Health
Vol.5 No.9(2013), Article ID:37153,10 pages DOI:10.4236/health.2013.59206
Cytokine gene expression in human hepatocytes infected with dengue virus serotype 3 (strain-16562)
![]()
1Center for Vaccine Development, Institute of Molecular Bioscience, Mahidol University, Nakorn Pathom, Thailand
2Department of Biology, Faculty of Science, Silpakorn University, Nakorn Pathom, Thailand; *Corresponding Author: jundee04@gmail.com, jundee@su.ac.th
3Department of Parasitology, Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand
4Department of Biopharmacy, Faculty of Pharmacy, Silpakorn University, Nakorn Pathom, Thailand
Copyright © 2013 Sutee Yoksan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 1 July 2013; revised 1 August 2013; accepted 1 September 2013
Keywords: Dengue virus; Hepatocyte; Cytokines; Type I interferon; Pattern recognition receptors
ABSTRACT
Liver is a site of viral replication and liver dysfunction is a characteristic of severe dengue infection. To understand these mechanisms, we analyzed the response of a hepatic cell linage, HepG2 to infection with dengue 3 virus (strain 16562). Steady state levels of mRNA accumulation were assessed for 14 genes involved in modulation of the host immune responses, at 6, 24 and 48 hpi, by quantitative reverse transcription real-time PCR (qRT-PCR). Fourteen genes showed altered expression upon infection with D3V including; cytokines/chemokines (IL-1β, IL-6, IL- 8, RANTES, MCP-2, IL-2Rα and TGF-βIIIR), type I interferon (IFN-α and IFN-β), and pattern-recognition receptors (TLR3, TLR8, RIG-1, MDA5 and MyD88). Although these genes are associated with mechanism of innate immune response and anti-viral activity, their altered expression does not inhibit D3V (strain 16562) growth kinetics and virus yield in HepG2 cells. Gene expression in liver may explain pathological changes associated with dengue virus infection.
1. INTRODUCTION
Dengue virus (DV) infection causes a life-threatening illness and it is considered to be the major human arbovirosis, affecting 50 - 200 million people and leading to about 20 thousand deaths annually in most tropical urban centers [1]. DV is a small, enveloped virus with a positive, single-stranded RNA genome and is classified as a member of the genus Flavivirus within the Flaviviridae family [2]. DV has four genetically distinct serotypes and it is transmitted to humans through the bite of an infected mosquito from the genus Aedes [3,4].
Infection with any of the four dengue viruses may cause a wide spectrum of clinical features from asymptomatic disease, an undifferentiated febrile illness, dengue fever, dengue haemorrhagic fever (DHF)/dengue shock syndrome (DSS). Patients with DHF present haemorrhagic tendencies, plasma leakage, thrombocytopenia, and hemoconcentration [5]. Hepatic dysfunction in DV infection is demonstrated by hepatomegaly and increases in transminase levels [6]. Clinical severe liver involved severe bleeding [5]. Analyses of liver autopsies of individual with DHF revealed extensive areas of tissue damage with foci of necrosis, and apoptosis [7]. Viral antigens were detected near the lesion areas, suggesting an association between virus replication and hepatic damage. Hepatomegaly, liver enzyme abnormality [8], occasional fulminant hepatic failure [9,10] and histological changes [11] also illustrate the influence of DV infection on liver function.
For a successful resolution of infection, efficient activation of innate/inflammatory and acquired immunity are required to block pathogen replication and invasion, as well as to promote tissue clearance of the pathogens and/ or infected cells. The most prominent pro-inflammatory cytokines IL-1, IL-6, and IL-8 are produced early after pathogen recognition and play a pivotal role in eliciting the innate response as well as in priming and coordinating the adaptive immune response. However, if production is impaired, the innate response will be delayed and inefficient in clearing the pathogen. On the other hand, if production is not controlled by feed-back mechanisms, the persistence of cytokines will increase tissue damage and worsen the severity of the disease. Thus, quantitative time-related changes in cytokine gene expression during infection can be studied as functional markers of protective immunity and/or of the outcome of the disease [1, 12,13].
Pattern-recognition receptors (PRRs) are key proteins of the innate immune response that recognize conserved molecular patterns from a wide variety of microbial pathogens, including bacteria, fungi, protozoa and viruses [14]. Distinct classes of innate PRRs have been identified, the toll-like receptors (TLRs), the retinoic acid-inducible gene I (RIG-I)-like helicases (RLHs), the nucleotidebinding oligomerization domain (NOD)-like receptor (NLR) proteins and the peptidoglycan recognition proteins (PGRPs). TLRs and RIG-I-like receptor (RLRs) are responsible for triggering antiviral innate immune response [15]. Virus recognition by the innate immune system is a first host defense to prevent viral invasion or replication before more specific protections by the adaptive immune system are generated [14]. Accordingly, TLR3 and TLR8 recognize the distinct type of virally derived nucleic acids [16]. Recognitions of RIG-I and MDA5 (Melanoma Differentiation-Associated gene 5) are associated with the induction of inflammatory genes, dendritic cell migration, maturation, and activation of the adaptive immune response, which contribute to the elimination of the infection [12,14]. Few reports demonstrate the involvement of PRRs in DV recognition [1,12,17-21]. However, no previous study demonstrates the response of hepatocytes after infection with D3V.
To gain a better understanding of the innate immune response and adaptive immune response to DV infection, we investigated the cytokines/chemokines, type I IFN and PRRs gene expression changes of hepatic cell linage, HepG2 infected with dengue virus serotype 3 (strain 16562) after 6, 24 and 48 hour post-infection (hpi) by qRT-PCR.
2. MATERIALS AND METHODS
2.1. Cell Culture and Virus Infection
Human hepatocellular carcinoma cells (HepG2/C3A, CRL-10741, Lot 58483225, ATCC, USA) were cultured on antibiotic-free Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen Life Technologies, CA, USA) supplemented with 5 mM glucose, 2 mM L-glutamine, 1% nonessential amino acids (NEA), and 10% fetal bovine serum in a CO2 humid incubation chamber at 37˚C. Cells were seeded at a density of 105 cell per mL, and after 2 days, when they reached 80% - 90% confluence, cells were either mock-infected or infected with D3V strain 16562 (MOI of 1 plaque forming units per cell). After 1 h, cells were washed and incubated with fresh medium for the indicated periods. For preparations of virus stocks, D3V was propagated in Aedes albopictus (C6/36) cells utilizing antibiotic-free MEM medium supplemented with 2 mM L-glutamine, 1% NEA, and 10% FBS in 28˚C.
2.2. Viral Titration by Plaque Assay
Viral titration was undertaken by plaque assay using Rhesus monkey kidney (LLC-MK2) cells. Cells were seeded in 6-well plates in DMEM (Invitrogen Life Technologies) supplemented with 10% inactivated FBS and 100 U/ml penicillin and 100 mg/ml streptomycin. Cells were maintained in a humidified incubator at 37˚C, 5% CO2 for 3 day until they reached confluence. The medium was removed and the monolayer was infected with 200 µl of serially diluted virus samples. The plates were incubated for 1.5 h at 37˚C for adsorption. Subsequently, 4 ml of 2% SeaKem® LE agarose (Lonza, St. Louis, MO, USA) mixed with Earl’s balance salt solution supplemented with 2% inactivated FBS and 100 U/ml penicillin and 100 mg/ml streptomycin were added to each well. The plates were maintained in a humidified incubator at 37˚C, 5% CO2 for 5 days. After this period, the cells were fixed with 10% formaldehyde at room temperature and the agarose plugs were removed. Plaques were visualized by staining with 1.25% crystal violet in 20% ethanol solution.
2.3. Viral Quantification by Taqman Real-Time RT-PCR
Viral RNA from D3V-infected C6/36 cell culture was isolated by using a PureLink Viral RNA/DNA Kit (Invitrogen, CA) according to the manufacturer’s instructions, and viral RNA levels were quantified by real-time RTPCR. Analysis was performed on Chromo4 Real-Time System (Bio-Rad, CA, USA). Primers were specific for a 73-bp region of the D3V prM gene [22]. The probe was labeled with the reporter dye Texas red at the 5’ end and the quencher dye BHQ2 at the 3’ end. Viral RNA copy numbers in the samples were calculated from a standard curve generated by using pre-quantified D3V-specific plasmid standards with known copy numbers.
2.4. Cytokine Gene Quantification by Real-Time PCR
RNA from uninfected or D3V-infected HepG2 cells was extracted using PureLink RNA Mini Kit (Invitrogen Life Technologies) according to the manufacturer’s instructions. The final number of cells used for RNA extraction was approximately 1 × 106 at each indicated times. Two µg of the total RNA were reverse transcribed to cDNA using the SuperScript® III First-Strand Synthesis System (Invitrogen Life Technologies) according to manufacturer’s instruction. The cDNA was diluted 1:10 and 1 µl of each sample was submitted to amplify with SensiFASTTM SYBR No-ROX Kit (Bioline, UK). The reaction was carried out for the selected genes using gene specific primers (Table 1) on Chromo 4 Real-Time System (Bio-Rad). The samples were subjected to a initial denature step at 95 for 2 min, followed by 40 cycles of denature at 95˚C for 5 s, annealing at 60˚C for 10 s, and extension with data collection at 72˚C for 20 s. After each run, melting curves were acquired by stepwise increase of the temperature from 65˚C to 95˚C with a heating rate of 1˚C per min to ensure that a single product was amplified in the reaction. Amplification efficiencies were determined for all genes by standard cures which were established by serial dilutions of pre-quantified genes-specific plasmid standards with known copy numbers. Each of the amplifications was triplicates. Each gene was normalized relative to control of housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GPDH) mRNA. The fold-changes in expression of gene-specific mRNA between samples and control (mock) were calculated.
2.5. Statistical Analysis
Gene expression of cytokines, antiviruses, and toll-like receptors evaluated in mock-infected and D3V-infected HepG2 cells were compared by t-test. Values of p < 0.05 were considered to be statistically significant. The statistical program SPSS 11.5 (IBM, NY, USA) was used.
3. RESULTS
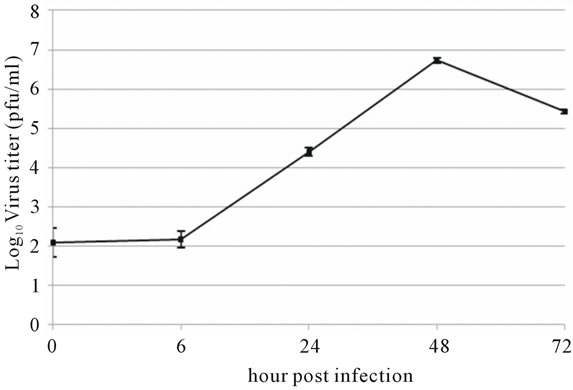
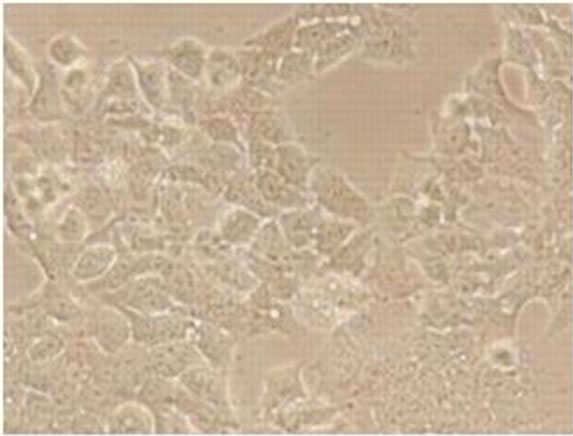
Viral RNA replication in HepG2 cells infected with D3V (strain 16562) for 72 h was determined by plaque assay of the virus released in the cell culture medium (Figure 1(a)). Dengue viral replication in HepG2 cells was also determined by quantitative real-time RT-PCR, showing increasing viral RNA production until 48 h of infection (Figure 1(b)). The results showed that new extracellular viral progeny reached maximum at 48 h. After 48 h, a degree of cell death becomes apparent and the experiment was terminated at 72 h (Figure 2).
To assess changes in cellular gene expression upon D3V infection, HepG2 cell line was infected with D3V at MOI of 1. Total cellular RNA was extracted from infected and mock-infected cells at 6, 24 and 48 hpi. Steady state levels of mRNA accumulation were determined for 14 genes (Table 1) by means of real-time PCR followed by melting curve analysis. Normalization of gene expression was carried out by assessing mRNA accumulation of a housekeeping gene (GPDH) in infected and mock-infected cell cultures. The normalized mRNA expression level of a cellular gene in D3V-infected cells
 (a)
(a)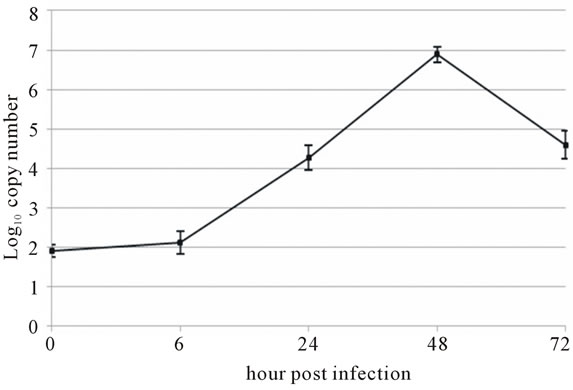 (b)
(b)
Figure 1. Growth characteristics of D3V (strain-16562) replicated on hepatic cell linage, HepG2. HepG2 cells were infected at an MOI of 1. At times post-infection, (a) D3V-infected supernatant were collected and titrated on LLC-MK2 for virus yield, (b) viral RNA was extracted from the infected supernatant and subjected to quantitative real-time RT-PCR using D3V specific primer and probe to quantify viral genome. Data are means and standard deviation of three independent experiments.
was considered significant when it departed from its level in mock-infected cells two-folds, in either up-regulation or down-regulation. The approach enabled the identification of 14 genes differentially expressed in D3V-infected HepG2 cells (Figures 3 and 4).
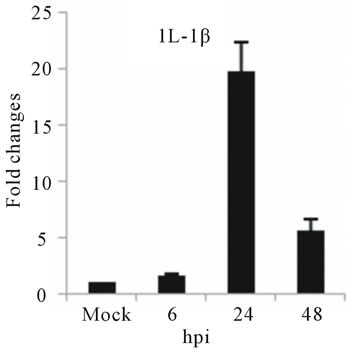
The highest sustained level of mRNA accumulation upon infection was observed for cytokine genes. The expression of seven cytokine genes showed significant upregulation of IL-1β, and IL-6 at 24 and 48 hpi (Figures 3(a) and (b)). IL-1β expression reached a 20-fold increase at 24 hpi and 5-fold increases at 48 hpi, the highest mRNA accumulation observe in this study (Figure 3(a)). Cytokine receptor IL-2Rα mRNA level showed a moderate accumulation at 24 and 48 hpi (Figure 3(c)), while TGF-βIIIR expression downregulated up to 10-fold at 48 hpi (Figure 3(d)). D3V infection significant affected
 (a)
(a)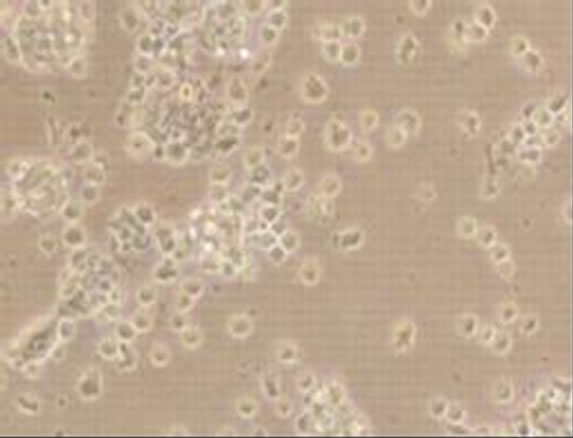 (b)
(b)
Figure 2. Morphology of HepG2 cells infected with D3V (strain 16562). Monolayers of HepG2 cells were adsorbed with D3V (MOI of 1) or mock-infected for 1 h at 37˚C, (a) mockinfected HepG2 cells at 72 h post-infection and (b) D3V-infected HepG2 cells at 72 h post-infection. Each figure is representative of one out of three independent experiments. Images were taken with an original magnification of ×400.
the expression of several chemokine genes. Chemokine IL-8, MCP-2 and RANTES were upregulated at 24 or 48 hpi (Figures 3(e)-(g)).
Changes in the expression of type I interferon were observed in D3V-infected HepG2 cells. IFN-α and IFN-β were affected upon D3V infection. While IFN-α mRNA level increased during course of infection times, IFN-β mRNA levels accumulated by 24 hpi and decreased by 48 hpi (Figures 4(a) and (b)), respectively.
As with type I interferon response, expression of pattern recognition receptors (PRRs) is modulated rapidly in response to pathogens. We observed that TLR3 and TLR8 expression were upregulated at toward 48 hpi for 5-fold and 3-fold, respectively (Figures 4(c) and (d)). RIG-I, MDA5, and MyD88 mRNA expression was also investigated during HepG2 infection with D3V. The analysis of RIG-I gene expression profile showed a 6-fold increase at 48 hpi (Figure 4(e)). MDA5 mRNA level increased toward 48 hpi up to 9-fold (Figure 4(f)). On the other hand, MyD88 mRNA level decreased for 3-fold at 48 hpi (Figure 4(g)).
4. DISCUSSION
Hepatocyte dysfunction both in vitro and in vivo are a characteristic of dengue virus infection, especially in the severe cases for which fulminant hepatic failure has been reported [10]. Analyses of liver autopsies from individuals with DHF revealed extensive areas of tissue damage, with foci of necrosis and apoptosis and viral antigens were detected in Kupffer cells and hepatocytes near the damaged areas, suggesting an association between virus replication and hepatic damage [23].
To further characterize the nature of host cell responses to infection by D3V, we used qRT-PCR to assess changes in expression of 14 cellular genes (Table 1) involved in modulation of the host immune response in human hepatocyte (HepG2) cells. Human hepatocyte is a DV secondary target cell and seems to play a role in the outcome of the infection [24-26] as well as severe forms of DHF/DSS [23,27,28].
In our study, IL-1β, and IL-6 cytokines showed significantly higher levels of expression upon infection (Figure 3(a)). IL-1β may be one of the factors that mediate dengue pathogenesis since it is a primary inducer of fever in cultured endothelial cells. IL-1β induced synthesis of prostaglandins E2 and I2, and platelet-activating factor, which were potent vasodialators and orchestrate a cascade of biochemical event that lead to vascular congestion, clot formation, cellular infiltration and endothelial leakage [29]. We found a significant increase in the expression of IL-1β mRNA induced by D3V at 24 and 48 hpi. This result was consistent with other reports in which IL-1β was found in both DF and DHF patients [2,30]. IL-6 is multifunction cytokine that plays a central role in host defense due to its wide range of immune and hepatopoietic activities and also its potential ability to induce the acute phase response of inflammation. Several reports demonstrated elevated level of IL-6 in DV infected patients and hepatic cells [31-33]. In agreement with our results, IL-6 mRNA level progressively increase during the course of infection can be correlated with the involvement of the liver in DHF and DSS in vivo. IL-6, as well as IL-1, is able to activate endothelial cells leading to the release of IL-8 and MCP, expression of adhesion molecules, and recruitment of leukocytes to inflammatory sites [34,35]. Both cytokines are potent inducers of vascular permeability and mediate pathologic changes including high fever, coagulation defects, and bleeding observed in infections with some single-stranded RNA viruses

Table 1. Primer sets of host genes used in this study.
[36].
The slight accumulation of IL-2Rα mRNA was observed by 24 hpi coincides with a down-regulation of TGF-βIIIR (Figures 3(c) and (d)). IL-2 Receptor is specific cell surface receptor on lymphocyte. IL-2Rα has been reported to be a marker of lymphocyte activation [37]. TGF-β has been shown to be associated with development of DHF/DSS [5]. While IL-2 mediates signaling through IL-2R, stimulating a set of complex signal transduction pathways resulting in cell proliferation, signaling through TGF-βIIIR-Smad results in inhibition of cellular proliferation, enhancement of matrix accumulation, and suppression of inflammation [38]. The putative shift we have observed, towards induction of cell proliferation suggested by IL-2R up-regulation coupled with TGF-β IIIR down-regulation, may stimulate virus replication. In our study was consistent with stimulation of PBMCs by IL-2, the ligand for IL-2 receptor, induces replication of a variety of HIV strains [39].
The elevated levels of IL-8, and RANTES mRNAs was also observed in infected HepG2 cells (Figures 3(e) and (f)). IL-8 is another important cytokine that acts as chemoattractant cytokine in inflammatory processes. Moreover, IL-8 also destroys tight-junction and cytoskeleton reorganization, resulting in increased vascular permeability [40]. Its relation to TLR pathway in DV infection has also been shown in HEK 293 cells that expressed TLR3, 7, 8 and enables IL-8 secretion after virus recognition [19]. As observed for IL-6, a correlation between IL-8 levels and the severity of dengue diseases has also been demonstrated [41]. RANTES is chemokine capable of recruiting lymphocyte and NK cells to the sites of inflammation [42]. Our results showed a strong induction in RANTES expression in D3V infected HepG2 cells (Figure 3(f)), in accordance with other reports which showed the up-regulation of its transcription and secretion in hepatic cells infected with D2V [1]. MCP-2 is upregulated during HepG2 cells infected with D3V (Figure 3(g)). MCP-2, like RANTES signal through CCR5, CCR2, and CCR3 receptors contributing to vascular permeability [43]. Based on these observations, one can speculate that DV recognition during its replication in liver stimulate the expression of IL-1β, IL-6, IL-8, RANTES, and MCP-2 as well as IL-2Rα enhancing the overall cytokine production observed in the pathogenesis of dengue disease.
Type I IFN responses function as the first line of defense against viral infections. Several viruses have been shown to inhibit type I IFN production in infected cells. DV can target the IFN production pathway in infected cells as an additional immune evasion strategy. On the other hand, several DV proteins have been identified in recent years to be involved in the inhibition of type I IFN signaling in infected cells. By inhibiting this important pathway, the virus is able to stop the induction of hundreds of IFN inducible genes with antiviral functions that
 (a)
(a) (b)
(b)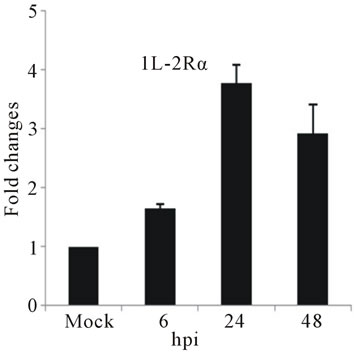 (c)
(c)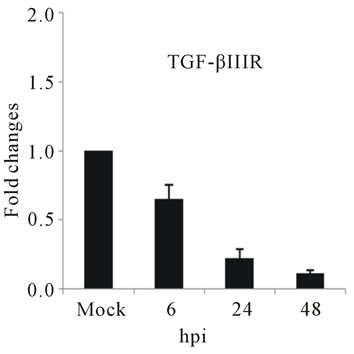 (d)
(d)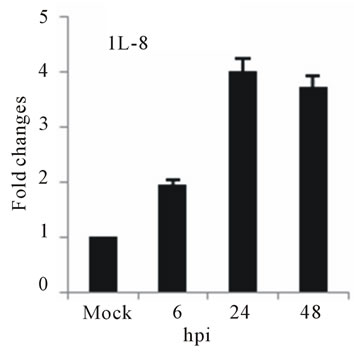 (e)
(e)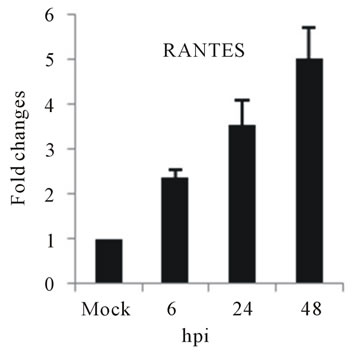 (f)
(f) (g)
(g)
Figure 3. Relative expression of cytokine genes in DEN-3 virus-infected HepG2 cells. Cells were mock-infected or infected with an MOI of 1, and total RNA was extracted at 6, 24 and 48 h post-infection and submitted to qRT-PCR to detect the mRNAs for IL-1β (a); IL-6 (b); IL-2Rα (c); TGF-βIIIR (d); IL-8 (e); RANTES (f); and MCP-2 (g). The results were normalized by GPDH expression and are presented as cytokine mRNA levels relative (-fold) to control (mock infected) samples. Results correspond to the average of at least three independent experiments.
may impair several aspects of the virus cycle [44]. A temporal pattern of accumulation of type I interferon mRNAs was observed upon infection of D3V-infected hepatocytes. IFN-α transcripts decreased more than 5- folds by 48 hpi (Figure 4(a)), while the opposite trend was observed for IFN-β mRNA, with a 2-fold decrease by 48 hpi (Figure 4(b)). Like many viruses, dengue inhibits IFN-α and IFN-β signaling by suppressing Jak-Stat activation, resulting in reduced host antiviral response [45]. The combination of a reduced host defense, increased uptake of the virus and delayed viral clearance likely synergizes to produce higher viremia resulting in a more severe outcome.
In our study, we observed toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), which were responsible for triggering antiviral innate immune responses [15]. TLR3 and 8 are implicated in nucleic acid sensing and usually are responsible for virus recognition [16]. RIG-I
 (a)
(a) (b)
(b)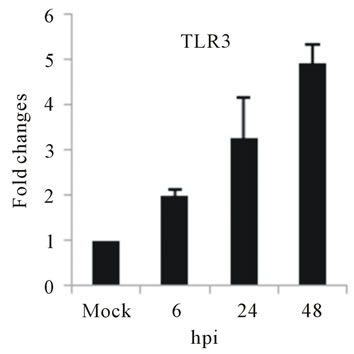 (c)
(c)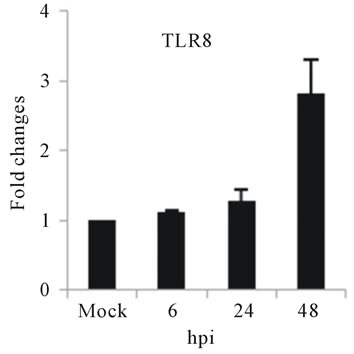 (d)
(d)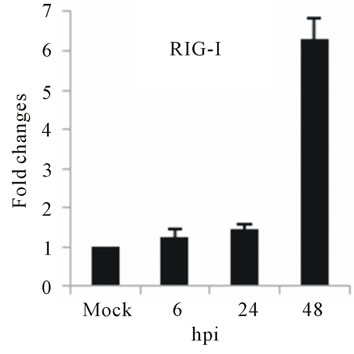 (e)
(e)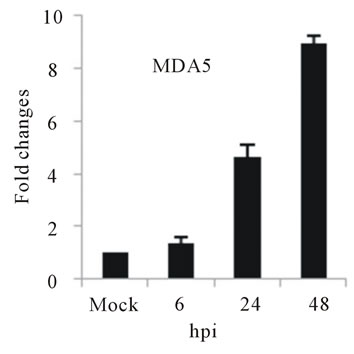 (f)
(f)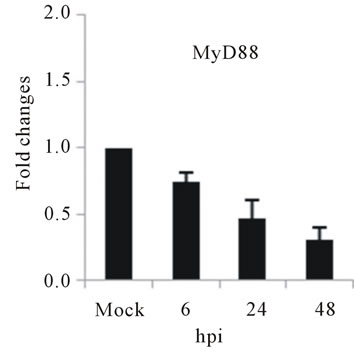 (g)
(g)
Figure 4. Relative expression of Type I interferon and PRRs genes in DEN-3 virus-infected HepG2 cells. Cells were mock-infected or infected with an MOI of 1, and total RNA was extracted at 6, 24 and 48 h post-infection and submitted to qRT-PCR to detect the mRNAs for IFN-α (a); IFN-β (b); TLR3 (c); TLR8 (d); RIG-I (e); MDA5 (f) and MYD88 (g). The results were normalized by GPDH expression and are presented as TLR mRNA levels relative (-fold) to control (mock infected) samples. Results correspond to the average of at least three independent experiments.
and MDA5 are cytoplasmic RNA helicases that sense dsRNA and can detect intracellular viral products [46]. We found that TLR3 mRNA expression progressively increased during the time course of D3V infection (Figure 4(c)), which coincided with the increase seen in the viral RNA (Figure 1). TLR3 detects double-stranded (ds) RNA which is produced only while the virus is replicating, when the positive-sense genomic RNA is transcribed in a negative-sense complementary strand that was indeed detected during the time course of infection of HepG2 cells (Figure 4(c)). On the other hand, an increase in TLR8 mRNA production is observed only at late stage of dengue virus infection (Figure 4(d)). Therefore, the difference profile of mRNA expression of these two TLRs may be explained by the fact that TLR3 sense dsRNA and TLR8 sense ssRNA [47].
RIG-I, MDA5 and MyD88 are PRRs located in the cytosol that, like TLR3, sense dsRNA [46]. RIG-I is a RNA helicase containing two caspase-recruiting domain (CARD)-like domains. Overexpression of RIG-I confers antiviral responses [46]. MDA5 is structurally similar to RIG-I, which also contains two CARD-like and a single helicase domains, and is suggested to mediate antiviral responses [46]. MyD88 is a central adapter shared by almost all TLRs. The association of TLRs and MyD88 recruits members of the interleukin-1 receptor-associated kinase (IRAK) family [46]. We found that the expression of both RIG-I and MDA5, but not MYD88 genes was greatly induced in HepG2 cells infected with D3V (Figures 4(e)-(g)). These results are in agreement with other studies which demonstrated that the involvement of RIGI and MDA5 in DV recognition in other non-immune cell lines [1,20,21,48] and suggest an important role of these two receptor in DV recognition by human liver cells.
5. CONCLUSION
The induction of IL-1β, IL-6, IL-8, RANTES, MCP-2, IFN-α and IFN-β as well as IL-2Rα genes constitutes another evidence that HepG2 cells respond to D3V through the activation of PRRs such as TLR3, TLR8, RIG-I and MDA5. In contrast, TGF-βIIIR and MYD88 down-regulation can be seen as a viral mechanism to escape from cellular defense response, as it is clear that viruses along their evolution develop ways to avoid host reaction [49]. Thus, it seems that both antiviral defense mechanisms and viral-induced strategies to assure replication are involved in interaction between D3V and hepatocytes and may contribute to pathogenesis. Therefore, the results presented here comprise part of the initial steps to guide the efforts toward the elucidation of the mechanisms triggered by D3V to activate innate immune response in liver cells.
6. ACKNOWLEDGEMENTS
We thank Assistant Professor Dr. Jarungsang Laksanaboonsong, Dean of Faculty of Science, Silpakon University at Sanarmchandra palace, Nakorn Pathom for providing laboratory facility. This work was supported by Grants No. RGP 2554-11 from Faculty of Science, Silpakorn University and partial funding from Dengue vaccine project, Mahidol University and Thailand Research Funding No. DIG5180004.
REFERENCES
- Conceição, T.M., El-Bacha, T., Villas-Bôas, C.S., Coello, G., Ramírez, J., Montero-Lomeli M. and Da Poian, A.T. (2010) Gene expression analysis during dengue virus infection in HepG2 cells reveals virus control of innate immune response. Journal of Infection, 60, 65-75. doi:10.1016/j.jinf.2009.10.003
- Rabablert, J. and Yoksan, S. (2009) Attenuated D2 16681- PDK53 vaccine: Defining humoral and cell-mediated immunity. Current Pharmaceutical Design, 15, 1203-1211. doi:10.2174/138161209787846865
- Hugo, L.E., Monkman, J., Dave, K.A., Wockner, L.F., Birrell, G.W., Norris, E.L., Kienzle, V.J., Sikulu, M.T., Ryan, P.A., Gorman, J.J. and Kay, B.H. (2013) Proteomic biomarkers for ageing the mosquito Aedes aegypti to determine risk of pathogen transmission. PLoS One, 8, e58656. doi:10.1371/journal.pone.0058656
- Vega-Rua, A., Zouache, K., Caro, V., Diancourt, L., Delaunay, P., Grandadam, M. and Failloux, A.B. (2013) High efficiency of temperate Aedes albopictus to transmit chikungunya and dengue viruses in the Southeast of France. PLoS One, 8, e59716. doi:10.1371/journal.pone.0059716
- Martina, B.E., Koraka, P. and Osterhaus, A.D. (2009) Dengue virus pathogenesis: An integrated view. Clinical Microbiology Review, 22, 564-581. doi:10.1128/CMR.00035-09
- Nagila, A., Permpongpaiboon, T., Tantrarongroj, S., Porapakkham, P., Chinwattana, K., Deakin, S. and Porntadavity, S. (2009) Effect of atorvastatin on paraoxonase1 (PON1) and oxidative status. Pharmacological Report, 61, 892- 898. http://www.if-pan.krakow.pl/pjp/pdf/2009/5_892.pdf
- Huerre, M.R., Lan, N.T., Marianneau, P., Hue, N.B., Khun, H., Hung, N.T., Khen, N.T., Drouet, M.T., Huong, V.T., Ha, D.Q., Buisson, Y. and Deubel, V. (2001) Liver histopathology and biological correlates in five cases of fatal dengue fever in Vietnamese children. Virchows Archives, 438, 107-115. doi:10.1007/s004280000329
- Mohan, B., Patwari, A.K. and Anand, V.K. (2000) Hepatic dysfunction in childhood dengue infection. Journal of Tropical Pediatrics, 46, 40-43. doi:10.1093/tropej/46.1.40
- Subramanian, V., Shenoy, S. and Joseph, A.J. (2005) Dengue hemorrhagic fever and fulminant hepatic failure. Digestive Diseases and Sciences, 50, 1146-1147. doi:10.1007/s10620-005-2722-6
- Ling, L.M., Wilder-Smith, A. and Leo, Y.S. (2007) Fulminant hepatitis in dengue haemorrhagic fever. Journal of Clinical Virology, 38, 265-268. doi:10.1016/j.jcv.2006.12.011
- Bhamarapravati, N. (1989) Hemostatic defects in dengue hemorrhagic fever. Reviews of Infectious Diseases, 11, S826-S829. http://www.jstor.org/stable/4454972
- Nasirudeen, A.M., Wong, H.H., Thien, P., Xu, S., Lam, K.P. and Liu, D.X. (2011) RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLOS Neglected Tropical Diseases, 4, 5. doi:10.1371/journal.pntd.0000926
- Nagila, A., Netsawang, J., Srisawat, C., Noisakran, S., Morchang, A., Yasamut, U., Puttikhunt, C., Kasinrerk, W., Malasit, P., Yenchitsomanus, P.T. and Limjindaporn, T. (2011) Role of CD137 signaling in dengue virus-mediated apoptosis. Biochemical and Biophysical Research Communications, 410, 428-433. doi:10.1016/j.bbrc.2011.05.151
- Seth, R.B., Sun, L. and Chen, Z.J. (2006) Antiviral innate immunity pathways. Cell Research, 16, 141-147. doi:10.1038/sj.cr.7310019
- Meylan, E. and Tschopp, J. (2006) Toll-like receptors and RNA helicases: Two parallel ways to trigger antiviral responses. Molecular Cell, 22, 561-569. doi:10.1016/j.molcel.2006.05.012
- Barton, G.M. (2007) Viral recognition by toll-like recaptors. Seminars in Immunology, 19, 33-40. doi:10.1016/j.smim.2007.01.003
- Wang, J.P., Liu, P., Latz, E., Golenbock, D.T., Finberg, R.W. and Libraty, D.H. (2006) Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. The Journal of Immunology, 177, 7114-7121. http://www.jimmunol.org/content/177/10/7114.long
- Sun, P., Fernandez, S., Marovich, M.A., Palmer, D.R., Celluzzi, C.M., Boonnak, K., Liang, Z., Subramanian, H., Porter, K.R., Sun, W. and Burgess, T.H. (2009) Functional characterization of ex vivo blood myeloid and plasmacytoid dendritic cells after infection with dengue virus. Virology, 383, 207-215. doi:10.1016/j.virol.2008.10.022
- Tsai, Y.T., Chang, S.Y., Lee, C.N. and Kao, C.L. (2009) Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cellular Microbiology, 11, 604- 615. doi:10.1111/j.1462-5822.2008.01277.x
- Loo, Y.M., Fornek, J., Crochet, N., Bajwa, G., Perwitasari, O., Martinez-Sobrido, L., Akira, S., Gill, M.A., GarcíaSastre, A., Katze, M.G. and Gale Jr., M. (2008) Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. Journal of Virology, 82, 335-345. doi:10.1128/JVI.01080-07
- Chang, T.H., Liao, C.L. and Lin, Y.L. (2006) Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes and Infection, 8, 157-171. doi:10.1016/j.micinf.2005.06.014
- Johnson, B.W., Russell, B.J., Lanciotti, R.S. (2005) Serotype-specific detection of dengue viruses in a fourplex real-time reverse transcriptase PCR assay. Journal of Clinical Microbiology, 43, 4977-4983. doi:10.1128/JCM.43.10.4977-4983.2005
- Limonta, D., Capó, V., Torres, G., Pérez, A.B. and Guzmán, M.G. (2007) Apoptosis in tissues from fatal dengue shock syndrome. Journal of Clinical Virology, 40, 50-54. doi:10.1016/j.jcv.2007.04.024
- Paes, M.V., Pinhão, A.T., Barreto, D.F., Costa, S.M., Oliveira, M.P., Nogueira, A.C., Takiya, C.M., Farias-Filho, J.C., Schatzmayr, H.G., Alves, A.M. and Barth, O.M. (2005) Liver injury and viremia in mice infected with dengue-2 virus. Virology, 338, 236-246. doi:10.1016/j.virol.2005.04.042
- Seneviratne, S.L., Malavige, G.N. and de Silva, H.J. (2006) Pathogenesis of liver involvement during dengue viral infections. Transactions of the Royal Society of Tropical Medicine and Hygiene, 100, 608-614. doi:10.1016/j.trstmh.2005.10.007
- Pattanakitsakul, S.N., Rungrojcharoenkit, K., Kanlaya, R., Sinchaikul, S., Noisakran, S., Chen, S.T., Malasit, P. and Thongboonkerd, V. (2007) Proteomic analysis of host responses in HepG2 cells during dengue virus infection. Journal of Proteome Research, 6, 4592-4600. doi:10.1021/pr070366b
- Silva, B.M., Sousa, L.P., Gomes-Ruiz, A.C., Leite, F.G., Teixeira, M.M., da Fonseca, F.G., Pimenta, P.F., Ferreira, P.C., Kroon, E.G. and Bonjardim, C.A. (2011) The dengue virus nonstructural protein 1 (NS1) increases NF-κB transcriptional activity in HepG2 cells. Archives of Virology, 156, 1275-1279. doi:10.1007/s00705-011-0969-0
- Assunção-Miranda, I., Amaral, F.A., Bozza, F.A., Fagundes, C.T., Sousa, L.P., Souza, D.G., Pacheco, P., BarbosaLima, G., Gomes, R.N., Bozza, P.T., Da Poian, A.T., Teixeira, M.M. and Bozza, M.T. (2010) Contribution of macrophage migration inhibitory factor to the pathogenesis of dengue virus infection. The FASEB Journal, 24, 218- 228. doi:10.1096/fj.09-139469
- Warke, R.V., Xhaja, K., Martin, K.J., Fournier, M.F., Shaw, S.K., Brizuela, N., de Bosch, N., Lapointe, D., Ennis, F.A., Rothman, A.L. and Bosch, I. (2003) Dengue virus induces novel changes in gene expression of human umbilical vein endothelial cells. Journal of Virology, 77, 11822-11832. doi:10.1128/JVI.77.21.11822-11832.2003
- Rabablert, J., Wasi, C., Kinney, R., Kasisith, J., Pitidhammabhorn, D. and Ubol, S. (2007) Attenuating characteristics of DEN-2 PDK53 in flavivirus-naïve peripheral blood mononuclear cells. Vaccine, 25, 3896-3905.
- Leong, A.S., Wong, K.T., Leong, T.Y., Tan, P.H. and Wannakrairot, P. (2007) The pathology of dengue hemorrhagic fever. Seminars in Diagnostic Pathology, 24, 227-236. doi:10.1053/j.semdp.2007.07.002
- Restrepo, B.N., Isaza, D.M., Salazar, C.L., Ramírez, R., Ospina, M. and Alvarez, L.G. (2008) Serum levels of interleukin-6, tumor necrosis factor-alpha and interferongamma in infants with and without dengue. Revista da Sociedade Brasileira de Medicina Tropical, 41, 6-10. doi:10.1590/S0037-86822008000100002
- Lei, H.Y., Yeh, T.M., Liu, H.S., Lin, Y.S., Chen, S.H. and Liu, C.C. (2001) Immunopathogenesis of dengue virus infection. Journal of Biomedical Science, 8, 377-388. doi:10.1007/BF02255946
- Gabay, C. (2006) Interleukin-6 and chronic inflammation. Arthritis Research & Therapy, 8, S2-S3. doi:10.1186/ar1917
- Lipsky, P.E. (2006) Interleukin-6 and rheumatic diseases. Arthritis Research & Therapy, 8, S2-S4. doi:10.1186/ar1918
- Bray, M. (2005) Pathogenesis of viral hemorrhagic fever. Current Opinion in Immunology, 17, 399-403. doi:10.1016/j.coi.2005.05.001
- Dejica, D. (2001) Serum soluble IL-2 receptor as a marker of lymphocyte activation in some autoimmune diseases. Effect of immunosuppressive therapy. Romanian Archives of Microbiology and Immunology, 60, 183-201. http://www.ncbi.nlm.nih.gov/pubmed/12165973
- Blobe, G.C., Liu, X., Fang, S.J., How, T. and Lodish, H.F. (2001) A novel mechanism for regulating transforming growth factor beta (TGF-beta) signaling. Functional modulation of type III TGF-beta receptor expression through interaction with the PDZ domain protein, GIPC. The Journal of Biological Chemistry, 276, 39608-39617. doi:10.1074/jbc.M106831200
- Kinter, A.L., Poli, G., Fox, L., Hardy, E. and Fauci, A.S. (1995) HIV replication in IL-2-stimulated peripheral blood mononuclear cells is driven in an autocrine/paracrine manner by endogenous cytokines. The Journal of Immunology, 154, 2448-2459. http://www.jimmunol.org/content/154/5/2448.abstract
- Talavera, D., Castillo, A.M., Dominguez, M.C., Gutierrez, A.E. and Meza, I. (2004) IL8 release, tight junction and cytoskeleton dynamic reorganization conducive to permeability increase are induced by dengue virus infection of microvascular endothelial monolayers. Journal of General Virology, 85, 1801-1813. doi:10.1099/vir.0.19652-0
- Bozza, F.A., Cruz, O.G., Zagne, S.M., Azeredo, E.L., Nogueira, R.M., Assis, E.F., Bozza, P.T. and Kubelka, C.F. (2008) Multiplex cytokine profile from dengue patients: MIP-1beta and IFN-gamma as predictive factors for severity. BMC Infectious Diseases, 8, 86. doi:10.1186/1471-2334-8-86
- Middleton, J., Patterson, A.M., Gardner, L., Schmutz, C. and Ashton, B.A. (2002) Leukocyte extravasation: Chemokine transport and presentation by the endothelium. Blood, 100, 3853-3860. doi:10.1182/blood.V100.12.3853
- Hellier, S., Frodsham, A.J., Hennig, B.J., Klenerman, P., Knapp, S., Ramaley, P., Satsangi, J., Wright, M., Zhang, L., Thomas, H.C., Thursz, M. and Hill, A.V. (2003) Association of genetic variants of the chemokine receptor CCR5 and its ligands, RANTES and MCP-2, with outcome of HCV infection. Hepatology, 38, 1468-1476. doi:10.1016/j.hep.2003.09.027
- Morrison, J., Aguirre, S. and Fernandez-Sesma, A. (2012) Innate immunity evasion by Dengue virus. Viruses, 4, 397-413. doi:10.3390/v4030397
- Muñoz-Jordan, J.L., Sánchez-Burgos, G.G., Laurent-Rolle, M. and García-Sastre, A. (2003) Inhibition of interferon signaling by dengue virus. Proceedings of the National Academy of Sciences of the United States of America, 100, 14333-14338. doi:10.1073/pnas.2335168100
- Kawai, T. and Akira, S. (2008) Toll-like receptor and RIG-I-like receptor signaling. Annals of the New York Academy of Sciences, 1143, 1-20. doi:10.1196/annals.1443.020
- Akira, S. and Takeda, K. (2004) Toll-like receptor signalling. Nature Reviews Immunology, 4, 499-511.
- Ramirez-Ortiz, Z.G., Warke, R.V., Pacheco, L., Xhaja, K., Sarkar, D., Fisher, P.B., Shaw, S.K., Martin, K.J. and Bosch, I. (2006) Discovering innate immunity genes using differential display: A story of RNA helicases. Journal of Cellular Physiology, 209, 636-644. doi:10.1002/jcp.20797
- Gale Jr., M. and Foy, E.M. (2005) Evasion of intracellular host defence by hepatitis C virus. Nature, 436, 939-945. doi:10.1038/nature04078