinopathies in India is 4.2%. With a population of over 1 billion and a birth rate of 28 per 1000, there are over 42 million carriers and over 12,000 infants are born each year with a major and clinical significant haemoglobinopathy. Out of these, clinically significant sickle cell anaemia and β-tha- lassaemic disorders account for almost equal numbers. Keywords: Haemoglobinopathies; Sickle Cell Anaemia; Thalassaemia; Central India; Prevention; Management 1. Introduction The haemoglobinopathies-sickle cell anaemia, thalassae- mia and other abnormal haemoglobins contribute to ge- netic diseases and imbalance health profile of a nation in general and Vidarbha region in particular. The haemo- globinopathies are a group of inherited conditions that result in the synthesis of either a globins chain with an abnormal structure or reduced synthesis of a globins chain with normal structure leading to chain imbalance. The inherited disorders of haemoglobin are prevalent largely in tropical countries including India. The inher- ited genetic diseases of haemoglobin are controlled by a single gene that transmits from parents to offspring from one generation to another affecting millions of people throughout the world. Sickle cell anaemia (SCA) and thalassaemia (βT) are such genetic disorders caused by point mutation, which are of major concern from the point of view of public health policy. Haemoglobin, a component of red blood cells, carries oxygen from the lungs to different body organs and tissues and brings carbon dioxide back to the lungs. For the first time the presence of Sickle cell gene (HbS) in India was detected in Irula boy in Nilgiri hills in 1952 [1]. This deleterious gene was later on found in many parts of the country. Linus Pualing and his co-workers in 1950 obtained abnormal haemoglobin by electrophoresis in which the proteins with the same molecular weight but different charges migrate at different rates. J. Ingram in 1957 obtained the molecular change in the haemoglobin molecule of sickle cell anaemia. Pattern of Inheritance Copyright © 2012 SciRes. OJBD  Incidence of Sickle Cell Anaemia and Thalassaemia in Central India 72 Carrier Affected Carrier SS AS A A A A AA AS AS AS AS AS SS SS trait N ormal N ormal N ormal trait trait traitanaemia anaemia At present about 5 percents of the world’s population are carriers of a potentially pathological haemoglobin gene (heterozygote condition). Every year about 300,000 infants worldwide are born with thalassaemia syndrome (30 percents) and sickle cell anaemia (70 percents). Globally, the percentage of carriers of thalassaemia is greater than that of carriers of SCA, but because of the high frequency of the sickle cell gene in certain regions the number of affected birth is higher than with thalas- saemia. While the general incidence of β-thalassaemia trait and sickle cell haemoglobinopathy varies between 3 and 17 percents and 1 and 44 percents respectively, be- cause of high consanguinity and caste and area endog- amy some communities show high incidence making the disease a major public and genetic health problem in In- dia [2,3]. Earlier reports show a very high frequency of sickle cell trait (> 20 percents) among the Mahar, Kurmi, Panka, Otkar, Pardhan, Pawara, Bhil etc. [4-20]. β-tha- lassaemia is a major monogenic single gene disorder re- sulting from a reduced or absent synthesis of β-globin ch- ain. This mutant gene is common in communities like, Sindhi, Parsee and Lohana and different ethnic groups of Punjabi, Bengali, Gujarati etc. [21-24]. There are five haplotypes which are responsible for SCA namely, Sene- gal, Bantu, Benin, Asian and Camroon across the world. 2. Material and Method A systematic mass screening camps were carried out in various schools and at the community level during which 6463 individuals comprising 3468 males and 2995 fe- males from four districts of Vidarbha region were screen- ed for haemoglobin S and haemoglobin βT using solu- tions of qualitative solubility test and NESTROFT re- spectively. Figure 1(a) shows the area from where the data have been collected. Necessary consents as prereq- uisite were obtained from individuals before subjecting them to the tests. 20 μl blood samples were drawn from finger prick for each test and mixed thoroughly with so- lutions. Result for haemoglobin S variant was noted down after 3 minutes while for haemoglobin β-variant it was taken after about 20 - 25 minutes. The sample with turbidity and opaque was considered positive for sickle cell and thalassaemia [25]. 2 ml intravenous blood sam- ple was drawn in B.D. vacutainer and brought to the DNA lab at CRC, Nagpur for further analysis. Haemato- logical indices were measured using calibrated ERMA particle counter. Laboratory investigations were carried out following standard procedure as described by Dacie and Lewis [25]. All the samples were subjected to haemoglobin electro- phoresis using cellulose acetate membrane in alkaline TEB buffer at pH 8.9 for pattern confirmation. The known samples (control) of HbS and HbβT along with present samples were run for electrophoresis. A2 fraction of adult haemoglobin was estimated by elution method of 413 nm using spectrophotometer as well as HPCL. Fig- ure 1(b) shows the mobility of pattern for different hae- moglobin variants. A value of more than 3.5 percents for A2 was considered as the cut off point for determination of β-thalassaemia trait. Foetal haemoglobin was esti- mated by the method of Betke et al. [26]. Only male samples were subjected to for G6PD defi- ciency status by florescent spottest [27]. 3. Results Total samples of 6463 individuals have been screened for sickle cell anaemia and thalassaemia with the qualitative solubility test and NESTROFT respectively. Out of which 374 (5.78 percents) and 145 (2.24 percents) indi- viduals were shown to be positive for HbS for HbβT re- spectively. Table 1 portrays the frequency for HbS and HbβT var- ies from 0.16 - 33.33 percents and 0.72 - 9.27 percents respectively. A very high frequency of HbS is recorded among the Pardeshi (25 percents) followed by the Pard- han (20.31 percents). A moderate frequency has been seen among the Dhiwar and the Bania (9.09 per- AS A AA Z SS (a) (b) Figure 1. (a) Map of Maharashtra showing study area; (b) Position of Hb variants. Copyright © 2012 SciRes. OJBD 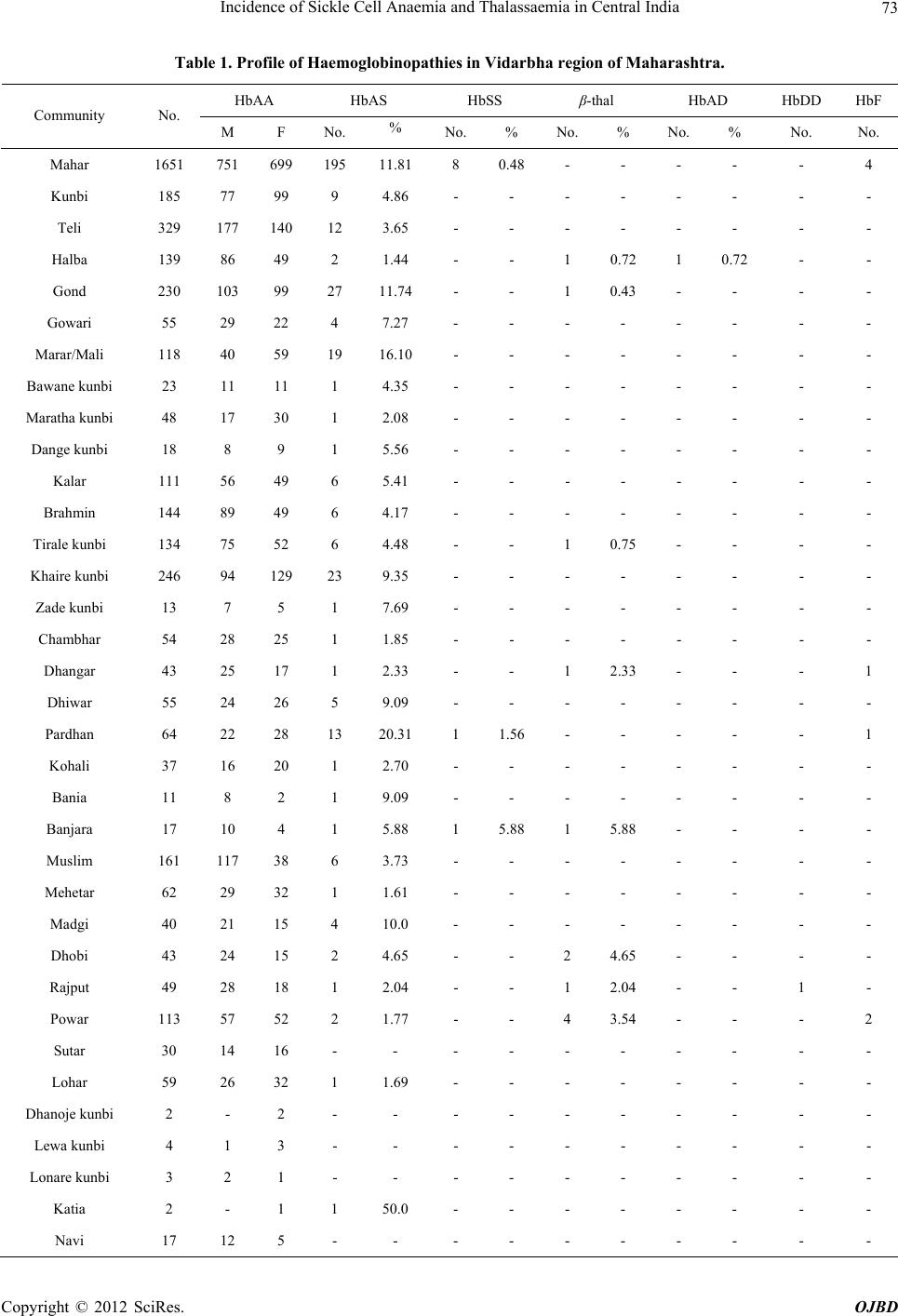 Incidence of Sickle Cell Anaemia and Thalassaemia in Central India Copyright © 2012 SciRes. OJBD 73 Table 1. Profile of Haemoglobinopathies in Vidarbha region of Maharashtra. HbAA HbAS HbSS β-thal HbAD HbDD HbF Community No. M F No. % No. % No. % No. % No. No. Mahar 1651 751 699 195 11.81 8 0.48 - - - - - 4 Kunbi 185 77 99 9 4.86 - - - - - - - - Teli 329 177 140 12 3.65 - - - - - - - - Halba 139 86 49 2 1.44 - - 1 0.72 1 0.72 - - Gond 230 103 99 27 11.74 - - 1 0.43 - - - - Gowari 55 29 22 4 7.27 - - - - - - - - Marar/Mali 118 40 59 19 16.10 - - - - - - - - Bawane kunbi 23 11 11 1 4.35 - - - - - - - - Maratha kunbi 48 17 30 1 2.08 - - - - - - - - Dange kunbi 18 8 9 1 5.56 - - - - - - - - Kalar 111 56 49 6 5.41 - - - - - - - - Brahmin 144 89 49 6 4.17 - - - - - - - - Tirale kunbi 134 75 52 6 4.48 - - 1 0.75 - - - - Khaire kunbi 246 94 129 23 9.35 - - - - - - - - Zade kunbi 13 7 5 1 7.69 - - - - - - - - Chambhar 54 28 25 1 1.85 - - - - - - - - Dhangar 43 25 17 1 2.33 - - 1 2.33 - - - 1 Dhiwar 55 24 26 5 9.09 - - - - - - - - Pardhan 64 22 28 13 20.31 1 1.56 - - - - - 1 Kohali 37 16 20 1 2.70 - - - - - - - - Bania 11 8 2 1 9.09 - - - - - - - - Banjara 17 10 4 1 5.88 1 5.88 1 5.88 - - - - Muslim 161 117 38 6 3.73 - - - - - - - - Mehetar 62 29 32 1 1.61 - - - - - - - - Madgi 40 21 15 4 10.0 - - - - - - - - Dhobi 43 24 15 2 4.65 - - 2 4.65 - - - - Rajput 49 28 18 1 2.04 - - 1 2.04 - - 1 - Powar 113 57 52 2 1.77 - - 4 3.54 - - - 2 Sutar 30 14 16 - - - - - - - - - - Lohar 59 26 32 1 1.69 - - - - - - - - Dhanoje kunbi 2 - 2 - - - - - - - - - - Lewa kunbi 4 1 3 - - - - - - - - - - Lonare kunbi 3 2 1 - - - - - - - - - - Katia 2 - 1 1 50.0 - - - - - - - - Navi 17 12 5 - - - - - - - - - - 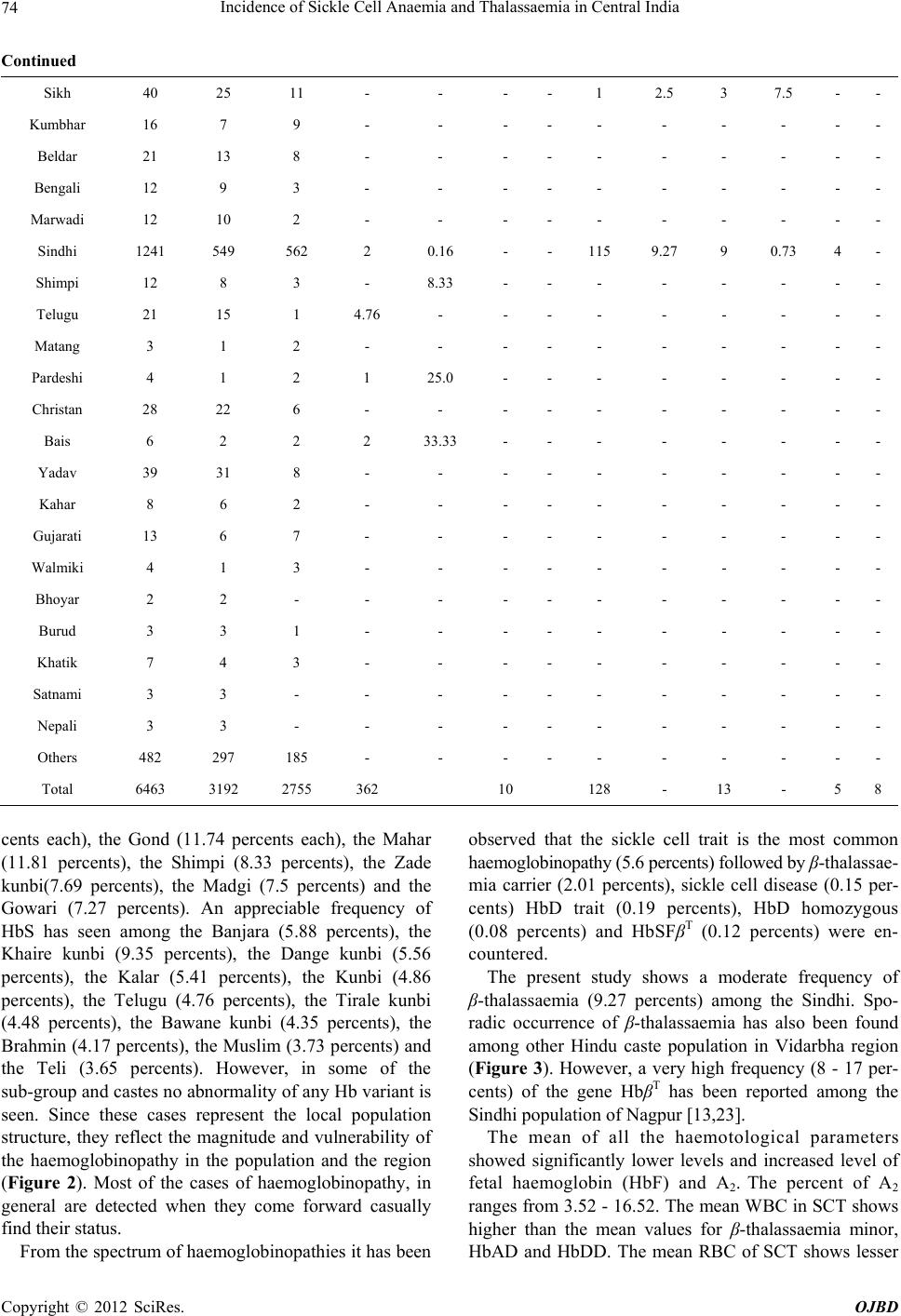 Incidence of Sickle Cell Anaemia and Thalassaemia in Central India 74 Continued Sikh 40 25 11 - - - - 1 2.5 3 7.5 - - Kumbhar 16 7 9 - - - - - - - - - - Beldar 21 13 8 - - - - - - - - - - Bengali 12 9 3 - - - - - - - - - - Marwadi 12 10 2 - - - - - - - - - - Sindhi 1241 549 562 2 0.16 - - 115 9.27 9 0.73 4 - Shimpi 12 8 3 - 8.33 - - - - - - - - Telugu 21 15 1 4.76 - - - - - - - - - Matang 3 1 2 - - - - - - - - - - Pardeshi 4 1 2 1 25.0 - - - - - - - - Christan 28 22 6 - - - - - - - - - - Bais 6 2 2 2 33.33 - - - - - - - - Yadav 39 31 8 - - - - - - - - - - Kahar 8 6 2 - - - - - - - - - - Gujarati 13 6 7 - - - - - - - - - - Walmiki 4 1 3 - - - - - - - - - - Bhoyar 2 2 - - - - - - - - - - - Burud 3 3 1 - - - - - - - - - - Khatik 7 4 3 - - - - - - - - - - Satnami 3 3 - - - - - - - - - - - Nepali 3 3 - - - - - - - - - - - Others 482 297 185 - - - - - - - - - - Total 6463 3192 2755 362 10 128 - 13 - 5 8 cents each), the Gond (11.74 percents each), the Mahar (11.81 percents), the Shimpi (8.33 percents), the Zade kunbi(7.69 percents), the Madgi (7.5 percents) and the Gowari (7.27 percents). An appreciable frequency of HbS has seen among the Banjara (5.88 percents), the Khaire kunbi (9.35 percents), the Dange kunbi (5.56 percents), the Kalar (5.41 percents), the Kunbi (4.86 percents), the Telugu (4.76 percents), the Tirale kunbi (4.48 percents), the Bawane kunbi (4.35 percents), the Brahmin (4.17 percents), the Muslim (3.73 percents) and the Teli (3.65 percents). However, in some of the sub-group and castes no abnormality of any Hb variant is seen. Since these cases represent the local population structure, they reflect the magnitude and vulnerability of the haemoglobinopathy in the population and the region (Figure 2). Most of the cases of haemoglobinopathy, in general are detected when they come forward casually find their status. From the spectrum of haemoglobinopathies it has been observed that the sickle cell trait is the most common haemoglobinopathy (5.6 percents) followed by β-thalassae- mia carrier (2.01 percents), sickle cell disease (0.15 per- cents) HbD trait (0.19 percents), HbD homozygous (0.08 percents) and HbSFβT (0.12 percents) were en- countered. The present study shows a moderate frequency of β-thalassaemia (9.27 percents) among the Sindhi. Spo- radic occurrence of β-thalassaemia has also been found among other Hindu caste population in Vidarbha region (Figure 3). However, a very high frequency (8 - 17 per- cents) of the gene HbβT has been reported among the Sindhi population of Nagpur [13,23]. The mean of all the haemotological parameters showed significantly lower levels and increased level of fetal haemoglobin (HbF) and A2. The percent of A2 ranges from 3.52 - 16.52. The mean WBC in SCT shows higher than the mean values for β-thalassaemia minor, HbAD and HbDD. The mean RBC of SCT shows lesser Copyright © 2012 SciRes. OJBD 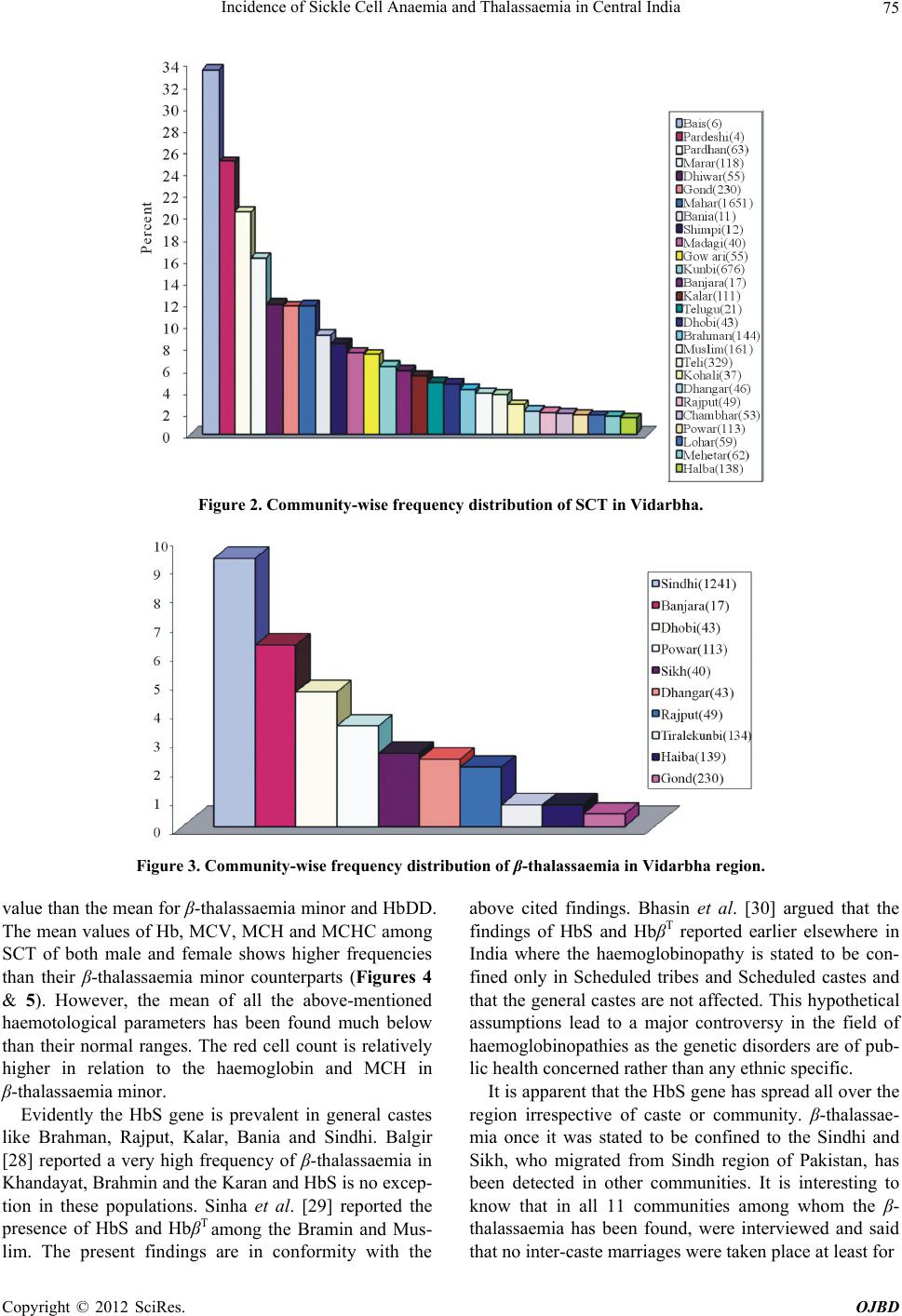 Incidence of Sickle Cell Anaemia and Thalassaemia in Central India 75 Figure 2. Community-w ise frequency distr i bution of SCT in Vidarbha. Figure 3. Community-wise fr eque nc y distribution of β-thalassaemia in Vidarbha region. value than the mean for β-thalassaemia minor and HbDD. The mean values of Hb, MCV, MCH and MCHC among SCT of both male and female shows higher frequencies than their β-thalassaemia minor counterparts (Figures 4 & 5). However, the mean of all the above-mentioned haemotological parameters has been found much below than their normal ranges. The red cell count is relatively higher in relation to the haemoglobin and MCH in β-thalassaemia minor. Evidently the HbS gene is prevalent in general castes like Brahman, Rajput, Kalar, Bania and Sindhi. Balgir [28] reported a very high frequency of β-thalassaemia in Khandayat, Brahmin and the Karan and HbS is no excep- tion in these populations. Sinha et al. [29] reported the presence of HbS and HbβT among the Bramin and Mus- lim. The present findings are in conformity with the above cited findings. Bhasin et al. [30] argued that the findings of HbS and HbβT reported earlier elsewhere in India where the haemoglobinopathy is stated to be con- fined only in Scheduled tribes and Scheduled castes and that the general castes are not affected. This hypothetical assumptions lead to a major controversy in the field of haemoglobinopathies as the genetic disorders are of pub- lic health concerned rather than any ethnic specific. It is apparent that the HbS gene has spread all over the region irrespective of caste or community. β-thalassae- mia once it was stated to be confined to the Sindhi and Sikh, who migrated from Sindh region of Pakistan, has been detected in other communities. It is interesting to know that in all 11 communities among whom the β- thalassaemia has been found, were interviewed and said that no inter-caste marriages were taken place at least for Copyright © 2012 SciRes. OJBD 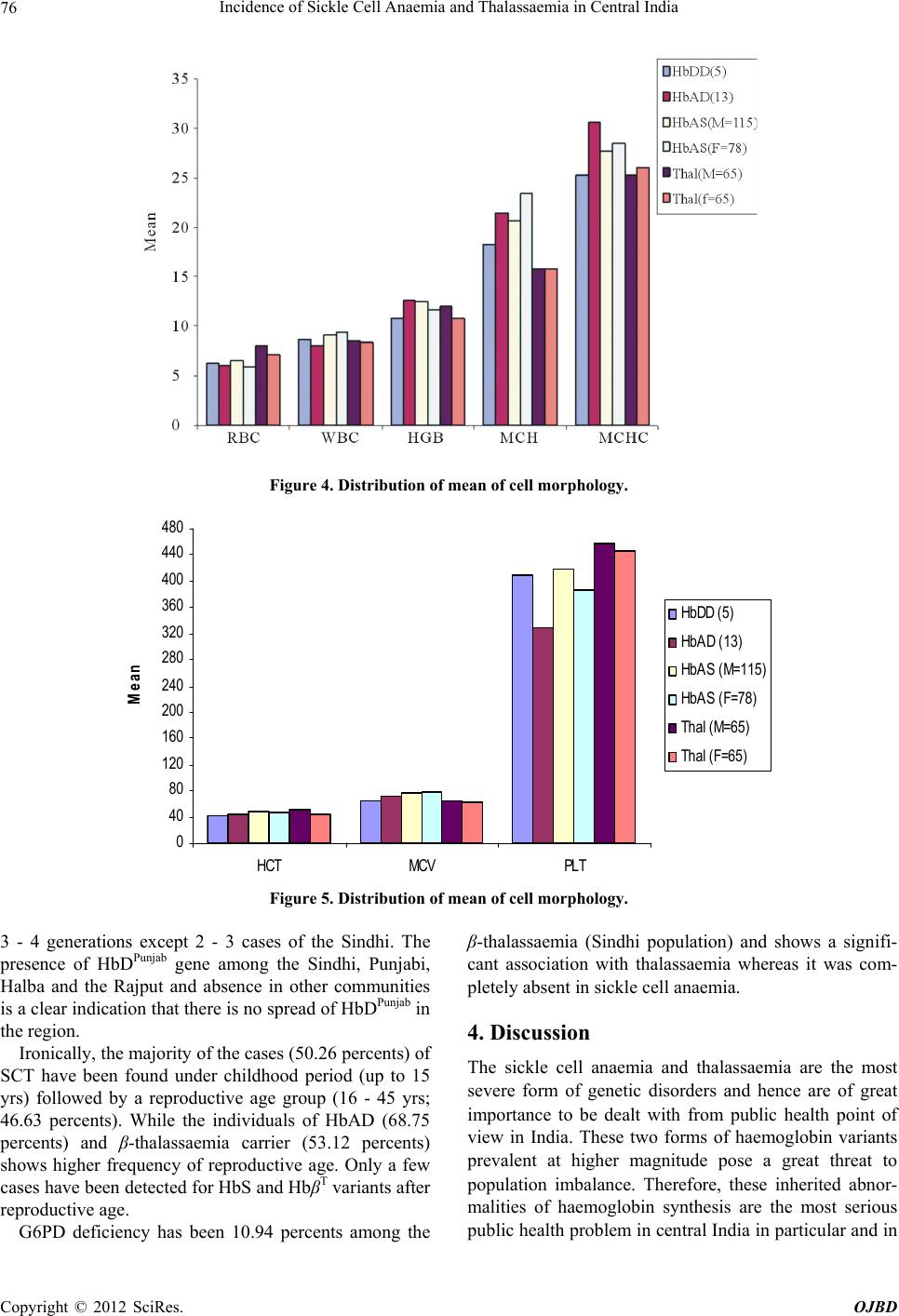 Incidence of Sickle Cell Anaemia and Thalassaemia in Central India 76 Figure 4. Distribution of mean of cell morphology. 0 40 80 120 160 200 240 280 320 360 400 440 480 HCT MCVPLT Mean HbDD (5) HbAD (13) HbAS (M=115) HbAS (F=78) Thal (M=65) Thal (F=65) Figure 5. Distribution of mean of cell morphology. 3 - 4 generations except 2 - 3 cases of the Sindhi. The presence of HbDPunjab gene among the Sindhi, Punjabi, Halba and the Rajput and absence in other communities is a clear indication that there is no spread of HbDPunjab in the region. Ironically, the majority of the cases (50.26 percents) of SCT have been found under childhood period (up to 15 yrs) followed by a reproductive age group (16 - 45 yrs; 46.63 percents). While the individuals of HbAD (68.75 percents) and β-thalassaemia carrier (53.12 percents) shows higher frequency of reproductive age. Only a few cases have been detected for HbS and HbβT variants after reproductive age. G6PD deficiency has been 10.94 percents among the β-thalassaemia (Sindhi population) and shows a signifi- cant association with thalassaemia whereas it was com- pletely absent in sickle cell anaemia. 4. Discussion The sickle cell anaemia and thalassaemia are the most severe form of genetic disorders and hence are of great importance to be dealt with from public health point of view in India. These two forms of haemoglobin variants prevalent at higher magnitude pose a great threat to population imbalance. Therefore, these inherited abnor- malities of haemoglobin synthesis are the most serious public health problem in central India in particular and in Copyright © 2012 SciRes. OJBD  Incidence of Sickle Cell Anaemia and Thalassaemia in Central India 77 India in general reflecting the genetic heterogeneity of the population. The earlier researchers have shown a complete ab- sence of gene HbS in Muslims [31-34]. On the contrary to this, the present findings show the presence of gene HbS among the Muslim (3.73 percents) and is in agree- ment with earlier findings [28,29]. A very high frequency among the Pardhan was reported [9,11,12,35]. The pre- sent findings are in agreement with the findings [4] where they had reported a very high frequency of HbS among the Mahar and the Kunbi. Sickle cell is prevalent in Maharashtra, Madya Pradesh, Orissa, Andhra Pradesh, Gujarat, Tamil Nadu, Karnataka, Kerala and Uttar Pradesh [36]. The HbS gene is prevalent among the gen- eral caste including Brahmins and is in conformity with the findings reported [28,29]. The presence of the deleterious gene HbS in some groups and the complete absence in some groups indi- cates that the independent mutation might have taken place during early life of human being. It is evident from the literature that the several ethnic groups with varied genetic elements have been assimilated into the main- stream, resulting in population diversity with the passage of time [37]. This situation leads to the parallel diver- gence of sub groups of the same community that the one group with the deleterious gene emerged while another group of the same ethnic elements evolved unaffected during the course of time. Initially the mutation might have originated at first and gradually reached a high fre- quency in these populations. Sometime migration may also one of the reasons to carry mutant allele into other population, which ultimately reached a high frequency. The subdivision of the population of India by geographic, linguistic, religious caste and other barriers has resulted in the existence and perpetuation of thousands of distinct highly inbred communities [38]. This remarkable genetic heterogeneity is a distinctive feature of the Indian popu- lation accounts for uneven and variable distribution of the haemoglobinopathies [39]. Diffusion of HbS muta- tion from the Middle East region to India by Arab or Muslim expansion was proposed by Livingstone [40]. In most cases, HbS mutation in the Middle East and in India shares a common haplotype but the high prevalence of HbS among the tribal and scheduled caste populations of India and its relatively less among the Muslims does not convince the suggestion. A considerable high level of HbF in few cases of sickle cell patient and β-thalassaemia carrier provides a protective mechanism in ameliorating the quality of life. A few incidences of splenomegaly have been observed in sickle cell patient, HbSβT and β-thalassaemia major. The persistence splenomegaly was greater in HbSFβT patients probably related to the raised HbF level found in Indian. Higgs et al. [41] reported a significantly greater persis- tence of splenomegaly in Jamaican. The effect of α-thalassaemia is not large enough to be noticeable in Indian populations [42] but this test was not done in the present study. The spread of Hbβ gene in the region is due to gene flow or migration of people from north-west pre-inde- pendent India ie, Sindh region of Pakistan. It is evident from the present findings that the β haemoglobin is in abundance in endemic form in the present population from where they had spread to other part of the central India. It is interesting to note that rapid proliferations of ab- normal haemoglobins S and HbβT in central India is due to castes and area endogamy and lack of medical facili- ties. These many aspects are the root causes for increase- ing complexions of SCD and β-thalassaemia in central India. The HbSβ-thalassaemia patients had more severe dis- ease with lower Hb level, MCV and MCH than their counterparts having HbSS. The red cell count was rela- tively higher among HbSβ patients in relation of the haemoglobin and MCH in carriers of the thalassaemia may be due to production of extra microcytes. In the present study the detection of HbAD (5 percents) in the Sikh is in agreement with the earlier reports [21,24,43]. 5. Conclusions Haemoglobinopathies are the most common monogenic disorder affecting the millions of population worldwide. The geographical distribution of HbS and HbβT variants in India is not uniform as the prevalence varies from 2 - 22 percent and 1 - 15 percent respectively in different region of the country. In central India the incidence of β-thalassaemia has been mainly attributed to its high prevalence in the migrant population of Sindhi origin. Screening of healthy population is required to determine the carrier rates and gene frequencies in this region. Be- cause of the complications associated with haemoglobi- nopathies and frequent health crisis these genetic disor- ders are becoming a growing health care problem in all regions of the developing country. The urgent attention and need of the hour is to launch community based mass screening programme at large level to target the high risk population so that implementation and monitoring can be done to reduce the prevalence of haemoglobinopathies in the country. The misbelieve/wrong notions that has been deeply rooted in the society is that, the presence of sickle cell gene among the lower castes is due to food habits as the people used to eat the flesh of dead animals during ear- lier days. Historically, the man used to eat animal flesh and tubers for his survival when he evolved until he learnt to produce food. Each and everybody were solely Copyright © 2012 SciRes. OJBD  Incidence of Sickle Cell Anaemia and Thalassaemia in Central India 78 dependent upon the nature, sharing a common platform and consumed same thing more or less equally whatever was available during that time. So it is worth mentioning here that every small group of population has something or the other genetic disorder as there is no association between caste or creed and disease. Therefore it is inap- propriate to say that SCA is confined to the lower caste and tribal groups. Earlier researchers were mostly con- fined to tribal groups and hence they did not have any other option but to write about scheduled tribe. From the present study it may be stated that the presence of HbS among higher communities pave the way of transmitting this gene in to lower castes during ancient period as the higher caste people used to sexually exploit socially and economically backward populations. To suppress this fact, there was planned and a thoughtful view of earlier re- searchers [10,44] who stigmatised the Mahar from cen- tral India with the high rate of sickling so that the label of high SCA could remains with them. With the advent of modern technology it has been cleared now that the ear- lier food habit has nothing to do with the occurrence of sickle cell or thalassaemia in man. Occurrence of delete- rious gene HbS and HbβT among the people of different communities is due to alteration at the molecular level in the genetic material (DNA) what is called mutation. Sickle cell anaemia and thalassaemia are the best exam- ples of point mutation as these disorders are caused by a single gene which is transmitted through parents to off- spring from generation to generations. And this mutation has taken place independently in different communities in different areas across the world during the course of human evolution. In present study the presence of HbS gene in almost all castes and communities demonstrate that sickle cell anaemia is no longer confined to specific ethnic groups; instead it is widely distributed in all tribal, scheduled caste, backward and higher caste populations’ native to this region. Similarly, sporadic occurrence of HbβT, HbD in the present study suggests the spread of HbβT well beyond the Sindh and Punjab regions to central India. The high magnitude of HbS and βT appears to contribute significantly to the load of haemoglobinopathies in this region which ultimately going to be a great challenge imbalancing the genetic constitution and threat to these populations in this region. Since many of the sicklers live up to 40 - 42 years with active reproductive life and car- riers lead a normal life like healthy ones, they enhance the chance of propagation of deleterious genes of sickle cell anaemia, thalassaemia and other abnormal haemo- globin genes for generations together. It is not unrealistic to state that the presence of HbS, βT and HbD in hitherto unreported communities from this region is due to of lack of research, under-diagnosis due to suspicion, prevailing bias and prejudices, stigma, complexity, defaming atti- tude, lack of awareness and absence of diagnostic facili- ties at prenatal stage. In Maharashtra, the incidence of HbβT has been mainly attributed to its high prevalence in the migrant population of Sindhi. The sporadic occurrence of HbβT among other communities is the indication of either gradual spread of β-gene due to hybridization or due to independent muta- tion during the course of evolution which needs further indepth research at the molecular level to decipher the reason. The most effective approach to tackle the menace of haemoglobinopathies, it is essential to offering genetic counselling, proper health education, sensitization to the individual concern, prenatal diagnosis and selective ter- mination of pregnancy of the affected foetus are the re- medial measures for prevention and management of haemoglobinopathies in India to see better tomorrow and healthy nation. 5.1. Prevention 1) Identification of groups at high risk through mass screening at the community level. 2) Carrier couples detected are informed of the genetic risk and advice for prenatal diagnosis. 3) Screening and counselling can lead to a significant reduction in affected births. 4) Strategy for prevention-provision of prenatal diag- nosis for risk couple and screening of People of premari- tal/reproductive age. 5) Genetic counselling is essential to protect the auton- omy of the individual. It should be sensitive to the cul- tural, religious and ethical views of the individual. 5.2. Management 1) Public education, genetics, detection of genetic risk the community, family history and premarital genetic counselling. 2) Nationwide programme for prevention and treat- ment. 3) A barrier to implementing effective haemoglobi- nopathy services is lack of awareness about genetic dis- eases. Improve understanding and awareness at commu- nity level. 4) Medical education and training courses should in- clude modules on genetic counselling, the application of genetics to public health and the associated ethical, legal and social issues. 5) Due to lack of sufficient data on the epidemiology of haemoglobinopathies, research and surveillance are important for planning and evaluation of intervention. 6) Heamoglobinopathies are becoming challenging task to introduce genetic approaches to control other chronic Copyright © 2012 SciRes. OJBD  Incidence of Sickle Cell Anaemia and Thalassaemia in Central India 79 childhood diseases. Before formulating are effective pro- gramme, health authorities, health professionals and ex- pert should perceive haemoglobin disorder as a public health problem. 6. Acknowledgements Author expresses his deep concern and gratitude to the Director, Anthropological Survey of India, and Govt. of India for sanctioning the project—haemoglobinopathies in central India, Prof. C.S. Singhrol, Prof. Moyna Chak- ravarty, and Prof. Anjali Kurane for offering their valu- able suggestions, people who donated blood and col- leagues who helped in analyzing blood samples in DNA Lab of ASI, Nagpur. REFERENCES [1] H. Lehman and M. Cutbush, “Sickle Cell Trait in South- ern India,” British Medical Journal, Vol. 1, No. 4755, 1952, pp. 404-405. doi:10.1136/bmj.1.4755.404 [2] R. S. Balgir, “The Burden of Haemoglobinopathies in India and the Challenges Ahead,” Current Science, Vol. 79, 2000, pp. 1536-1547. [3] R. S. Balgir, “The Genetic Burden of Haemoglobinopa- thies with Special Reference to Community Health in In- dia and the Challenges Ahead,” Indian Journal of Hema- tology and Blood Transfusion, Vol. 20, 2002, pp. 2-7. [4] B. N. Shukla and B. R. Solanki, “Sickle Cell Anaemia in Central India,” The Lancet, Vol. 271, No. 7015, 1958, p. 297. doi:10.1016/S0140-6736(58)91035-3 [5] V. V. Deshmukh, “Deficiency of Erythrocyte Glucose-6- Phosphate Dehydrogenase and Sickle-Cell Trait: A Sur- vey at Aurangabad, Maharashtra,” Indian Journal of Medical Research, Vol. 56, No. 6, 1968, pp. 821-825. [6] L. A. Ghatge, “Haemoglobins in Kurmi, Pradhan, PK Community of Madhya Pradesh,” Journal of the Medical Research, Vol. 66, No. 2, 1977, p. 260. [7] R. S. Negi, “Population Dynamics of Sickle Cell Traits Distribution in India,” Ph.D. Thesis (Unpublished), Uni- versity of Calcutta, Calcutta, 1976. [8] S. L. Kate, “Health Problem of Tribal Population Groups from the State of Maharashtra,” Indian Journal of Medi- cal Science, Vol. 55, No. 2, 2001, pp. 99-108. [9] N. M. Blake, A. Ramesh, M. Vijaykumar, J. S. Murthy and K. K. Bhatia, “Genetic Studies on Some Tribes of the Telangana Region, Andhra Pradesh, India,” Acta Anthro- pogenetica, Vol. 5, No. 1, 1981, pp. 41-56. [10] S. R. Das, N. Kumar, P. N. Bhattacharjee and D. B. Sas- try, “Blood Groups (ABO, MN, & Rh), ABH Secretion, Sickle Cell, PTC Taste and Colour Blindness in the Ma- har of Nagpur,” Journal of the Royal Anthropological In- stitute of Great Britain and Ireland, Vol. 91, No. 2, 1961, pp. 345-355. doi:10.2307/2844419 [11] H. P. Banker, S. L. Kate, G. D. Mokanshi, V. A. Kheed- kar and M. A. Phadke, “Distribution of Sickle Cell Hae- moglobin among Different Tribal Groups in Maharashtra State,” Indian Journal of Haematology, Vol. 2, 1984, p. 4. [12] J. D. Goud and P. R. Rao, “Genetic Studies among the Five Tribal Populations of Andhra Pradesh, South India,” Anthropologischer Anzeiger, Vol. 37, No. 1, 1979, pp. 1-9. [13] M. Jain, K. Das, P. B. S. V. Padmanabham, P. Dhar and V. R. Rao, “A1A2BO, Rh (D) Blood Groups and Haemo- globinopathies among Neo-Buddhist (Mahar) of Nagpur City,” Man in India, Vol. 84, No. 1-2, 2003, pp. 77-83. [14] B. P. Urade, “Haemoglobinopathies in Vidarbha Region of Maharashtra,” National Conference on Prevention of Beta-Thalassaemia in India, Anthropological Survey of India, Kolkata, 7-9 March 2008. [15] B. P. Urade and S. K. Mallick, “Incidence of Sickle Cell Anaemia in Chhattisgarh—A Review,” National Seminar, School of Studies in Anthropology, Pt. R.S.S. University, Raipur, 2003. [16] B. P. Urade and S. K. Mallick, “Incidence of Sickle Cell Anaemia among the Mahar of Raipur, Chhattisgarh,” Na- tional Seminar, School of Studies in Anthropology, Pt. R.S.S. University, Raipur, 25-27 February 2004. [17] B. P. Urade and M. Chakravarty, “Haemoglobinopathies of Central and North-Eastern India,” National Seminar, School of Studies in Anthropology, Pt. R.S.S. University, Raipur, 28-30 November 2006. [18] B. P. Urade, M. Chakravarty and S. K. Mallick, “Sickle Cell Anaemia—A Genetically Handicap Disease,” In: R. K. Pathak, A. K. Sinha, B. G. Banerjee, R. N. Vasishat and C. J. Edwin, Eds., Bio-Social Issues in Health, North- ern Book Centre, New Delhi, 2008, pp. 47-55. [19] B. P. Urade, J. Verma and V. R. Rao, “Haemoglobi- nopathies in Maharashtra—Current Scenario and Chal- lenges Ahead,” National Seminar on Human Genomic and Cultural Diversity at School of Studies in Anthro- pology, Pt. R.S.S. University, Raipur, 2-4 March 2009. [20] G. Chakravarty and B. P. Urade, “Haematological Profile in Sickle Cell Anaemia and Thalassaemia Patients in Vi- darbha Region of Maharashtra,” National Seminar on Human Genomic and Cultural Diversity at School of Stu- dies in Anthropology, Pt. R.S.S. University, Raipur, 2-4 March 2009. [21] J. K. Siddoo, S. K. Siddoo, W. H. Chase, L. M. Dean and W. H. Perry, “Thalassaemia in Sikhs,” Blood, Vol. 11, No. 3, 1956, pp. 197-210. [22] R. S. Balgir, “Genetic Epidemiology of the Three Pre- dominant Abnormal Haemoglobins in India,” Journal of the Associations of Physicians of India, Vol. 44, No. 1, 1996, pp. 5-28. [23] A. Jawahirani, M. Mamtani, K. Das, V. Rughwani and H. Kulkarni, “Prevalence of β-Thalassaemia in Subcastes of Indian Sindhi—Results from a Two Phase Survey,” Pub- lic Health, Vol. 121, No. 3, 2007, pp. 193-198. doi:10.1016/j.puhe.2006.10.017 [24] N. Saha and B. Banerjee, “Incidence of Abnormal Hae- moglobins in Punjab,” Calcutta Medical Journal, Vol. 62, 1965, pp. 82-86. [25] J. V. Dacie and S. M. Lewis, “Practical Haematology,” Churchill Livingstone, Edinburgh, 1977. [26] K. Betke, H. R. Marti and I. Schlicht, “Estimation of a Copyright © 2012 SciRes. OJBD  Incidence of Sickle Cell Anaemia and Thalassaemia in Central India Copyright © 2012 SciRes. OJBD 80 Small Percentage of Foetal Haemoglobin,” Nature, Vol. 184, 1959, pp. 1877-1878. doi:10.1038/1841877a0 [27] K. G. Beutler, J. C. Kaplan, G. W. Lohr, B. Ramot and W. N. Valentine, “International Committee for Standardiza- tion in Haematology; Recommended Screening Test G6PD Deficiency,” British Journal of Haematology, Vol. 43, No. 3, 1979, pp. 43-46. [28] R. S. Balgir, “Scenario of Haemoglobin Variants in Cen- tral-East Coast of India,” Current Science, Vol. 90, No. 12, 2006, pp. 1651-1657. [29] S. Sinha, A. Kumar, V. Gupta, S. Kumar, V. P. Singh and R. Raman, “Haemoglobinopathies-Thalassaemias and Ab- normal Haemoglobins in Eastern Uttar Pradesh and Ad- joining Districts of Neighbouring States,” Current Sci- ence, Vol. 87, No. 6, 2004, pp. 775-780. [30] M. K. Bhasin, H. Walter and H. Danker-Hopfe, “Glucose- 6-Phosphate Dehydrogenase Deficiency and Abnormal Haemoglobins (S and E) in People of India,” Journal of Human Ecology, Vol. 3, 1994, pp. 131-159. [31] M. Vijaykumar, K. C. Malhotra, H. Walter, K. Gilbert, P. Landenberg, A. Dannewitz, A. Sorensen, R. Chakraborty, A. P. Reddy and B. N. Mukherjee, “Genetic Studies among the Siddis of Karnataka, India. A Migrant Population from Africa,” Zeitschrift fur Morphologie und Anthro- pologie, Vol. 77, No. 2, 1987, pp. 97-121. [32] A. C. Gorakshakar, M. S. Sathe, S. R. Shirsat and H. M. Bhatia, “Genetic Studies in Ratnagiri and Sindhudurg Districts of Maharashtra—Incidence of ABO, Rh (D), Ina Antigens, G-6PD Deficiency and Abnormal Haemoglo- bins,” Journal of the Indian Anthropological Society, Vol. 22, 1987, pp. 38-46. [33] S. M. A. Hakim, A. J. Baxi, V. Balkrishnan, K. V. Kul- karni, S. S. Rao and H. T. Jhala, “Haptoglobin, Transfer- rin and Abnormal Haemoglobin in Indian Muslims,” In- dian Journal of Medical Research, Vol. 60, No. 5, 1972, pp. 699-703. [34] N. R. Saha, L. Kirk, S. Shanbhag, S. R. Joshi and H. M. Bhatia, “Population Genetic Studies in Kerala and the Nilgiris (South West Asia),” Human Heredity, Vol. 26, 1976, pp.175-197. doi:10.1159/000152802 [35] V. R. Rao and H. M. Bhatia, “Quantitative Assessment of HbS in Sickle Cell Heterozygotes among Some Tribes in Maharashtra,” Indian Journal of Medical Research, Vol. 87, 1988, pp. 257-261. [36] M. B. Agarwal, “The Burden of Haemoglobinopaties in India—Time to Wake Up?” Journal of the Associations of Physicians of India, Vol. 53, 2005, pp. 1017-1018. [37] R. V. Russel and H. Lal, “Tribes and Castes of the Central Provinces of India,” Vol. 4, McMillon Company, London, 1916. [38] G. M. Brittenham, “Globin Gene Variant and Polymor- phism in India,” In: W. P. Winter, Ed., Haemoglobin Variants in Human Populations, CRC Press Inc., Florida, Vol. 2, 1988, pp. 79-109. [39] C. S. Iswad and S. N. Naik, In: H. M. Bhatia and V. R. Rao, Eds., Genetic Atlas and Indian Tribes, ICMR, Bomba, 1986. [40] F. B. Livingstone, “Who Gave Whom Haemoglobin S— The Use of Restriction Cite Haplotype Variation for the Interpretation of the Evolution of the βS Globin Gene,” American Journal of Human Biology, Vol. 1, No. 3, 1989, pp. 289-302. doi:10.1002/ajhb.1310010309 [41] D. R. Higgs, B. E. Aldridge, J. Lamb, J. B. Clegg, D. J. Weatherall, R. H. Hayes, Y. Grandison, Y. Lowrie, K. P. Mason, B. E. Searjeant and G. R. Searjeant, “The Interac- tion of Alpha Thalassaemia and Homozygous Sickle Cell Disease,” The New England Journal of Medicine, Vol. 306, No. 24, 1982, p. 1441. doi:10.1056/NEJM198206173062402 [42] A. E. Kulozik, B. C. Kar, R. K. Satapathy, B. E. Searjeant, G. R. Searjeant and D. J. Weatherall, “Foetal Haemoglo- bin Levels and βS Globin Haplotypes in an Indian Popula- tion with Sickle Cell Disease,” Blood, Vol. 69, 1987, p. 1742. [43] G. W. G. Bird and H. Lehmann, “Haemoglobin D in In- dia,” British Medical Journal, Vol. 1, 1956, p. 514. doi:10.1136/bmj.1.4965.514 [44] I. Karve and V. M. Dandekar, “Anthropometric Meas- urement of Maharashtra, Deccan College Monograph,” Series No. 8, 1951. |