Journal of Cancer Therapy
Vol.4 No.1(2013), Article ID:28177,8 pages DOI:10.4236/jct.2013.41039
Long-Term Results of Ewing’s Sarcoma—In a Single Institution
![]()
Department of Orthopedic Surgery, Kosin University Gospel Hospital, Busan, Korea.
Email: *shchung@kosin.ac.kr
Received December 18th, 2012; revised January 20th, 2013; accepted January 28th, 2013
Keywords: Ewing’s Sarcoma; Treatment Result; Prognostic Factors
ABSTRACT
Purpose: This study analyzed oncological and functional outcomes of treatment for Ewing’s sarcoma, as well as its significant risk factors through long-term follow up. Objective and Method: Between September 1990 and April 2009, 20 cases that were diagnosed and treated as Ewing’s sarcoma in Kosin University Gospel Hospital were entered onto the study. Mean follow-up period was 45.4 (12 - 108) months. There were 7 cases of male and 13 cases of female, and mean age was 19.9 (5 - 48) years old. Retrospective review was done about treatment outcomes, complications, and significant risk factors. Results: In terms of oncologic results, there were 9 cases of CDF (continuous disease free), 1 case of NED (no evidence of disease), 4 cases of AWD (alive with disease), 5 cases of DOD (dead of disease), and 1 case of DWOD (dead with other disease). Five-year overall survival rate of all the patients was 70.0% and event-free survival rate was 50.0%. The mean MSTS (Musculoskeletal Tumor Society) score was 15.9 (53%) points at last follow-up. Among prognostic factors of age at diagnosis, Enneking stage, size of tumor, site of primary lesion, and distant metastasis, 5-year survival rate of groups without metastasis were 90.9%, nevertheless 44.4% in other group with the metastasis showing statistical significance (p = 0.020). Postoperative complications were 3 cases of infection, each 2 cases of ankylosis and metal failure, and each 1 case of leg length discrepancy, periprosthetic fracture, and local recurrence. Conclusion: Five-year survival rate of this study was similar to that of multicenter studies in America and Europe. Among the prognostic factors, distant metastasis was proven to be most significant. Enneking stage, size of tumor and site of primary lesion are also important and could be statistically significant if with more cases.
1. Introduction
Ewing’s sarcoma, which was first described by James Ewing in 1921, is a rare bone tumor that accounts for less than 10% of whole primary malignant bone tumor. However, this tumor is known to be second most common malignant bone tumor in children and adolescents, next to osteosarcoma. This tumor is shown to be one of the poorest prognostic ones, and its frequency in East Asia is rare [1].
In this report, we retrospectively studied 20 patients who were diagnosed as Ewing’s sarcoma and treated at our institution in last 20 years, analyzed and report their oncologic and functional results, complications, and prognostic factors.
2. Objective and Methods
Between September 1990 and April 2009, twenty patients who were diagnosed as Ewing’s sarcoma and treated at Kosin University Gospel Hospital, Busan, Republic of Korea, and possible to be followed up for at least 12 months, were elected for this study. Mean follow-up period was 45.4 (12 - 108) months (Table 1). Among the patients, surgery, radiation therapy, and chemotherapy were all performed in 8 patients, surgery and chemotherapy in 9 patients, radiation therapy and chemotherapy in 2 patients, and only chemotherapy in 1 patient.
For staging, simpla X-ray, chest CT (computed tomography), bone scan, CT or MRI (magnetic resonance imaging) of lesion site, PET-CT (Positron emission tomography-computed tomography) were performed with hematologic tests which were needed for neoadjuvant chemotherapy.
Staging was determined by determined Enneking classification, functional evaluation by MSTS (Musculoskeletal Tumor Society) score, survival rate by KaplanMeier method, and comparison of survival rates among different groups by Log-rank method [2,3].
3. Characteristics of the Patients
Among the whole 20 patients, there were 7 men and 13
Table 1. Demographic characteristics of 20 patients with Ewing’s sarcoma.
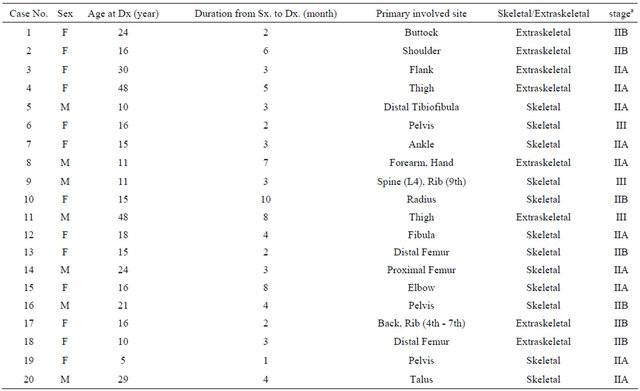
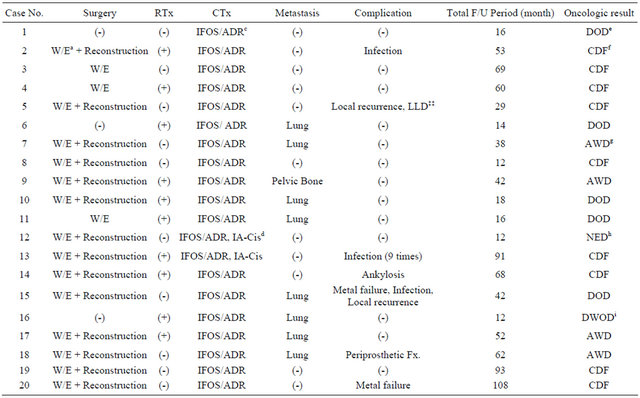
aW/E: Wide excision, bStage: Enneking’s staging system of malignant bone tumors, cIFOS/ADR: Ifosfamide(HOLOXAN®) 14 g/m2/Doxorubicin (A.D. MYCIN®) 90 mg/m2, dIA-Cis: Intraarterial Cisplatin(UNISTIN®) 120 mg/m2, eDOD: Dead of disease, fCDF: continuous disease free, gAWD: Alive with disease, hNED: No evidence of disease, iDWOD: Dead with other diasese, ‡‡LLD: Limb length discrepancy.
women. Mean age at diagnosis was 19.9 (5 - 48) years old, mostly in adolescents and young adults. Mean period from onset of the symptoms until time at diagnosis was 4.2 (1 - 10) months. For primary lesion sites, there were 5 cases in thigh, 4 in pelvis, 3 in lower leg, each 2 cases in spine and forearm, and each 1 case in shoulder, foot, elbow, and flank. 12 cases were osseous-type, and the other 8 cases were extraosseous. Enneking stage at the time of the diagnosis was IIA in 10 cases, IIB in 7 cases, and III in 3 cases.
4. Surgery
Surgical treatment was performed in 17 of 20 patients. Among them, 14 patients underwent wide excision following limb salvage surgery together and only wide excision was performed in 3 patients. For the method of reconstruction in limb salvage surgery, there were 8 cases of autologous bone grafts, 4 cases of tumor prosthesis insertions, and 2 cases of allogenous bone grafts. Surgery was not performed in 3 patients because they had lung metastasis before the surgery so radiation therapy and chemotherapy was firstly performed, but expired due to general condition aggravation.
5. Radiation Therapy
Radiation therapy was performed in 10 patients at primary lesion site before the surgery. There were 4 cases at thigh, each 2 cases at pelvis and spine, and each 1 case at shoulder and forearm. Mean radiation dosage was 45.1 (15 - 60) Gy. In 2 cases, lung metastasis was found and aggravated during neoadjuvant chemotherapy and radiation therapy, and finally expired.
6. Chemotherapy
Neoadjuvant chemotherapy was performed in all 20 cases, but 17 of them completed whole adjuvant chemotherapy. As combination regimen, IFOS/ADR (Ifosfamide (HOLOXAN®) 14 g/m2, Doxorubicin (A.D.MYCIN®) 90 mg/m2) was used in all the patients, 2 cycles preoperatively and another 2 cycles postoperatively. And in 2 of them, preoperative intraarterial cisplatin (UNISTIN®) 120 mg/m2 was additionally used, in order to diminish volume of the tumor for surgical resection. In principle, 2 cycles of neoadjuvant chemotherapy following surgery following another 2 cycles of adjuvant chemotherapy was supposed to be applied, however, in case of showing poor response or stage III at diagnosis, up to 12 cycles were elongated.
7. Results
7.1. Oncologic Results
Oncologically, there were 9 cases of CDF (continuous disease free), 1 case of NED (no evidence of disease), 4 cases of AWD (alive with disease), 5 cases of DOD (dead of disease), and 1 case of DWOD (dead with other disease). By Kalplan-Meier method, 5-year overall survival rate (OS) of whole 20 cases was 70.0%, and eventfree survival rate (EFS) was 50.0%.
Based on the age at the time of the diagnosis, 12 cases were less than 18 years old, 6 were 18 - 30 years old, and 2 cases were more than 31 years old. 5-year survival rates of each group were 75.0%, 66.7%, 50.0%, showing slight decrement as the patients got older, but it was not statistically significant (p = 0.258).
Based on Enneking state at the time of the diagnosis, there were 10 cases of stage IIA, 7 cases of IIB, and 3 cases of III. Each 5-year survival rates were 90.0%, 57.1%, 33.3%, showing decreased survival rate with advanced state, but no statistical significance (p = 0.122).
Based upon size (maximal diameter) of the tumor, 7 cases were less than 8 cm and 13 cases were more than 8 cm. Each 5-year survival rates were 85.7% and 61.5%, but it was also not statistically significant (p = 0.089).
Analyzed by division into extremities and trunks for primary lesion site, there were 13 cases at extremities and 7 cases at trunks. Each 5-year survival rates were 76.9% and 57.1%, showing lower survival rate when occurred at the trunk, but it also didn’t show statistical significance (p = 0.085).
Distant metastasis occurred in 9 cases; 8 cases at lung and 1 case at pelvic bone. Mean period from the date of the diagnosis until the metastasis found was 12.6 months. There were metastasis already existed when diagnosed in 3 cases, and metastasis newly occurred during preoperative treatment in 6 cases. For treatment of the metastatic lesions, among 3 cases of stage III which already had distant metastasis when diagnosed, 2 patients were performed combination treatment of surgery, radiation therapy, and chemotherapy and the other 1 patient were performed radiation therapy and chemotherapy. Among 6 cases who were found distant metastasis during neoadjuvant chemotherapy, 4 patients were performed elongated chemotherapy up to 12 cycles, 1 patient got lobectomy for lung lesion because of good general condition, and the other 1 patient got only conservative treatment due to poor general condition but ultimately expired. 5-year survival rates were 90.0% without distant metastasis and 44.4% with distant metastasis, and it showed statistical significance (p = 0.016) (Figure 1).
7.2. Functional Results
Mean MSTS score of 14 cases who were survived until final follow-up was 19.3 (14 - 25) points (64.3 (46.7 - 83.3)%). Devided by tumor lesion sites, mean 20.0 points (66.7%) when occurred at pelvis, 20.7 points (69.0%)
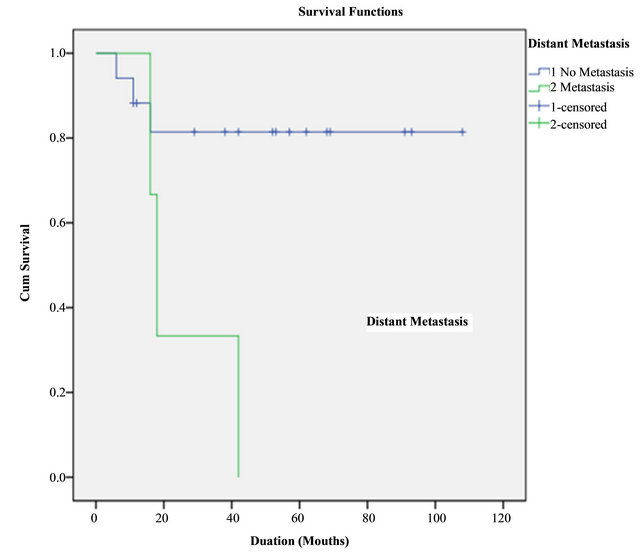
Figure 1. Survival rate according to distant metastasis, (1: no metastasis, 2: metastasis).
at other trunks except pelvis, 22.5 points (75.0%) at upper extremities, and 17.9 points (59.7%) at lower extremities, which appears that the patients with lesion at lower extremities showed relatively lower satisfaction.
8. Complications
Postoperative complications occurred at 6 cases; 3 cases of infection, each 2 cases of ankylosis and metal failure, and each 1 case of limb length discrepancy, periprosthetic fracture, and local recurrence. One patient supervened limb length discrepancy and local recurrence, and another one supervened infection, ankylosis and metal failure. Local recurrence occurred at ankle in 1 case, and it took 13 months from the date of surgery until recurrence found. In this case, wide excision for recurred lesion following additional chemotherapy was performed.
9. Discussion
Classic treatment modality for primary lesion of Ewing’s sarcoma had been local therapy, such as radiation therapy or surgery alone. This method was somewhat effective for local sites, but its prognosis was very poor; most patients expired due to distant metastasis. However, with development of surgical technique and discovery of risks for complications after radiation therapy, dependence on radiation therapy was decreased [4,5]. When radiation therapy is needed, dosage at least 40 Gy is known to be able to improve the result of local treatment, especially more effective in the tumor at axial skeletal such as skull base or sacrum [6]. In this study, mean radiation dose of 45.1 Gy was applied at 10 cases. 5 cases were performed at axial skeleton such as pelvis and spine, and the other 5 cases at proximal thigh and shoulder. When the mass was difficult to be surgically resected because of their huge size at pelvis, or to prevent postoperatively microscopic metastasis, radiation therapy was performed with chemotherapy. The 5-year survival rate of this group was 60.0%, lower than that of whole group’s 70.0%, which is thought to be due to their primary lesion sites’ at trunk or extremities near trunk, not because of low effectiveness of radiation therapy itself.
General purpose of surgery for Ewing’s sarcoma is two; treatment of the tumor and preservation of local site’s function as much as possible. In early phase, indications for surgical treatment were limited; for tumors of extremities which were too large for radiation therapy or pathologic fractures, and most of them were amputations [7]. Until 1960’s, main treatment modalities were radiation therapy or surgical resection alone, and 5-year survival rate at that time was only around 10% and most of the patients expired within 2 years from the time of the diagnosis, usually due to distant metastasis [8]. However, since application of limb salvage surgery in 1970’s, 90% - 95% of the patients nowadays are performed limb salvage surgery, and treatment results were much more improved with adaptation of chemotherapy. Current treatment policy is firstly induction of remission of the tumor by combination chemotherapy and radiation therapy, and resection of primary lesion by surgery, then maintaining consistent chemotherapy to prevent metastasis or recurrence [9,10].
The effect of surgical treatment on the survival rate was not confirmed yet, but was proven to be significantly effective for lowering rates of local recurrence [7]. And with intensive chemotherapy and radiation therapy together, better survival rates would be expected. In our result, 5-year survival rate of 17 cases who were performed chemotherapy, radiation therapy, and surgery together was 82.4%, showing better prognosis than whole 20 patients’ survival rate of 70.0% (Figure 2). Therefore, if possible, aggressive surgical treatment would be desirable.
Pelvis is the most common sites for Ewing’s sarcoma, but the prognosis is much poorer than those at other sites, because of lack of main anatomical barrier against dissemination of the tumor, difficult for local treatment due to many surrounding organs and neurovascular bundles, and more common recurrence than other sites [11]. However, Puri et al. recommended aggressive surgical treatment for nonmetastatic Ewing’s sarcoma at pelvic bone, because surgical resection as much as possible can lower local recurrence rates and provides excellent functional results, only if tumor resection margin is thought to be able to get obtained through preoperative imaging studies [12]. In this study, however, surgery was performed in only 1 of 4 patients at pelvis, because 2 of 3 patients expired before the surgery due to discovery and aggravation of lung metastasis during the neoadjuvant chemotherapy, and the other 1 patient refused the surgery.
Chemotherapy was milestone for the treatment of Ewing’s sarcoma. After introduction of the chemotherapy, rates of metastasis were decreased and if showing response to chemotherapy, the size of primary lesion was decreased enough for performing limb salvage surgery. Thus this provided an important opportunity for improving the tumors’ survival rates, and nowadays, the chemotherapy became a mainstream for treatment methods of Ewing’s sarcoma. After Rosen et al. had performed combination chemotherapy of VacCD (vincristine, actinomycin-D, Cyclophosphamide, and doxorubicin) in 1974 [13], many institutions demonstrated clinical efficacy of VAcCD therapy. In early and middle 2000s, Grier et al. and Ferrari et al. proved that better prognosis could be obtained by adding ifosfamide and etoposide to VAcCD therapy, by reporting 5-year survival rates of 72% and 70% each [14,15]. And this regimen became current standard chemotherapy protocol for Ewing’ sarcoma [16]. In addition, Womer et al. demonstrated that treatment result was better when the chemotherapy cycle had been performed with 2-week interval, rather than previous 3-week interval [17].
In our institution, ifosfamide(14 g/m2) and doxorubicin (90 mg/m2) were alternatively administered each 2 cycles preoperatively and postoperatively for a total of 4 cycles with 2-week intervals. In case of showing poor response or presenting stage III, the regimen was elongated up to 12 cycles. The 5-year over survival rate (OS) was 70.0% and event-free survival rate (EFS) was 50.0%, which is similar to those of massive retrospective studies in USA or Europe’s. Rodriguez et al. reported 5-year overall survival rate of 63.5% and event-free survival rate of
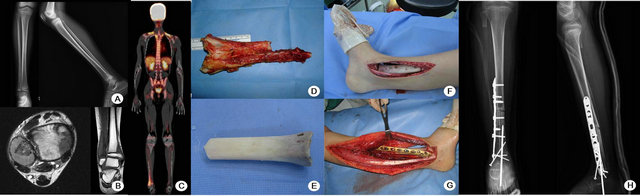
Figure 2. A 10 year-old male patient. (A - C) Preoperative image studies show suspicious malignant bone tumor at right distal tibiofibula. He was diagnosed as Ewing’s sarcoma after open bone biopsy. (D - G) After wide excision, the involved bone was wide excised and then, pasteurizated and autografted. (H) Post-operative radiographic results were good in this patient.
55.1% in 220 patients [18]. And Bacci et al. reported 5-year event-free survival rate of 56.9% in 579 patients [19]. The other result in Republic of Korea was also similar; Lee et al. reported 5-year overall survival rate of 58.9% and event-free survival rate of 52.6% in 76 patients [20].
As prognostic factors of Ewing’s sarcoma, age at diagnosis, Enneking’s stage, size of the tumor, location of primary lesion, distant metastasis et al. are suggested. Among them, distant metastasis is the most important single prognostic factor, especially poorest prognosis if distant metastasis was already existed when diagnosed [21,22].
For the age at diagnosis, Lee et al. reported that younger patients, especially younger than 18 years old showed better prognosis [23]. However, Bacci et al. claimed that that was not a significant prognostic factor [24]. In our study, also, older patients showed relatively poorer survival rates, but it was not statistically significant (p = 0.258).
For the stage, Simpson et al. reported that higher Enneking stage at the diagnosis resulted higher expire rate, thus this would be a significant prognostic factor [25]. In our study, increased Enneking stage at the diagnosis of IIA, IIB, and III showed proportionally decreased 5-year survival rate of 90.0%, 66.7%, and 50.0%, but this was not statistically significant (p = 0.122).
About the size of the tumor, Cotterill et al. based on volume of 100 ml [26], and Lee et al. based on maximal diameter of 8 cm [23], and they reported that larger tumors than these figures showed poorer prognosis than smaller ones’. In this study, based on maximal diameter, 5-year survival rate of 13 cases of more than 8 cm was 61.5%, which was poorer than with the other 7 cases’ 85.7% of less than 8 cm, but it didn’t show statistical significance (p = 0.089).
For the location of the primary lesion, randomized study by IESS-1 demonstrated that it is a statistically significant prognostic factor, and many other studies commonly reported that 5-year survival rates of tumors at pelvic bone were 15% - 35%, which is significantly poorer than those of tumors except at pelvic bone were 30% - 77% [27]. Bacci et al. also demonstrated that tumors at pelvic bone showed poorest prognosis [28]. Our study also reports that 5-year survival rate of 7 cases at trunk was 57.1%, which was much lower than 76.9% of the other 13 cases’ at extremities, but this also wasn’t statistically significant (p = 0.085). However, when focused on 4 cases at the pelvis, 5-year survival rate was 25.0%, which was the poorest and consistent with the findings of the other studies.
In our study, 5-year survival rate of 4 cases at upper extremities were 50%, which were relatively poorer than other sites’ survival rates. The reason for this result is thought to be due to relatively longer period from the onset of the symptoms until the time of the diagnosis; the whole patients’ took 4.2 months but the group of upper extremities took 7.8 months to be diagnosed as Ewing’s sarcoma. This phenomenon could be due to relatively later onset of the symptoms, causing pulmonary metastasis already when diagnosed as the tumor. Simpson et al. reported that Ewing’s sarcoma at upper extremities longer durations of the symptoms are related to lower survival rates, because of advanced Enneking stage at the diagnosis [25]. Therefore, when meeting patients who complains of pain at upper extremities, more frequent visit to clinic should be recommended, and if malignancy is suspected, immediate studies should be performed to diagnose as soon as possibleFor distant metastasis, Lee et al. reported that the metastasis is one of the most important prognostic factors [23]. In our study, it was confirmed that 5-year survival rate of 9 cases with distant metastasis was 44.4%, which was significantly poorer than 5-year survival rate of the other 11 cases’ 90.9% without distant metastasis (p = 0.016). In our institution’s treatment experience, pulmonary metastasis would have been disappeared when they responded well to neoadjuvant chemotherapy, therefore it is expected that aggressive treatment could improve the prognosis of this tumor.
Among these prognostic factors, Enneking’s stage at diagnosis, size of the tumor, location of primary lesion were not statistically significant because of not enough numbers of the patients. However, with more cases, these factors could also be significant enough for meaningful prognostic factors.
For recent decades, with successful results of multimodal therapies, long-term survival rate of Ewing’s sarcoma has been improved. Recent 5-year survival rates of local Ewing’s sarcoma reached approximately at 70%, and 10-year survival rates reached approximately at 50% [24]. However, 5-year survival rates of metastatic or recurrent Ewing’s sarcoma still remain around 25% - 29% [29]. In this study, 9 cases got distant metastasis and their 5-year survival rate was 44.4%, therefore we think that researches to improve its prognosis should be progressed more.
Also, Ewing’s sarcoma shows high recurrence rate of 30% - 40%, even after combination treatment of surgery, radiation therapy, and chemotherapy [30]. Therefore longer term follow up must be done to diagnose and treat this tragedy.
10. Conclusions
As a result of analyzing 20 patients who were diagnosed and treated as Ewing’s Sarcoma in Kosin University Gospel Hospital for last 20 years, 5-year OS was 70.0% and EFS was 50.0%, which shows similar results with mass studies in USA and Europe. Confirmed statistically significant prognostic factor was distant metastasis (p = 0.016). Enneking stage at the diagnosis, size of the tumor, and location of the tumor didn’t show statitstical significance due to lack of the cases, but these also can be meaningful if with larger amount of the cases in the future.
In order to improve the prognosis of Ewing’s sarcoma, prompt diagnosis with intensive, consistent chemotherapy, radiation therapy, and surgical treatment should be combined. And thorough and long-term follow-up must be done, even after termination of treatment, because of showing higher recurrence rate than other malignancies. Also, we expect improved treatment results of Ewing’s Sarcoma, if more prospective multicenter research in Korea would be processed.
REFERENCES
- Y. Iwamoto, “Diagnosis and Treatment of Ewing’s Sarcoma,” Japanese Journal of Clinical Oncology, Vol. 37, No. 2, 2007, pp. 79-89. doi:10.1093/jjco/hyl142
- W. F. Ennecking, W. K. Dunham, M. C. Gebhardt, M. Malawer and D. J. Prichard, “A System for the Evaluation of Reconstructive Procedures after Surgical Treatment of Tumors of the Musculoskeletal System,” Clinical Orthopaedics and Related Research, Vol. 286, 1993, pp. 241- 246.
- E. L. Kaplan and R. Meier, “Nonparametric Estimation from Incomplete Observation,” Journal of the American Statistical Association, Vol. 53, No. 282, 1958, pp. 457- 481. doi:10.1080/01621459.1958.10501452
- S. Laskar, I. Mallick, T. Gupta and M. A. Muckaden, “Post-Operative Radiotherapy for Ewing Sarcoma: When, How and How Much?” Pediatric Blood & Cancer, Vol. 51, No. 5, 2008, pp. 575-580. doi:10.1002/pbc.21657
- D. J. Indelicato, S. R. Keole, A. H. Shahlaee, W. Shi, C. G. Morris and R. B. Marcus Jr., “Definitive Radiotherapy for Ewing Tumors of Extremities and Pelvis: Long-Term Disease Control, Limb Function, and Treatment Toxicity,” International Journal of Radiation Oncology, Biology and Physics, Vol. 72, No. 3, 2008, pp. 871-877. doi:10.1016/j.ijrobp.2008.02.023
- S. Donaldson, “Ewing Sarcoma: Radiation Dose and Target Volume,” Pediatric Blood & Cancer, Vol. 42, No. 5, 2004, pp. 471-476. doi:10.1002/pbc.10472
- M. Bernstein, H. Kovar, M. Paulussen, et al., “Ewing’s Sarcoma Family of Tumors: Current Management,” The Oncologist, Vol. 11, No. 5, 2006, pp. 503-519.
- B. Padhye and G. McCowage, “Chemotherapy Regimens in Newly Diagnosed and Recurrent Ewing Sarcoma in Children and Young Adults,” Cancer Forums, Vol. 34, No. 3, 2010, pp. 1-7.
- S. A. Khan and S. Rastogi, “Surgery in Ewing’s SarcomaMyth or Reality,” Indian Journal of Medical Pediatric Oncology, Vol. 25, No. 2, 2004, pp. 58-63. doi:10.1097/BLO.0b013e318059b8c9
- T. A. Damron, W. G. Ward and A. Stewart, “Osteosarcoma, Chondrosarcoma, and Ewing’s Sarcoma: National Cancer Data Base Report,” Clinical Orthopaedics and Related Research, Vol. 459, 2007, pp. 40-47.
- S. Bhagat, H. Sharma, D. S. Pillai and M. J. Jane, “Pelvic Ewing’s Sarcoma: A Review from Scottish Bone Tumour Registry,” The Journal of Orthopaedic Surgery, Vol. 16, No. 3, 2008, pp. 333-338.
- A. Puri, A. Gulia, N. A. Jambhekar and S. Laskar, “Results of Surgical Resection in Pelvic Ewing’s Sarcoma,” Journal of Surgical Oncology, Vol. 106, No. 4, 2012, pp. 417-422.
- G. Rosen, N. Wollner, C. Tan, et al., “Proceedings: Disease-Free Survival in Children with Ewing’s Sarcoma Treated with Radiation Therapy and Adjuvant Four-Drug Sequential Chemotherapy,” Cancer, Vol. 33, No. 2, 1974, pp. 384-393. doi:10.1002/1097-0142(197402)33:2<384::AID-CNCR2820330213>3.0.CO;2-T
- H. E. Grier, M. D. Krailo and N. J. Tarbell, “Addition of Ifosfamide and Etoposide to Standard Chemotherapy for Ewing’s Sarcoma and Primitive Neuroectodermal Tumor of Bone,” The New England Journal of Medicine, Vol. 348, 2003, pp. 694-701. doi:10.1056/NEJMoa020890
- S. Ferrari, E. Palmerini, M. Alberghini, et al., “Vincristine, Doxorubicin, Cyclophosfamide, Actinomycin D, Ifosfamide, and Etoposide in Adult and Pediatric Patients with Nonmetastatic Ewing Sarcoma. Final Results of a Monoinstitutional Study,” Tumori, Vol. 96, No. 2, 2010, pp. 213-218.
- M. Paulussen, S. Bielack, H. Jurgens and P. G. Casali, “Ewing’s Sarcoma of the Bone: ESMO Clinical Recommendations for Diagnosis, Treatment and Follow-Up,” Annals of Oncology, Vol. 20, No. 4, 2009, pp. 140-142. doi:10.1093/annonc/mdn103
- R. B. Womer, D. C. West, M. D. Krailo, P. S. Dickman and B. Pawel, “Randomized Comparison of Every-TwoWeek vs. Every-Three-Week Chemotherapy in Ewing Sarcoma Family Tumors (ESFT),” Journal of Clinical Oncology, Vol. 26, No. 20, 2008, pp. 504-512.
- C. Rodriguez-Galindo, T. Liu, M. J. Krasin, et al., “Analysis of Prognostic Factors in Ewing Sarcoma Family of Tumors: Review of St. Jude Children’s Research Hospital Studies,” Cancer, Vol. 110, No. 2, 2007, pp. 375-384. doi:10.1002/cncr.22821
- G. Bacci, A. Longhi, S. Ferrari, M. Mercuri, M. Versari and F. Bertoni, “Prognostic Factors in Non-Metastatic Ewing’s Sarcoma Tumor of Bone: An Analysis of 579 Patients Treated at a Single Institution with Adjuvant or Neoadjuvant Chemotherapy between 1972 and 1998,” Acta Oncologica, Vol. 45, No. 4, 2006, pp. 469-475. doi:10.1080/02841860500519760
- J. A. Lee, D. H. Kim, J. B. Cho, et al., “Treatment Outcome of Korean Patients with Localized Ewing Sarcoma Family of Tumors: A Single Institution Experience,” Japanese Journal of Clinical Oncology, Vol. 41, No. 6, 2011, pp. 776-782. doi:10.1093/jjco/hyr033
- G. C. Rodriguez, F. Navid, T. Liu, C. A. Billups, B. N. Rao and M. J. Krasin, “Prognostic Factors for Local and Distant Control in Ewing Sarcoma Family of Tumors,” Annals of Oncology, Vol. 19, No. 4, 2008, pp. 814-820. doi:10.1093/annonc/mdm521
- B. M. Seddon and J. S. Whelan, “Emerging Chemotherapeutic Strategies and the Role of Treatment Stratification in Ewing Sarcoma,” Pediatr Drugs, Vol. 10, No. 2, 2008, pp. 93-105. doi:10.2165/00148581-200810020-00004
- J. Lee, B. H. Hoang, A. Ziogas and J. A. Zell, “Analysis of Prognostic Factors in Ewing Sarcoma Using a Population-Based Cancer Registry,” Cancer, Vol. 116, No. 8, 2010, pp. 1964-1973. doi:10.1002/cncr.24937
- G. Bacci, A. Longhi, F. Fagioli, A. Briccoli, M. Versari and P. Picci, “Adjuvant and Neoadjuvant Chemotherapy for Osteosarcoma of the Extremities: 27-Year Experience at Rizzoli Institute, Italy,” European Journal of Cancer, Vol. 41, No. 18, 2005, pp. 2836-2845. doi:10.1016/j.ejca.2005.08.026
- P. M. Simpson, R. Reid and D. Porter, “Ewing’s Sarcoma of the Upper Extremity: Presenting Symptoms, Diagnostic Delay and Outcome,” Sarcoma, Vol. 9, No. 1-2, 2005, pp. 15-20. doi:10.1080/00207540500050113
- S. J. Cotterill, S. Ahrens, M. Paulussen, et al., “Prognostic Factors in Ewing’s Tumor of Bone: Analysis of 975 Patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group,” Journal of Clinical Oncology, Vol. 18, No. 17, 2000, pp. 3108-3114.
- F. Fiorenza and L. Jeys, “Ewing’s Sarcoma of BoneMini-Symposium,” Journal of Orthopaedic Trauma, Vol. 24, No. 5, 2010, pp. 342-345.
- G. Bacci, S. Ferrari, F. Bertoni, et al., “Prognostic Factors in Nonmetastatic Ewing’s Sarcoma of Bone Treated with Adjuvant Chemotherapy: Analysis of 359 Patients at the Istituto Ortopedico Rizzoli,” Journal of Clinical Oncology, Vol. 18, No. 1, 2000, pp. 4-11.
- V. Subbiah, P. Anderson, A. J. Lazar, E. Burdett, K. Raymond and J. A. Ludwig, “Ewing’s Sarcoma: Standard and Exterimental Treatment Options,” Current Treatment Options in Oncology, Vol. 10, No. 1-2, 2009, pp. 126-140. doi:10.1007/s11864-009-0104-6
- L. M. Barker, T. W. Pendergrass, J. E. Sanders and D. S. Hawkins, “Survival after Recurrence of Ewing’s Sarcoma Family of Tumors,” Journal of Clinical Oncology, Vol. 23, No. 19, 2005, pp. 4354-4362.
NOTES
*Corresponding author.