American Journal of Plant Sciences
Vol.5 No.18(2014), Article ID:49281,12 pages
DOI:10.4236/ajps.2014.518297
Mutational Analyses of a Fork Head Associated Domain Protein, DAWDLE, in Arabidopsis thaliana
Lakshmi Ayiloor Narayanan1,2, Dipaloke Mukherjee3, Shuxin Zhang4, Bin Yu4, David Chevalier1,5*
1Department of Biological Sciences, Mississippi State University, Starkville, Mississippi, USA
2Department of Basic Sciences, CVM, Mississippi State University, Starkville, Mississippi, USA
3Department of Food Science, Nutrition and Health Promotion, Mississippi State University, Starkville, Mississippi, USA
4Center for Plant Science Innovation & School of Biological Sciences, University of Nebraska-Lincoln, Lincoln, Nebraska, USA
5Department of Biological Sciences, East Georgia State College, Swainsboro, Georgia, USA
Email: *dchevalier@ega.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 7 June 2014; revised 18 July 2014; accepted 18 August 2014
ABSTRACT
DAWDLE (DDL) gene encodes a protein that contains an N-terminal arginine-rich domain and a C-terminal Fork Head Associated (FHA) domain in Arabidopsis thaliana. DDL protein is believed to function in microRNA biogenesis by mediating the recruitment of pri-microRNA to DICER-LIKE 1 and also stabilizing the microRNA. The aim of this study was to conduct a structure-function analysis to identify the regions in DDL that are of functional significance. Targeted Induced Local Lesions in Genome screen was performed in the Columbia erecta-105 background of Arabidopsis resulting in the identification of eight point mutations spanning DDL. The mutants were characterized by phenotypic and molecular analyses based on the prior knowledge on ddl knockout mutants. Height of the plant, hypocotyl and root length, and fertility were measured for phenotypic characterization, and microRNA172 levels were measured to assess the mutation effect at the molecular level. Phenotypic and molecular analyses of the mutants revealed effects resulting in ddl phenotypes of varying degrees in different organs and each mutant displayed at least one phenotype studied. Reduction in fertility and increase in stem length were two phenotypes that most of the mutants consistently displayed. Identification and characterization of several key residues in the arginine rich region and FHA domain will serve as an important tool for elucidation of DDL signaling pathway.
Keywords:microRNA, DAWDLE, FHA Domain, Gene Mapping, Arabidopsis

1. Introduction
Fork Head Associated (FHA) domain is a highly conserved and modular protein interaction domain that was first identified in Fork Head transcription factors [1] . Not restricted to Fork Head transcription factors, they have their presence in more than 2000 proteins that represent a wide variety of prokaryotic and eukaryotic proteins with different functions [2] . FHA domain is unique for the fact that they are the only known phosphoproteinbinding domain that specifically recognizes the phospho-threonine (Thr) residues distinguishing them from phospho-serine (Ser). Other phospho binding domains like WW, and 14-3-3 recognize both phospho-Ser/Thr. Though FHA domain binds only phospho-Thr peptides, they have wide ligand specificity, and is one of the reasons for its presence in proteins with diverse functions [2] . FHA domain is present in several proteins that mediate signal transduction such as kinases, phosphatases, and transcription factors functioning in several cellular processes such as DNA damage response, cell cycle regulation or growth [2] .
DAWDLE (DDL) is one of the eighteen genes containing sequences coding for FHA domain in Arabidopsis. Two T-DNA insertion mutants ddl-1 and ddl-2 exhibit a developmentally delayed pleiotropic phenotype resulting in short root and hypocotyl, altered floral organs, distorted siliques and reduced seed production [3] . Pleiotropic phenotype of ddl is reminiscent of the phenotype exhibited by mutants belonging to the microRNA biogenesis pathway [4] . Previous studies have demonstrated a reduced accumulation of small RNAs including several microRNAs in ddl knockout mutants suggesting that DDL may have a crucial function in miRNA biogenesis [5] . miRNAs are endogenous 21 - 24 nucleotides RNAs that play important functions in gene regulation at the transcriptional and post transcriptional level in eukaryotes. They function by complementary base pairing with the target mRNA and cause mRNA degradation or inhibition of protein synthesis [6] [7] . They play a crucial role in regulating gene expression in eukaryotes and are in turn regulated temporally and spatially. Earlier studies have reported that DDL may be functioning in the recruitment of pri-miRNA transcript to DCL1 by an unknown mechanism for its further processing and in stabilizing pri-miRNAs [5] .
DDL consists of 314 amino acids. Its protein structure includes an N terminal arginine (Arg)-rich domain and C-terminal Fork Head Associated (FHA) domain. The Arg-rich domain bears a putative nuclear localization signal (NLS) and a putative region for RNA binding (Figure 1). Amino acid residues 218 to 282 towards the C terminus of the protein form the FHA domain of DDL [3] . A recent paper reporting the crystal structure of DDL FHA domain suggests that DDL has a canonical FHA domain to recognize phospho-threonine molecules, and the domain is folded into a 7-stranded β-sandwich architecture containing a conserved putative phospho-threonine recognition cleft between two pairs of β strands: β2-β3 and β4-β5. This conserved cleft comprises of Arg223 and Asn273 and recognizes an acidic glutamate molecule, a phospho-threonine mimic [8] . DDL was shown to localize to the nucleus and also bind to mRNA in vitro [3] [5] . DDL potentially interacts with DCL1 as shown in a yeast two-hybrid assay and a coimmunoprecipitation assay in Nicotiana benthamiana [5] [8] .
It is not yet understood which part of DDL is important and indispensable for its function. In order to understand the function of DDL and the contribution of different domains towards its function, it is necessary to characterize the protein. A structure-function study is a powerful methodology that can identify functionally significant amino acids in different domains of a protein. The aim of this study was to conduct a structure-function analysis of the DDL protein and to identify a few key amino acids for a better understanding of the regions of DDL that are important for its function. A thorough structure-function study was carried out by phenotypic and molecular analyses of eight ddl mutants. The study isolated several relevant amino acid residues in the FHA and Arg-rich domain that are important for the function of DDL.
2. Materials and Methods
2.1. Sequence Alignments of DDL and Its Orthologs in Plants and Animals
Using NCBI protein BLAST search (http://blast.ncbi.nlm.nih.gov/), orthologs of the DDL protein were identi-
 (a)
(a) (b)
(b)
Figure 1. Structure of DDL protein. (a) Protein sequence and (b) domain structure of DDL. Its N-terminal Arginine rich domain contains the putative Nuclear Localization Signal (NLS, highlighted red in the sequence) and RNA binding region, and Fork Head Associated (FHA, highlighted green in the sequence) domain is present towards the C-terminal end.
fied. Those proteins that had sequence similarity with the entire length of DDL were selected for sequence alignment. Protein sequence of each ortholog was retrieved from NCBI and they were aligned using the software program ClustalW [9] , with gap open and extension penalties of 10 and 0.1, respectively, and with BLOSUM as the amino acid substitution matrix. Sequences for all the DDL orthologs common to both the plant and animal species were aligned, and a separate alignment was created only for the plant species. Finally, BOXSHADE (http://www.ch.embnet.org/software/BOX_form.html) was used to highlight the highly conserved amino acids among the orthologs.
2.2. Construction of a Phylogenetic Tree
Aligned sequences were used to construct two Bayesian phylogenetic trees using the software program MrBayes (version 3.1.2) [10] (http://mrbayes.sourceforge.net/). The program is used to perform Bayesian inference of phylogenetic relationships, and uses a form of the Markov Chain Monte Carlo (MCMC) algorithm. A total of 50,000 generations of MCMC simulations were run with the first 1250 trees ignored as burn-ins. Eventually, all the trees were found to converge, indicated by final standard deviation of split frequency values of 0.006465 and 0.000157, for the tree constructed from all the sequences considered together and the one developed from only the sequences from the plant species, respectively.
2.3. EMS Mutagenesis and Targeted Induced Local Lesions in Genome (TILLING) Screen
EMS mutagenesis and TILLING screen were performed at the Fred Hutchinson Cancer Center, Seattle (http://tilling.fhcrc.org/). EMS mutagenesis was performed on Columbia er-105 seeds [11] . Mutations were identified in FHA domain and Arg-rich regions of DDL in two separate TILLING screens. Seeds from a segregating population for the mutation were ordered from the Arabidopsis Stock Center (ABRC).
2.4. In Silico Analysis of TILLING Mutants
To gain a basic understanding of the impact of each mutation on the protein function, an online tool screening for non-acceptable polymorphisms (SNAP) was used (http://rostlab.org/services/snap/). Wild type and their corresponding mutant sequences were given as the input. Results of SNAP run contain a binary prediction (neutral/ non-neutral), reliability index (RI range 0 - 9; low-high), and the expected accuracy as presented Table 1 [12] .
2.5. Identifying Homozygote Mutants
DNA was extracted using the modified Dellaporta method [13] . CAPS (Cleavage Amplification Polymorphism Sequence) or the dCAPS (derived CAPS) markers were used to isolate homozygote mutants in each TILLING lines [14] . The dCAPS primers were designed using the following website:
http://helix.wustl.edu/dcaps/dcaps.html [15] . List of primers used for genotyping is documented in Table2

Table 1. Output from SNAP run.
Table 2. Primers used for genotyping of TILLING alleles.
FP: forward primer; RP: reverse primer.
2.6. Phenotypic Analyses
Two back-crosses to the wild type parent er-105 were performed to reduce the effect from background mutations before analyzing the phenotype of the mutants.
For hypocotyl and root analyses, er-105, TILLING mutant, WS-2 (Wild type for ddl-1) and ddl-1 were grown on square plates containing 1/2 MS and 10 μM GA growth media. To promote synchronized germination, one hour of red light treatment followed by 23 hours of dark treatment was performed. Following this, they were allowed to grow vertically in a growth chamber under a controlled condition of light at 20˚C. After 12 days of growth, the hypocotyl and root lengths of the mutants and the corresponding wild type plants from the same plate were measured using ImageJ software (http://rsb.info.nih.gov/ij/index.html).
Heights were measured for individual plants grown on soil for 30 days.
The number of seeds was counted using a dissecting scope. Five siliques each from the same plants grown on soil were used for this measurement.
For all these quantification, ddl-1 and WS-2 were used as positive controls by planting them each time an analysis was being conducted. One-way ANOVA test using the software program package Statistical Analysis System (SAS) version 9.2 was used to study the statistical significance of the data [16] (α = 0.05).
2.7. Molecular Analyses
Quantitative analysis of the miRNA levels in ddl TILLING alleles was performed by RNA filter hybridization [17] . miRNA was isolated from the inflorescence of different genotypes and hybridized for miR172, a DCL1 dependent miRNA. ddl-1 and WS-2 were used as positive controls. Quantification of miRNA accumulation was performed using the ImageJ software (http://rsb.info.nih.gov/ij/index.html). All results were normalized to an internal control, U6 RNA, used in the experiment.
3. Results
3.1. Identification and Sequence Alignments of DDL and Its Orthologs in Plants and Animals
Protein BLAST of DDL against reference sequences resulted in a large number of proteins with high sequence homology especially in the FHA domain region. A total of fourteen proteins with a high degree of homology and query coverage of more than 80% were selected for further analyses (Table 3).
All the fourteen proteins from both plants and animals were aligned with DDL. Five of those proteins that belonged to the plant species were used in the alignment with only plant orthologs of DDL. ClustalW alignments of the combined plant and animal orthologs of DDL, and only plant orthologs of DDL are presented in the Figure 2(a) and Figure 2(b), respectively.
3.2. Phylogenetic Tree of DDL Orthologs
The tree constructed from the DDL ortholog sequences from both the plant and animal species was found to group the plants, insects, and mammals separately in three distinct clades (Figure 3(a)). In the tree, constructed from the DDL sequences from only the plants (Figure 3(b)), two distinct clades are visible. In the first one, the two species of Arabidopsis are grouped together, in the second, the other three species are clustered, with V. vinifera forming a separate branch. The values at each node represent the Bayesian posterior probablity values. All the values are >70% indicating that the branches are very well supported.
3.3. Isolation of New ddl Alleles
Only half of the M2 TILLING population was analyzed in the first TILLING screen, which targeted the FHA
Table 3 . List of DDL orthologs.
Table 4 List of mutants isolated from TILLING screen.
DDL orthologs from various plants and animal species with high (>80%) query coverage.
 (a)
(a) (b)
(b)
Figure 2. Sequence alignment of DDL and its orthologs. A box-shade image representing sequence alignments of DDL with (a) animal and plant and (b) plant only homologs. Black shading indicates the presence of strictly conserved amino acid in half or more than half the orthologs including DDL. If the orthologs have similar amino acid sequence in the alignment, gray shading is used. Strictly conserved consensus is represented by capital letter and by small letter amino acid codes otherwise. Position of TILLING mutations is indicated in the aligned sequences of DDL and its orthologs in plants and animals.
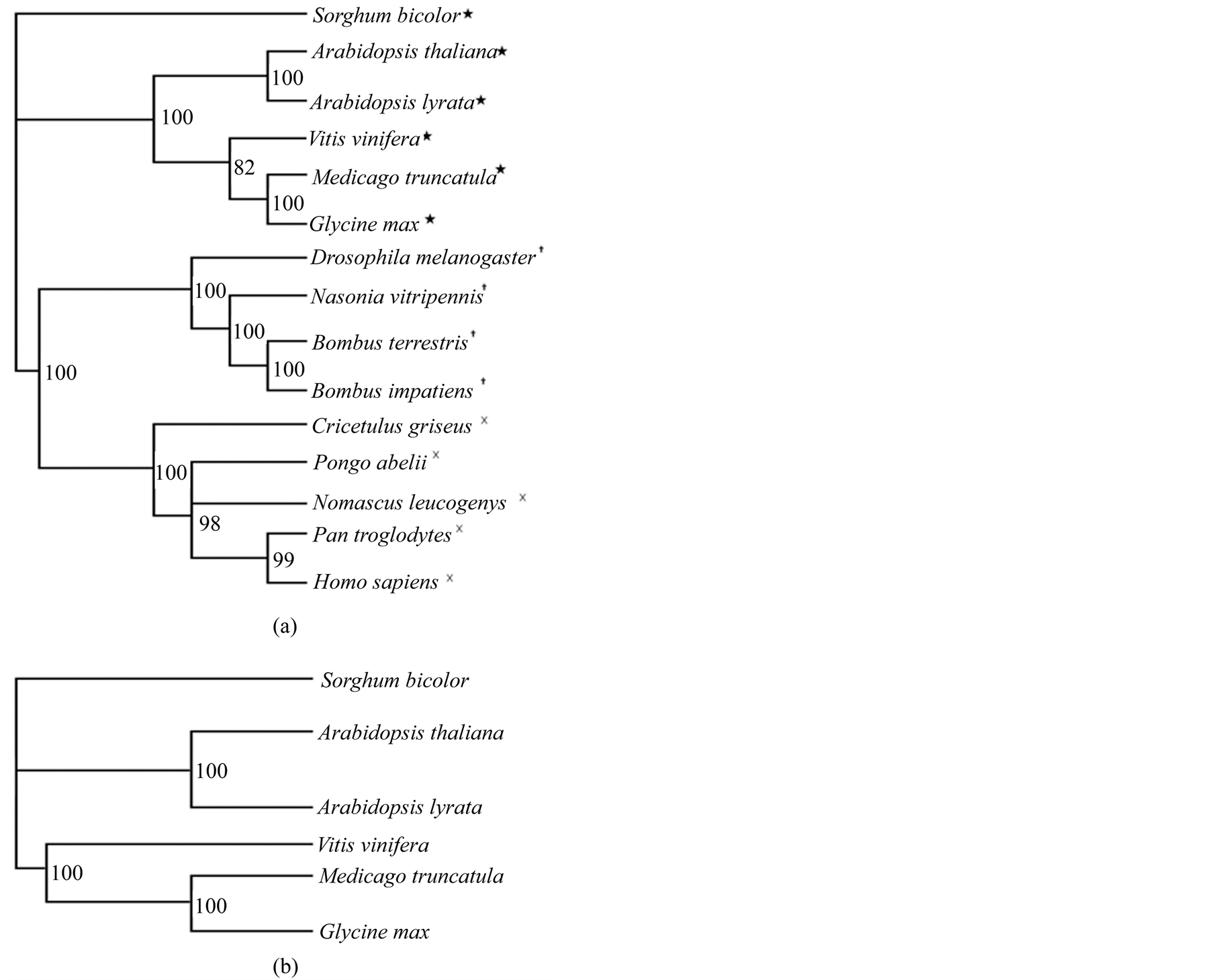
Figure 3. Phylogenetic trees built from the sequence alignment of DDL and orthologs. (a) Bayesian phylogenetic consensus tree constructed from DDL ortholog sequences showing the phylogenetic relationship between different plant (★), insect (┼), and animal species (×). (b) Bayesian phylogenetic consensus tree constructed based on DDL orthologs in various plant species. Sorghum bicolor is used as the outgroup in both the trees. The Bayesian posterior probability values are indicated at each node.
domain. This screen identified ten point mutations. Some mutations were either silent (no change in the amino acid sequence) or in intronic regions, thus not causing any change in the protein sequence. Three of the mutants that harbor a mutation in an exon and resulted in change in the DDL protein sequence were selected for further analyses. We have named these alleles ddl-3, ddl-4 and ddl-5 (Table 4).
The entire M2 TILLING population was analyzed in the second tilling screen, which targeted the argininerich region containing the putative nuclear localization signal. The screen identified twenty-six mutations of which only five harbor a mutation that maps in an exon and results in change in the DDL protein sequence. As seen in the first screen, the other mutations were either silent (no change in the amino acid sequence) or in intronic regions, thus not causing any change in the protein sequence. Mutants from the second screen are ddl-10, ddl-11, ddl-12, ddl-14 and ddl-15 (Table 4).
G222R (ddl-3) and S236F (ddl-5) mutations are harbored in the highly conserved amino acid residues in the FHA domain as seen in sequence alignment of DDL orthologs from plants and also in the combined plant and animal protein alignment. ddl-4 (P231L) mutation is also in a strictly conserved region in the orthologs from the plant sequence alignment. In the combined plant and animal ortholog alignment this amino acid is strictly conserved except in Drosophila. R45K (ddl-12) is highly conserved among plant orthologs. They also show a perfect conservation but in the insect species from the sequence alignment containing both plant and animal orthologs. V143I (ddl-15) is strictly conserved only in primates, and also aligns strictly with the ortholog in Arabi
dopsis lyrata, the closest relative of A. thaliana in the group, in the combined sequence alignment of both plant and animal orthologs. In the plant sequence alignment the region aligns with A. lyrata. However, there are amino acids residues similar to Valine (Leucine and Isoleucine) present in the region in both the alignments with the exception of insect species in the combined plant and animal sequence alignment. R43K (ddl-11) shows a weak conservation in alignment with plant orthologs (matching only with A. lyrata) and a relatively better conservation in the alignment of both animal and plant orthologs. E21K (ddl-10) and G75R (ddl-14) do not seem to belong to a conserved position in animal or plant orthologs except with A. lyrata and also in V. vinifera in case of ddl-10. All the mutations are indicated in the sequence alignment figures and presented in Figure 2(a) and Figure 2(b).
3.4. Phenotypic Analysis
After the identification of homozygote mutant following two backcrosses, the phenotype was quantified in order to determine the strength of each TILLING allele. Parameters for phenotypic analysis were selected based on the displayed phenotypes of ddl-1 and ddl-2 knockout mutants. Phenotypic analysis involved measuring the height of the plants on soil, measurement of root and hypocotyl length on vertical plates, and fertility assessment by counting the number of seeds/silique.
All parameters tested were compared to Columbia er-105 and results were subjected to one-way ANOVA analysis using SAS version 9.2 [16] (α = 0.05). ddl-1 and WS-2 plants were grown at the same time with each TILLING allele to analyze the strength of the allele and also served as a positive control. The results are shown in Figures 4(a)-(d).
ddl-3 and ddl-14 displayed a significant reduction in root length compared to er-105. ddl-4, ddl-5, ddl-10, ddl-12, and ddl-15 displayed a significant increase in root length compared to er-105. ddl-11 did not show any significant difference in root length compared to er-105.
ddl-3, ddl-10, and ddl-15 displayed significantly increased hypocotyl length and ddl-12, a significantly decreased hypocotyl length compared to er-105. Rest of the alleles did not show a significant change.
Among all the TILLING alleles that displayed an increased stem height, only ddl-14 was not significantly larger compared to er-105. Also, ddl-12 showed a significant reduction in stem length compared to er-105.
All TILLING alleles displayed a significant reduction in the average number of seeds formed per silique when compared to er-105. Among all TILLING alleles maximum reduction in seed number was observed in ddl-12 and they were reduced to 25% of WT er-105.
3.5. Molecular Analysis
The accumulation of miRNA172 in the inflorescence was tested in different TILLING alleles, er-105, ddl-1 and WS-2. ddl-3, ddl-5, ddl-14 and ddl-15 displayed a reduced accumulation of miR172 compared to er-105. A marginal increase in miRNA accumulation was seen in ddl-12. miR172 accumulation in ddl-5, ddl-10, ddl-11 did not show any considerable variation compared to er-105. Results of the miR172 accumulation analysis are depicted in Figure 5.
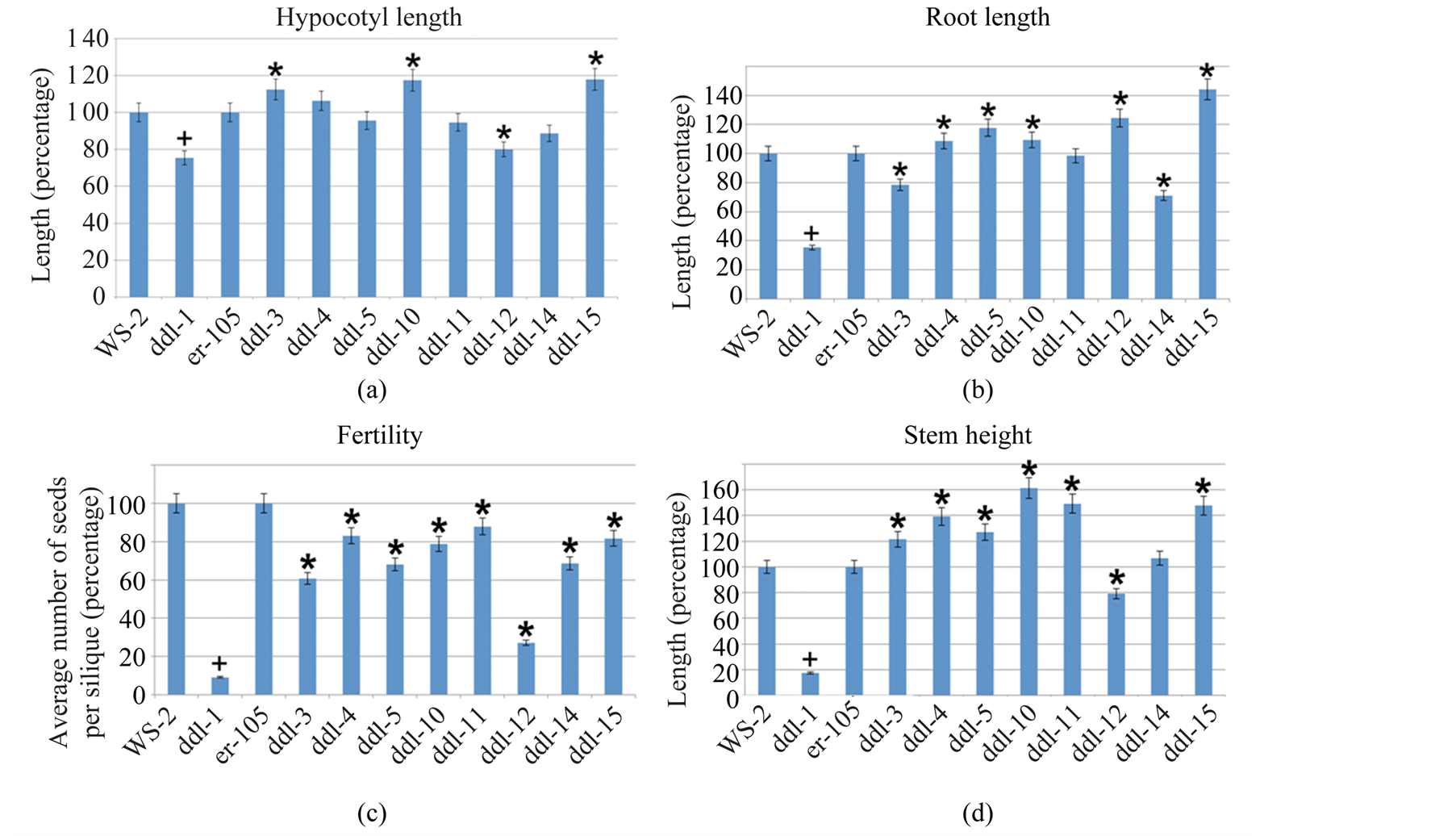
Figure 4. Phenotypic analyses of TILLING mutants. (a)-(d) Results from phenotypic analysis of hypocotyl, root, fertility and stem of TILLING alleles. Proc ANOVA procedure in Statistical Analysis Software was used for calculating the significance of the data. Twelve-day-old seedlings were used for root and hypocotyl analyses. Completely developed siliques were used to count the number of seeds. One-month-old plants were used for stem analysis. + Represents the significant difference between ddl-1 and WS-2 and * represents a significant difference between the TILLING allele and er-105. Minimum of 22 plants were used for hypocotyl and root analyses, 28 siliques were used for fertility analysis and 15 plants were used for stem height. Error bars represent 5% percentage error.
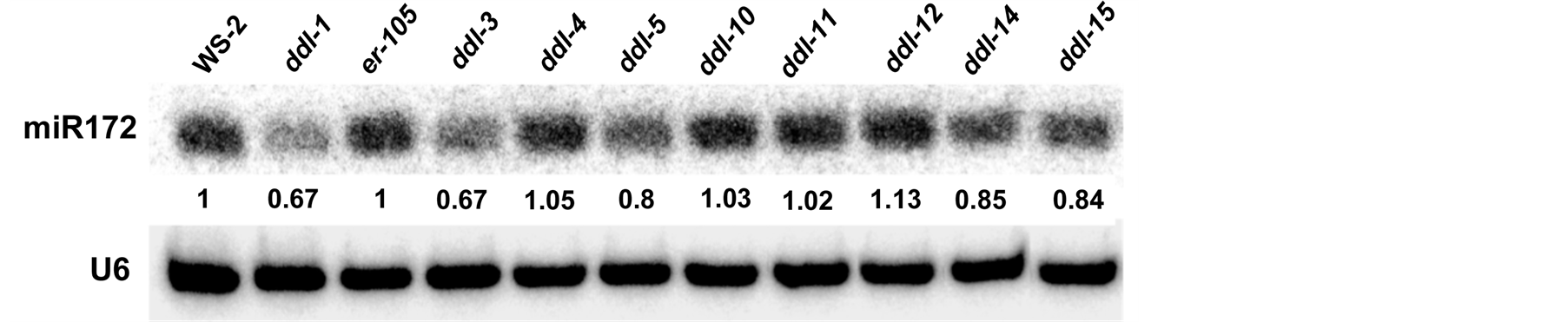
Figure 5. Small RNA blot of ddlTILLING mutants. Small RNA blot to quantify miRNA accumulation in different TILLING alleles. ddl-1 and WS-2 are used as positive controls. Quantification of miRNA accumulation was performed using the ImageJ software. The numbers represent the relative amount of miRNA present in TILLING alleles compared to er-105 and ddl-1 compared to WS-2. All results have been normalized to an internal control U6RNA used in the experiment.
4. Discussion
Structure-function study of DDL involved the characterization of eight allelic point mutants. Some of the mutations map to conserved regions of Arg-rich and FHA domain region of DDL and a few of them were differentially conserved in plants and animal orthologs. All the mutants displayed at least one of the phenotypes studied suggesting that Arg-rich region and FHA domain are required for its function. Phenotypic and molecular analyses of the mutants revealed effects resulting in ddl phenotypes of varying degrees in different organs. Table 5 summarizes results from the structure-function study of DDL. The same mutation that seems to have imparted a relatively strong ddl phenotype to one organ imparted a weak or no phenotype to some other organ. An amino acid change can result in protein misfolding and structural changes leading to alteration in activity of the protein.
In general, proteins are capable of interacting with different proteins in different tissues and organs. They in
Table 5 . Summary of phenotypic and molecular analyses.
teract with regulatory proteins that can enhance or repress its function specific to a particular organ. Amino acid changes can disrupt such interactions leading to lower or higher affinity interactions that are specific to organs or tissues. Disrupted interactions may lead to phenotypic variations depending on the function of the protein in specific regions of the organ. At the same time a large insertion, or deletion at various positions in the same protein can cause a reduced or no phenotypic effect. Hence, mutation effect is contextual based on its position in the protein sequence structure [18] .
While all the mutants had a significantly decreased fertility and most of them had a higher stem length compared to Col er-105, the effect of some of the substitutions seem to be interesting in terms of root length and microRNA level.
A mutation in G222R (ddl-3) in the FHA domain and G75R (ddl-14) in the putative NLS region significantly reduces the root length. G222R mutation is in a highly conserved region of DDL and its orthologs in plants and animals. This G222 is positioned in a crucial region adjacent and upstream of R223 that takes part in putative phospho-threonine binding. Upon mutation of R223 a reduced binding affinity to DCL1 was observed in vitro in Nicotina benthamiana [8] . G222 is also conserved in other FHA domain proteins such as KINASE ASSOCIATED PROTEIN PHOSPHATASE (KAPP) and RAD53. KAPP is a component of many receptor-like protein kinase signaling pathways. They interact with RLKs in a phosphorylation dependent manner through a kinase interacting (KI) domain that also contains an FHA domain. A mutation in this conserved G of the KAPP FHA domain prevents its binding capacity with the RLKs [19] .
G75R, although belonging to an important region, is not conserved except with its close relative Arabidopsis lyrata. While G222R results in an increased hypocotyl and stem length, G75R does not significantly increase or decrease the hypocotyl and stem length. At the molecular level, the mutations seem to decrease miR172 accumulation in the inflorescence.
miRNAs regulate early and late plant development, patterning, and differentiation. They themselves are spatially, and temporally regulated while being the master regulators of gene expression including the regulation of transcription factors [20] . Hence it is not surprising to see a pleiotropic phenotype in a mutant that has reduced expression of miRNAs.
The tested microRNA, miR172, is known to have important functions in the transition of juvenile to adult and then reproductive phases during the development of the plant. miR172 interacts with APETALA2, a floral organ identity gene, at different developmental stages of flower whorls and this is required for the development of floral identity [21] . Hence, a defective flower development due to reduced miR172 correlates with the observed reduction in seed set in G222R (ddl-3) and G75R (ddl-14) mutants. Reduced fertility in other mutants that do not show a reduction in miR172 may be due to an altered accumulation of some other miRNA that has not been tested, but may be involved in flower development.
G222R mutation is a non-neutral change and G75R is a neutral change as suggested by a web-based program, SNAP prediction [12] . However, experimentally both the mutations tend to have an effect on the plant phenotype. The nature of amino acid change from non-polar (G) to polar (R) could affect the structure of the protein. Although G is generally known to be present inside, or outside protein molecules, in this case it could be embedded within the protein and when an aliphatic polar amino acid like Arginine gets substituted, it would tend to stay on the surface of the protein, altering the protein structure and its binding partners. However, these are only speculations and require structural biology studies and functional assays such as target binding studies to prove it.
R45K (ddl-12), another residue of the NLS, is conserved in all the plant and animal orthologs except in insects. This mutation results in a significantly reduced stem length but has an opposite effect in the root. Another substitution in proximity to R45K is R43K (ddl-11) that is weakly conserved among plant orthologs and better conserved among the ones from the animals. R43K seems to increase the stem length but does not affect root length. R45K is predicted to be neutral change and R43K, a non-neutral change by the SNAP prediction software tool. Both R and K are polar basic amino acids, very similar in nature. But even a small change in size, or properties of an amino acid side chain can alter the protein function. They both display similar levels of miRNA accumulation, ddl-12 (R45K) even showing a slightly increased level by 13% compared to Col er-105.
The most striking phenotype is observed in fertility. All the mutations resulted in a significantly reduced fertility, the most dramatic phenotype observed in ddl-1 and ddl-2 knockout mutants. It is not clear if the reduced fertility is due to reduced accumulation of miRNAs or the disruption of an unknown secondary function that DDL could have had. If the reduced fertility is due to reduced accumulation of miRNAs, it requires testing of several other miRNAs functioning in flower development to correlate the phenotype at a molecular level.
A significantly higher stem length that was observed in all TILLING alleles except ddl-12 suggests that there is a disruption of protein-protein interactions in the stem.
5. Conclusion
Overall the variation in phenotypes observed for each substitution makes it very difficult to comment on the importance of different residues in relative terms. All the amino acid substitutions studied displayed at least one phenotype of knockout ddl mutant. These results suggest that the FHA, Arg-rich region and NLS are important for DDL function.
Acknowledgements
We greatly appreciate the help of Derick Reid (Department of Biological Sciences, Mississippi State University) in the phenotypic analysis of the ddl TILLING mutants.
References
- Li, J., Lee, G.I., Van Doren, S.R. and Walker, J.C. (2000) The FHA Domain Mediates Phosphoprotein Interactions. Journal of Cell Science, 113, 4143-4149.
- Mahajan, A., Yuan, C., Lee, H., Chen, E.S., Wu, P.Y., et al. (2008) Structure and Function of the Phosphothreonine-Specific FHA Domain. Science Signaling, 1, re12. http://dx.doi.org/10.1126/scisignal.151re12
- Morris, E.R., Chevalier, D. and Walker, J.C. (2006) DAWDLE, a Fork Head-Associated Domain Gene, Regulates Multiple Aspects of Plant Development. Plant Physiology, 141, 932-941. http://dx.doi.org/10.1104/pp.106.076893
- Morel, J.B., Godon, C., Mourrain, P., Béclin, C., Boutet, S., Feuerbach, F., et al. (2002) Fertile Hypomorphic ARGONAUTE (ago1) Mutants Impaired in Post-Transcriptional Gene Silencing and Virus Resistance. The Plant Cell, 14, 629-639. http://dx.doi.org/10.1105/tpc.010358
- Yu, B., Bi, L., Zheng, B., Ji, L., Chevalier, D., et al. (2008) The FHA Domain Proteins DAWDLE in Arabidopsis and SNIP1 in Humans Act in Small RNA Biogenesis. Proceedings of the National Academy of Sciences, 105, 10073-10078. http://dx.doi.org/10.1073/pnas.0804218105
- Axtell, M.J., Westholm, J.O. and Lai, E.C. (2011) Vive la Difference: Biogenesis and Evolution of microRNAs in Plants and Animals. Genome Biology, 12, 221. http://dx.doi.org/10.1186/gb-2011-12-4-221
- Voinnet, O. (2009) Origin, Biogenesis, and Activity of Plant microRNAs. Cell, 136, 669-687. http://dx.doi.org/10.1016/j.cell.2009.01.046
- Machida, S. and Yuan, A.Y. (2013) Crystal Structure of Arabidopsis thaliana Dawdle Fork Head-Associated Domain Reveals a Conserved Phospho-Threonine Recognition Cleft for Dicer-Like1 Binding. Molecular Plant, 6, 1290-1300. http://dx.doi.org/10.1093/mp/sst007
- Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Research, 22, 4673-4680. http://dx.doi.org/10.1093/nar/22.22.4673
- Huelsenbeck, J.P. and Ronquist, F. (2001) MRBAYES: Bayesian Inference of Phylogenetic Trees. Bioinformatics, 17, 754-755. http://dx.doi.org/10.1093/bioinformatics/17.8.754
- Till, B.J., Reynolds, S.H., Greene, E.A., Codomo, C.A., Enns, L.C., et al. (2003) Large-Scale Discovery of Induced Point Mutations with High-Throughput TILLING. Genome Research, 13, 524-530. http://dx.doi.org/10.1101/gr.977903
- Bromberg, Y. and Rost, B. (2007) SNAP: Predict Effect of Non-Synonymous Polymorphisms on Function. Nucleic Acids Research, 35, 3823-3835. http://dx.doi.org/10.1093/nar/gkm238
- Dellaporta, S.L., Wood, J. and Hicks, J.B. (1983) A Plant DNA Miniprepration: Version II. Plant Molecular Biology Reporter, 1, 19-21. http://dx.doi.org/10.1007/BF02712670
- Neff, M.M., Neff, J.D., Chory, J. and Pepper, A.E. (1998) dCAPS, a Simple Technique for the Genetic Analysis of Single Nucleotide Polymorphisms: Experimental Applications in Arabidopsis thaliana Genetics. The Plant Journal, 14, 387-392. http://dx.doi.org/10.1046/j.1365-313X.1998.00124.x
- Neff, M.M., Turk, E. and Kalishman, M. (2002) Web-Based Primer Design for Single Nucleotide Polymorphism Analysis. Trends in Genetics, 18, 613-615. http://dx.doi.org/10.1016/S0168-9525(02)02820-2
- SAS Institute Inc. (2008) SAS/STAT® 9.2 User’s Guide. SAS Institute Inc., Cary.
- Park, W., Li, J., Song, R., Messing, J. and Chen, X. (2002) CARPEL FACTORY, a Dicer Homolog, and HEN1, a Novel Protein, Act in microRNA Metabolism in Arabidopsis thaliana. Current Biology, 12, 1484-1495.http://dx.doi.org/10.1016/S0960-9822(02)01017-5
- Khan, S. and Vihinen, M. (2007) Spectrum of Disease-Causing Mutations in Protein Secondary Structures. BMC Structural Biology, 7, 56. http://dx.doi.org/10.1186/1472-6807-7-56
- Li, J., Smith, G.P. and Walker, J.C. (1999) Kinase Interaction Domain of Kinase-Associated Protein Phosphatase, a Phosphoprotein-Binding Domain. Proceedings of the National Academy of Sciences of the United States of America, 96, 7821-7826. http://dx.doi.org/10.1073/pnas.96.14.7821
- Válóczi, A., Várallyay, é., Kauppinen, S., Burgyán, J. and Havelda, Z. (2006) Spatio-Temporal Accumulation of microRNAs Is Highly Coordinated in Developing Plant Tissues. The Plant Journal, 47, 140-151.http://dx.doi.org/10.1111/j.1365-313X.2006.02766.x
- Zhu, Q.H. and Helliwell, C.A. (2011) Regulation of Flowering Time and Floral Patterning by miR172. Journal of Experimental Botany, 62, 487-495. http://dx.doi.org/10.1093/jxb/erq295
NOTES
![]()
*Corresponding author.