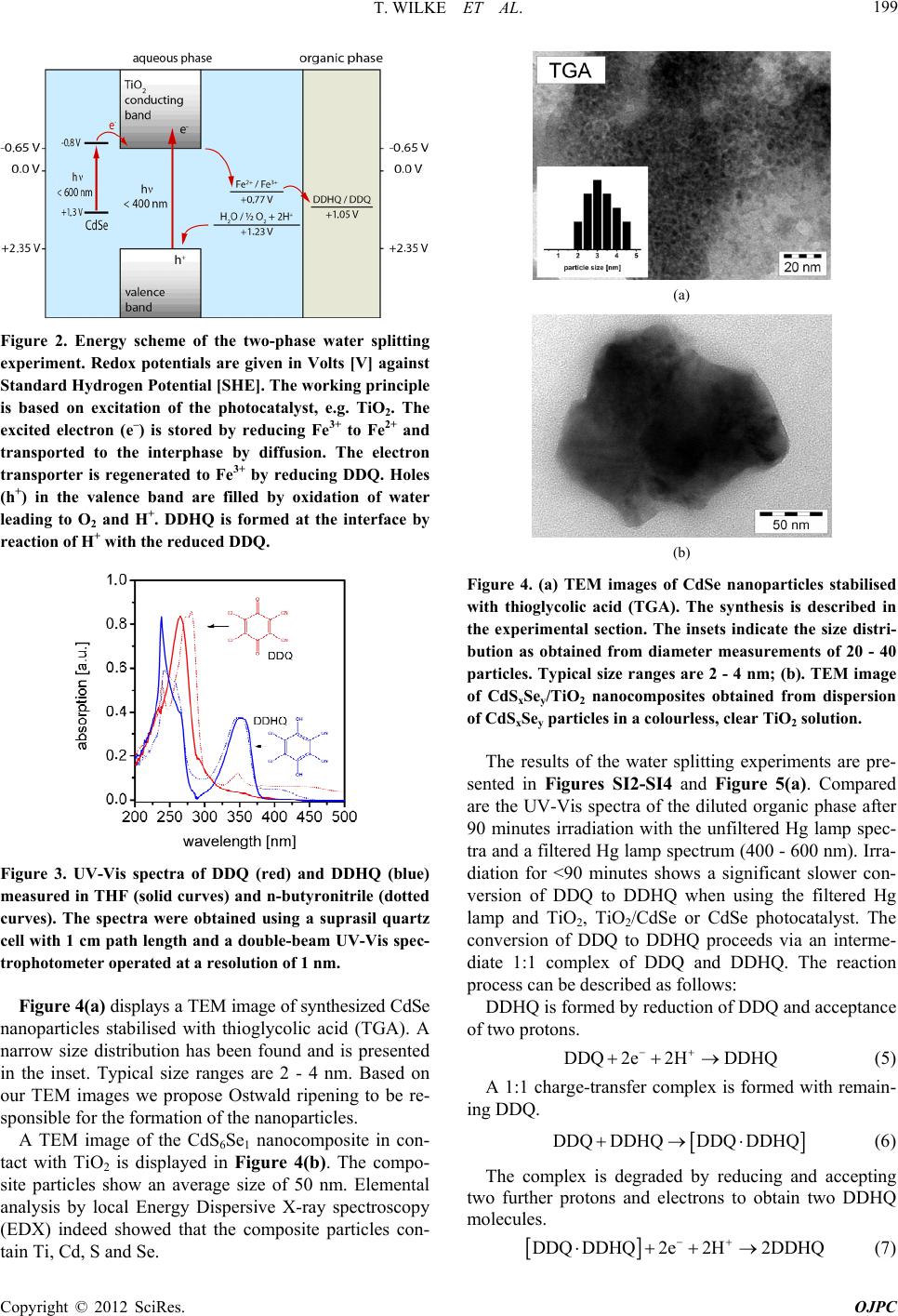
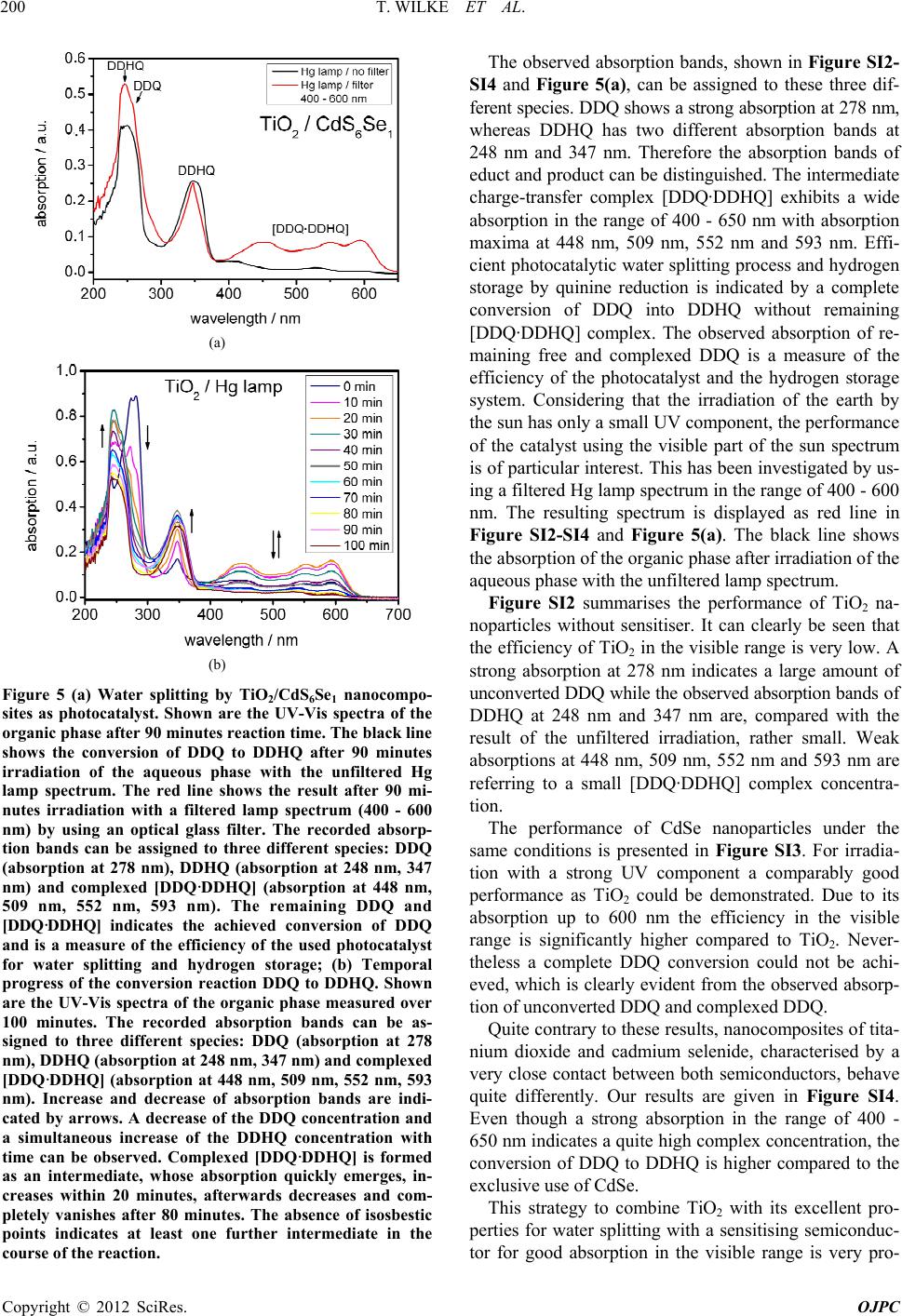
T. WILKE ET AL.
Copyright © 2012 SciRes. OJPC
202
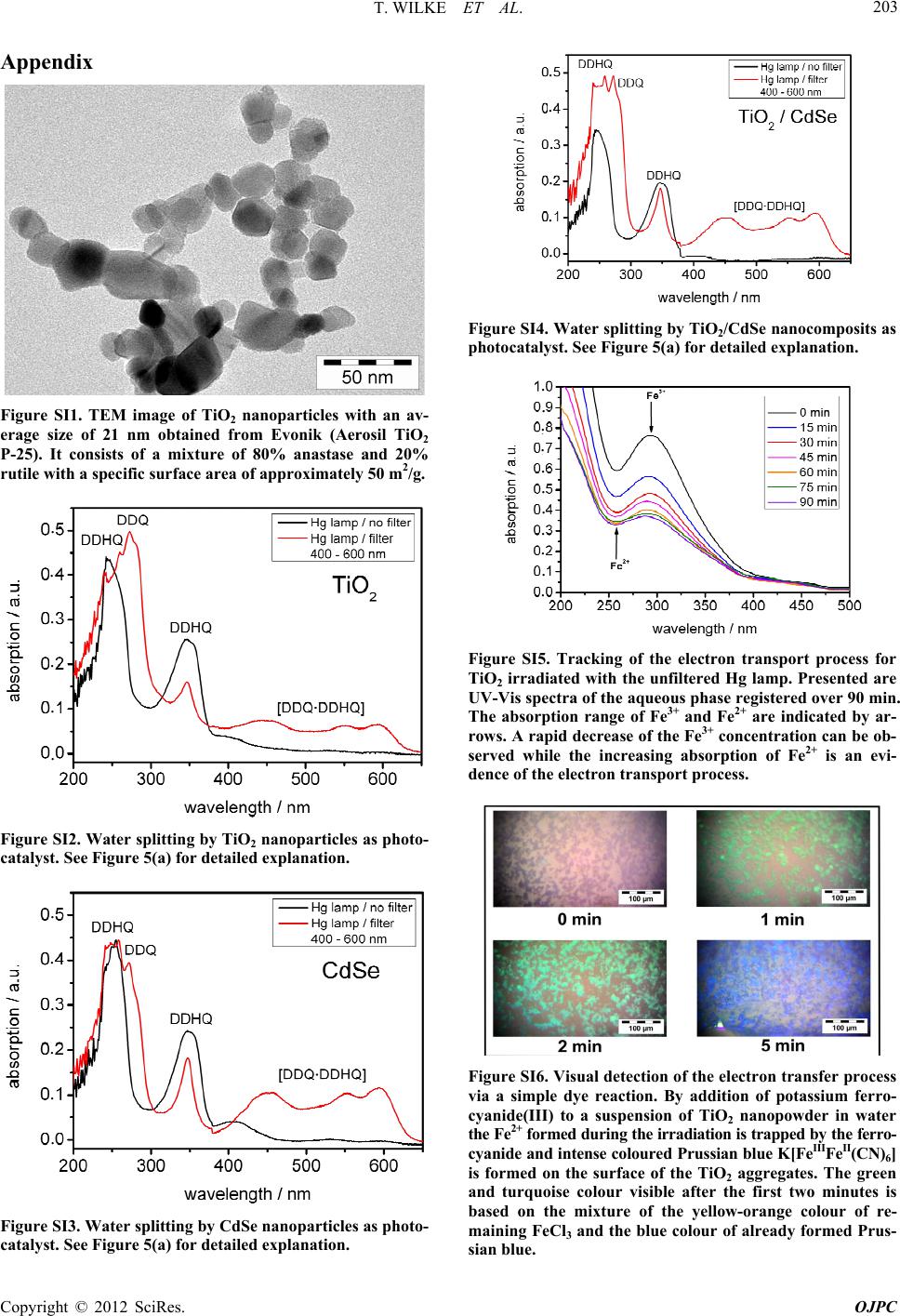
colour of remaining FeCl3 and the blue colour of already
formed Prussian blue. After 5 minutes of irradiation the
surface of the TiO2 aggregates appears intensive blue
coloured, see Supplementary Information.
It can be assumed that the surface of the TiO2 aggre-
gates is rough with different kind of edges. An enlarge-
ment of the light optical micrograph, which is presented
in Figure 7, indicates high active reaction areas, which
are visible as bright spots on the surface of the TiO2 ag-
gregates, mostly near edges. Edges therefore appear to be
the areas of higher photoreduction activity.
4. Conclusion
We presented an experimental setup to investigate the
performance of nanocrystalline semiconductors for photo-
catalytic water splitting. Chemical storage of the gener-
ated hydrogen by quinoid systems seems to be a good
alternative to evolution of gaseous H2 as it is easy to
handle and can be use as recyclable fuel for fuel cells, as
we show in a further publication [18].
5. Acknowledgements
The authors gratefully acknowledge Dr. Christian W.
Lehmann and his group (Max-Planck Institut für Koh-
lenforschung, Mülheim/Ruhr) for the implementation of
the TEM measurements and Dr. Daniel Ogermann (In-
stitute of Physical Chemistry, Heinrich-Heine-University,
Düsseldorf) for his assistance in preparation of the pho-
tocatalyst samples.
REFERENCES
[1] R. Pike and P. Earis, “Powering the World with Sun-
light,” Energy & Enviromental Sc ience, Vol. 3, No. 2, 2010,
p. 173. doi:10.1039/b924940k
[2] N. Kelly, T. Gibson and D. Ouwerkerk, “Generation of
High-Pressure Hydrogen for Fuel Cell Electric Vehicles
Using Photovoltaic-Powered Water Electrolysis,” Inter-
national Journal of Hydrogen Energy, Vol. 36, No. 24,
2011, pp. 15803-15825.
doi:10.1016/j.ijhydene.2011.08.058
[3] E. Durgun, S. Ciraci, W. Zhou and T. Yildirim, “Transi-
tion-Metal-Ethylene Complexes as High-Capacity Hydro-
gen-Storage Media,” Physical Review Letters, Vol. 97,
No. 22, 2006, pp. 1-4.
doi:10.1103/PhysRevLett.97.226102
[4] M. Grätzel, “Photoelectrochemical Cells,” Nature, Vol. 414,
No. 6861, 2001, pp. 338-344. doi:10.1038/35104607
[5] A. Hagfeldt and M. Grätzel, “Light-Induced Redox Reac-
tions in Nanocrystalline Systems,” Chemical Reviews,
Vol. 95, No. 1, 1995, pp. 49-68.
doi:10.1021/cr00033a003
[6] M. Kaneko and I. Okura, “Photocatalysis—Science and
Technology,” Springer, Heidelberg, 2002.
[7] D. Ogermann, T. Wilke and K. Kleinermanns, “CdSxSey/
TiO2 Solar Cell Prepared with Sintered Mixture Deposi-
tion,” Open Journal of Physical Chemistry, Vol. 2, No. 1,
2012, pp. 47-57. doi:10.4236/ojpc.2012.21007
[8] D. R. Cooper and N. M. Dimitrijevic, “Photosensitization
of CdSe/ZnS QDs and Reliability of Assays for Reactive
Oxygen Species Production,” Nanoscale, Vol. 2, No. 1,
2010, pp. 114-121. doi:10.1039/b9nr00130a
[9] I. Robel, M. Kuno and P. V. Kamat, “Size-Dependent
Electron Injection from Excited CdSe Quantum Dots into
TiO2 Nanoparticles,” Journal of the American Chemical
Society, Vol. 129, No. 14, 2007, pp. 4136-4137.
doi:10.1021/ja070099a
[10] M. Grätzel, “Dye-Sensitized Solar Cells,” Journal of Pho-
tochemistry and Photobiology C, Vol. 4, No. 2, 2003, pp.
145-153. doi:10.1016/S1389-5567(03)00026-1
[11] T. Meyer, D. Ogermann, A. Pankrath, K. Kleinermanns
and T. J. J. Müller, “Phenothiazinyl Rhodanylidene Mero-
cyanines for Dye-Sensitized Solar Cells,” The Journal of
Organic Chemistry, Vol. 77, No. 8, 2012, pp. 3704-3715.
doi:10.1016/10.1021/jo202608w
[12] R. Menzel, D. Ogermann, S. Kupfer, D. Weib, H. Görls,
K. Kleinermanns, L. González and R. Beckert, “4-Meth-
oxy-1,3-thiazole Based Donor-Acceptor Dyes: Charac-
terization, X-Ray Structure, DFT Calculations and Test as
Sensitizers for DSSC,” Dyes and Pigments, Vol. 94, No.
3, 2012, pp. 512-524. doi:10.1016/j.dyepig.2012.02.014
[13] T. Wilke and K. Kleinermanns, to be published, 2012.
[14] T. Ohno, K. Fujihara, K. Sarukawa, F. Tanigawa and M.
Matsumura, “Splitting of Water by Combining Two Photo-
catalytic Reactions through a Quinone Compound Dis-
solved in an Oil Phase,” Zeitschrift für Physikalische
Chemie, Vol. 213, No. 2, 1999, pp. 165-174.
doi:10.1524/zpch.1999.213.Part_2.165
[15] A. L. Rogach, A. Kornowski, M. Gao, A. Eychmüller and
H. Weller, “Synthesis and Characterization of a Size Se-
ries of Extremely Small Thiol-Stabilized CdSe Nano-
crystals,” The Journal Of Physical Chemistry B, Vol. 103,
No. 16, 1999, pp. 3065-3069. doi:10.1021/jp984833b
[16] G. A. Martínez-Castañón, M. G. Sánchez-Loredo, J. R. Mar-
tínez-Mendoza and F. Ruiz, “Synthesis of CdS Nanopar-
ticles: A Simple Method in Aqueous Media,” Azojomo,
Vol. 1, No. 1, 2005, pp. 1-2. doi:10.2240/azojomo0170
[17] J. Yu, X. Zhao, J. Du and W. Chen, “Preparation, Micro-
structure and Photocatalytic Activity of the Porous TiO2
Anatase Coating by Sol-Gel Processing”, Journal of Sol-
Gel Science and Technology, Vol. 17, No. 2, 2000, pp.
163-171. doi:10.1023/A:1008703719929
[18] T. Wilke, M. Schneider and K. Kleinermanns, “1,4-Hy-
droquinone is a Hydrogen Reservoir for Fuel Cells and
Recyclable via Water Splitting,” 2012.