Case Reports in Clinical Medicine
Vol.2 No.1(2013), Article ID:28937,4 pages DOI:10.4236/crcm.2013.21025
Concurrence of composite adrenal pheochromocytoma-ganglioneuroma and renal pelvic cancer: A case report
![]()
1Department of Surgery, Division of Urology, Tottori University Faculty of Medicine, Yonago, Japan; *Corresponding Author: khonda_khonda56@msn.com
2Department of Surgery Related, Division of Urology, Kobe University Graduate School of Medicine, Kobe, Japan
Received 28 January 2013; revised 28 February 2013; accepted 13 March 2013
Keywords: Pheochromocytoma; Ganglioneuroma; Renal Pelvic Cancer
ABSTRACT
Embryologically, chromaffin cells of the pheochromocytoma and ganglion cells of the ganglioneuroma are both derived from neural crest cells. Composite pheochromocytoma-ganglioneuroma (Pheo-GN) in a single adrenal gland is very rare. A case report of a patient with composite Pheo-GN of the adrenal gland and renal pelvic cancer is presented. Laparoscopic left adrenalectomy, nephroureterectomy and para-aortic lymphadenectomy were performed. This represents the first report of simultaneous surgical treatment for composite Pheo-GN and renal pelvic cancer.
1. INTRODUCTION
Pheochromocytoma and ganglioneuroma share a common embryologic origin, the neural crest. Composite pheochromocytoma-ganglioneuroma (Pheo-GN) in a single adrenal gland is very rare. Patients with Pheo-GN are usually diagnosed with pheochromocytoma through signs and symptoms, specific hormone tests such as catecholamines and/or images from CT and 123Ior 131I-metaiodobenzylguanidine (MIBG) scintigraphy. We present the clinical, labolatory, imaging and pathological findings in a 70-year-old man with Pheo-GN and renal pelvic cancer. The patient underwent transperitoneal laparoscopic adrenalectomy, radical nephroureterectomy and para-aortic lymphadenectomy. This represents the first report of simultaneous surgical treatment of composite Pheo-GN and renal pelvic cancer.
2. CASE REPORT
A 70-year-old man was referred to our hospital for examination of a left adrenal tumor, which had been incidentally found on abdominal computed tomography (CT) (Figure 1). He reported a 10-year history of diabetes that had been controlled with medication, but had no other clinical complaints. Abdominal contrast-enhanced CT revealed a 65 × 55-mm heterogeneously enhanced mass in the left adrenal gland and a 25 × 20-mm mass in the left renal pelvis. Serum catecholamine levels were normal, but urinary fractionated catecholamine levels were elevated. Preoperative work-up showed elevated 24-h levels of urine vanillylmandelic acid (18.8 mg/24h; reference range, 1.5 - 4.3 mg/24h), metanephrine (0.89 mg/ 24h; reference range, 0.05 - 0.20 mg/24h), normetanephrine (0.70 mg/24h; reference range, 0.10 - 0.28 mg/24h) and dopamine (2400 mg/24h; reference range, 100 - 1000 mg/24h). Subsequent 123I-MIBG scintigraphy showed moderately intense focal uptake in the left adrenal lesion. Conversely, ureteroscopic biopsy at the site of CT findings in the left renal pelvis revealed urothelial carcinoma. From these findings, the patient was diagnosed with pheo-

Figure 1. Abdominal enhanced CT revealing a 65 × 55-mm heterogeneously enhanced mass (arrow) in the left adrenal gland (a) and a 25 × 20-mm mass (arrow) in the left renal pelvis (b).
chromocytoma and renal pelvic cancer, and was prepared for surgical treatment by administration of Doxazosin, an α1-blocker. The patient underwent transperitoneal laparoscopic adrenalectomy, radical nephroureterectomy and para-aortic lymphadenectomy.
The adrenal tumor was well-circumstanced, firm, with mixed grayish-white and yellowish-white components. No necrosis or hemorrhage was observed. The renal pelvic tumor was a broad-based papillary tumor located at the superior calyx (Figure 2). Histologically, the adrenal tumor consisted of 2 irregularly distributed but distinct patterns. The first pattern showed the characteristic “Zellballen” architectural growth pattern in which neoplastic cells, with abundant basophilic cytoplasm, form rounded to oval nets surrounded by a fibrovascular network of sustentacular cells. This portion of the tumor stained with chromogranin A and synaptophysin, consistent with pheochromocytoma. The second pattern showed neoplastic cells arranged in bundles with small spindle-shaped nuclei and cytoplasm and consisting of ganglion cells embedded in fibrous stroma. This portion of the tumor with weakly stained by chromogranin A and synaptophysin, consistent with ganglioneuroma. The left adrenal tumor thus exhibited histological features of composite PheoGN (Figures 3 and 4). Pathological diagnosis of the renal pelvic tumor was urothelial carcinoma, infiltrating into the renal parenchyma (pT3), but with no lymph node metastases.
Postoperatively, diabetes treatment was discontinued. Three courses of combination chemotherapy comprising gemcitabine plus carboplatin were administered as adjuvant therapy. The patient is presently alive with no evidence of recurrence or metastatic disease as of 24 months postoperatively.
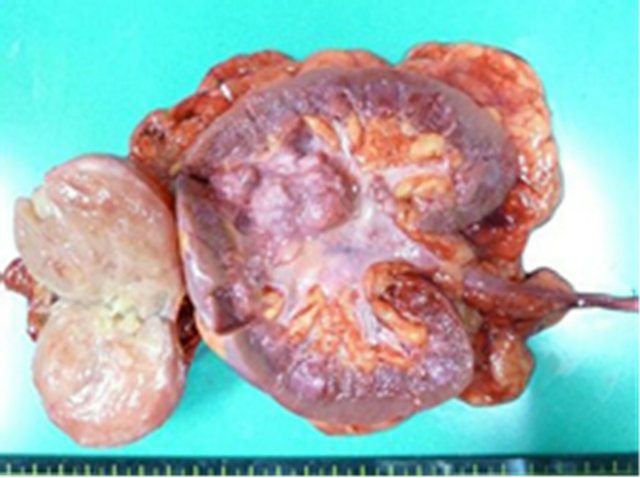
Figure 2. Gross findings of the left adrenal tumor and renal pelvic tumor. The cut surface of the adrenal tumor is a mixture of grayish-white and yellowish-white solid components. No necrosis or hemorrhage is apparent. The renal pelvic tumor is a papillary broad-based tumor located at the superior calyx.
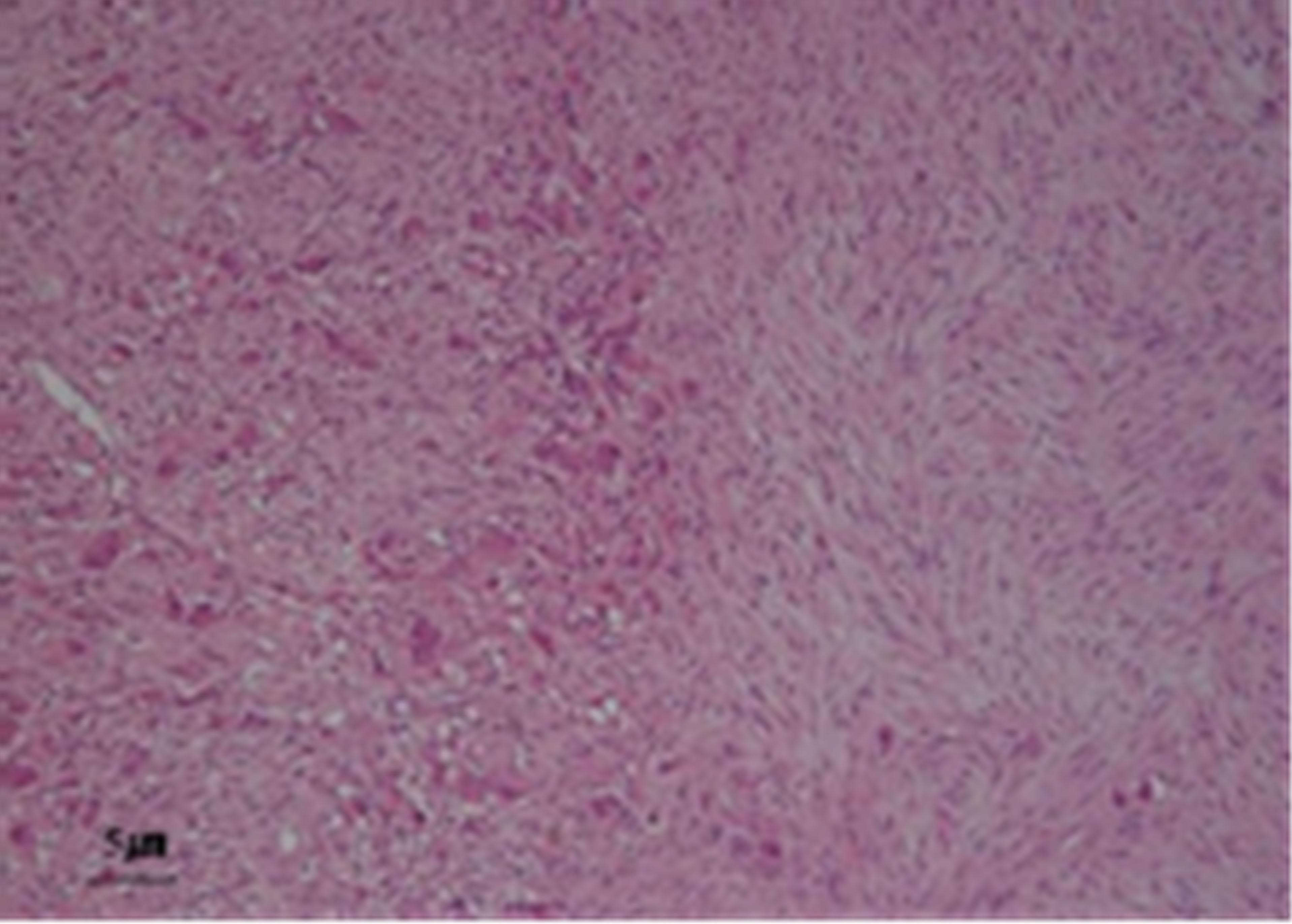
Figure 3. Hematoxylin-eosin stain of the adrenal gland tumor shows two separate components that merge from one into the other. One (left) shows features of pheochromocytoma. The other (right) displays features of ganglioneuroma.
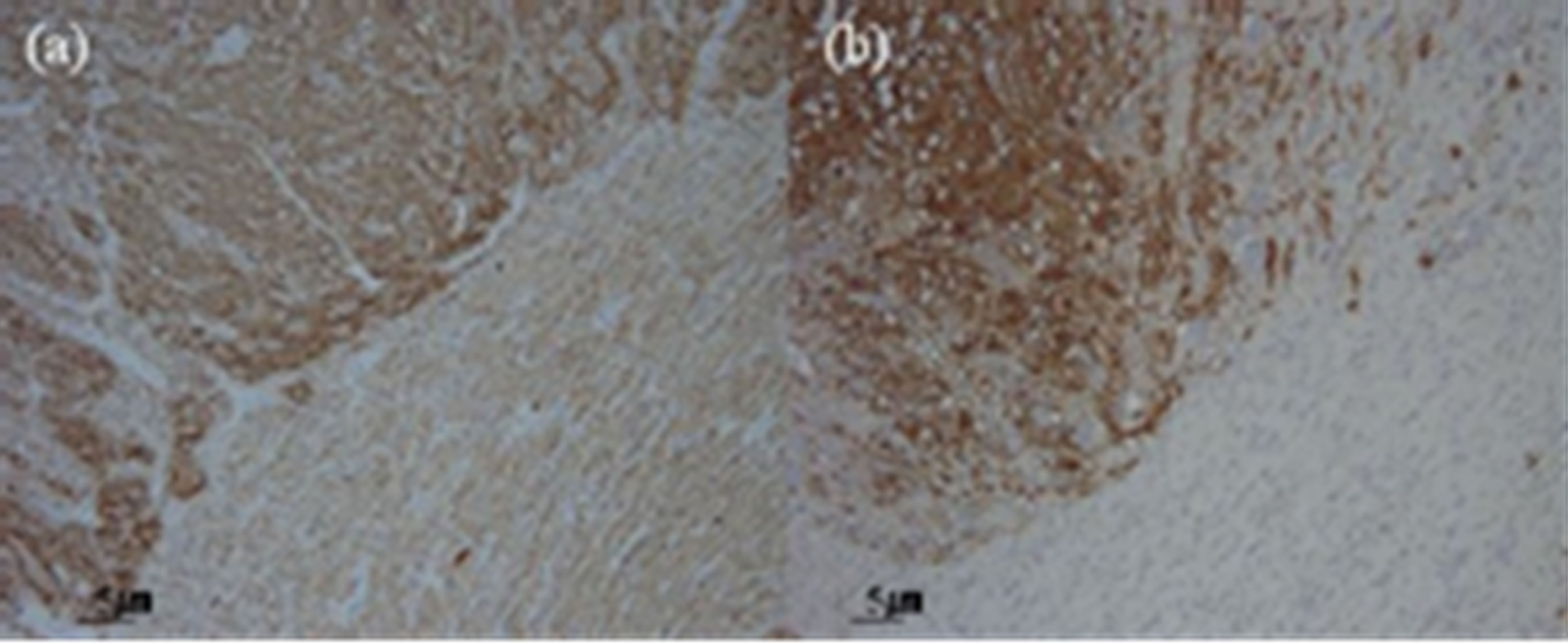
Figure 4. The pheochromocytoma component (upper left) of the tumor staining strongly for chromogranin A (a) and synaptophysin (b), and the ganglioneuroma component (lower right) of the tumor stains weakly.
3. DISCUSSION
Sympathogonia derived from certain neural crest cells differentiate into neuroblasts and pheochromoblasts. These neural crest-derived cells migrate ventrally and differentiate, giving rise to the cellular components of the sympathetic ganglia and adrenal medulla. The adrenal medulla is a complex organ populated by chromaffin cells, neurons, and glial cells. Any disturbance of migration or maldevelopment of the neural crest might result in the development of composite tumor [1-4]. The designation “composite pheochromocytoma” has been used to describe tumors with both components theoretically arising from a common embryological progenitor. Alternatively, if associated tumors have a different embryological origin from the pheochromocytoma, the term “mixed pheochromocytoma” is currently adopted [5]. Embryologically, chromaffin cells of the pheochromocytoma and ganglion cells of the ganglioneuroma are both derived from neural crest cells [2,6]. The coexistence of pheochromocytoma and ganglioneuroma is designated as composite Pheo-GN. Immunohistochemically, functional markers of chromogranin A and synaptophysin are useful to distinguish between pheochromocytoma and ganglioneuroma. Chromogranin A is localized in the matrix of secretory granules along with catecholamines, whereas synaptophysin is principally a membrane marker of small synaptic-like vesicles and is present at very low concentrations in the membranes of secretory granules. Both are strongly and diffusely expressed in pheochromocytoma cells. In contrast, in ganglioneuroma, staining is weak or focal and may display a linear or punctate distribution [2].
Khan et al. reported 45 cases of composite pheochromocytoma, 71% of which coexisted with ganglioneuroma. Preoperatively, functional evidence of Pheo-GN was found in 67% [2]. Since most ganglioneuromas do not secrete catecholamines or steroid hormones, patients may have no clinical manifestations. However, the excessive hormone levels secreted by adrenal pheochromocytoma stimulate adrenoceptor activation, including hypertension [7]. The most significant imaging feature of adrenal ganglioneuroma is an attenuation value of less than 40 HU on CT. However, significant enhancement on CT is frequently seen in adrenal pheochromocytoma [7]. As 131I-MIBG scintigraphy is highly sensitive and specific for detecting pheochromocytoma, but not for ganglioneuroma [7], patients with Pheo-GN are usually diagnosed with pheochromocytoma through signs and symptoms, specific hormone tests such as catecholamines and/or images from CT and 131I-MIBG scintigraphy. Preoperative diagnosis of composite Pheo-GN is extremely difficult.
Once pheochromocytoma has been diagnosed, the primary treatment is surgical resection after appropriate medical hormonal blockade. Laparoscopic adrenalectomy may be a better substitute due to the minimally invasive nature of the procedure. Moreover, this technique causes less fluctuations in blood pressure when pressure is exerted on the neoplasm during surgical manipulations and procedures. Via a transperitoneal approach, a clear anatomical landmark is visible and sufficient intraabdominal space for procedures is readily obtained [7]. Moreover, laparoscopic nephroureterectomy has been increasingly utilized as a surgical management strategy for upper urothelial carcinoma. The intermediate-term oncological outcomes are similar between laparoscopic and open approaches [8]. Roscigno et al. analyzed the effects of lymph node dissection on 1130 patients with stage T1-4 disease. Both cancer-specific survival and disease-free survival rates are improved in patients with Stage T2 or greater disease who undergo lymph node dissection [9]. In contrast, Kondo et al. showed that the number of lymph nodes removed was not necessarily associated with improved survival, but performing a complete template dissection imparted a survival advantage [10]. The role of routine lymphadenectomy at nephroureterectomy for upper tract urothelial carcinoma remains uncertain and controversial. The present patient underwent transperitoneal laparoscopic adrenalectomy, radical nephroureterectomy and para-aortic lymphadenectomy. Laparoscopic surgery is associated with lower morbidity rates, less postoperative pain, better esthetic results, and a briefer recovery period.
4. CONCLUSIONS
Preoperative diagnosis of composite Pheo-GN is extremely difficult. But surgical resection is the first choice of treatment for adrenal pheochromocytoma and adrenal ganglioneuroma as same as renal pelvic cancer without metastasis.
We have provided the case report of simultaneous surgical procedures for composite adrenal Phe-GN and renal pelvic cancer.
REFERENCES
- Lisewski, D., Ryan, S., Lim, E.M., Frost, F. and Nguyen, H. (2006) Concomitant compostite adrenal phoechromocytoma, multipte gastric stromal tumours and pseudohermaphrodism in a patient with von Recklinghausen’s disease. International Seminars in Surgical Oncology, 3, 11. doi:10.1186/1477-7800-3-11
- Khan, A.N., Solomon, S.S. and Childress, R.D. (2010) Composite pheochromocytoma-ganglioneuroma: A rare experiment of nature. Endocrine Practice, 16, 291-299.
- Lau, S.K., Chu, P.G. and Weiss, L.M. (2011) Mixed cortical adenoma and composite pheochromocytomaganglioneuroma: An unusual corticomedullary tumor of the adrenal gland. Annals Diagnostic Pathology, 15, 185-189. doi:10.1016/j.anndiagpath.2010.02.005
- Tischler, A.S. (2000) Divergent differentiation in neuroendocrine tumors of the adrenal gland. Seminars in Diagnostic Pathology, 17, 120-126.
- Juarez, D., Brown, R.W., Ostrowski, M., Reardon, M.J., Lechago, J. and Truong, L.D. (1999) Pheochromocytoma associated with neuroendocrine carcinoma. A new type of composite pheochromocytoma. Archives of Pathology & Laboratory Medicine, 123, 1274-1279.
- Mezitis, S.G., Geller, M., Bocchieri, E., Del Pizzo, J. and Merlin, S. (2007) Association of pheochromocytoma and ganglioneuroma: Unusual finding in neurofibromatosis type 1. Endocrine Practice, 13, 647-651.
- Shi, B.B., Li, H.Z., Chen, C., Rong, S., Fan, H., Wen, J. and Li, H.J. (2009) Differential diagnosis and laparoscopic treatment of adrenal pheochromocytoma and ganglioneuroma. Chinese Medical Journal, 122, 1790-1793.
- Ristau, B.T., Tomaszewski, J.J. and Ost, M.C. (2012) Upper tract urothelial carcinoma: Current treatment and outcomes. Urology, 79, 749-756. doi:10.1016/j.urology.2011.12.024
- Roscigno, M., Shariat, S.F., Margulis, V., et al. (2009) Impact of lymph node dissection on cancer specific survival in patients with upper tract urothelial carcinoma treated with radical nephroureterectomy. The Journal of Urology, 181, 2482-2489. doi:10.1016/j.juro.2009.02.021
- Kondo, T., Hashimoto, Y., Kobayashi, H., et al. (2010) Template-based lymphadenectomy in urothelial carcinoma of the upper urinary tract: Impact on patient survival. International Journal of Urology, 17, 848-854. doi:10.1111/j.1442-2042.2010.02610.x