Advances in Microbiology
Vol. 2 No. 4 (2012) , Article ID: 25953 , 10 pages DOI:10.4236/aim.2012.24075
A Novel Non Radioactive PCR-DNA Probe for the Detection of Aflatoxin Producing Aspergillus Species from Major Food Crops Grown in India*
Division of Microbiology, Defense Food Research Laboratory, Siddarthanagar, India
Email: #h_v_batra@rediffmail.com
Received September 13, 2012; revised October 18, 2012; accepted October 27, 2012
Keywords: AFB1-Aflatoxin B1; HPLC-High Performance Liquid Chromatography; Nor-1; Norsolorinic Acid Synthase; PCR-Polymerase Chain Reaction
ABSTRACT
In the present study, a novel non radioactive digoxigenen labelled PCR-DNA probe was developed targeting nor-1 gene to assess the contamination of aflatoxigenic Aspergillus species in food grain samples from Southern parts of India. The sensitivity of developed PCR-DNA probe was determined to be 10 pg of genomic DNA and 1 pg of purified PCR product. The specificity of the DNA probe was validated by testing against an array of Aspergillus, Fusarium and Penicillium spp. A total of 89 Aspergillus isolates were recovered from 152 grain samples of maize, paddy, and groundnut. Among them, maize had the highest (90%) incidence of toxigenic Aspergillus species. When developed PCR-DNA probe was evaluated onto pure cultures of toxigenic and nontoxigenic Aspergillus species, 60 samples were positive for the nor-1 gene probe. DNA probe results unequivocally matched with the HPLC analysis. In conclusion, the novel PCR-DNA probe developed in this study may find its application in rapid detection of aflatoxin-producing Aspergillus isolates from contaminated cereal grains.
1. Introduction
Aflatoxins are common mycotoxins in agriculture and food commodities causing serious health hazards to humans and animals leading to great economic losses. Among the aflatoxin group produced by Aspergillus species, Aflatoxin B1 is most potent carcinogen and it was classified as group 1 carcinogen by the International Agency for Research on cancer [1]. Although aflatoxinproduction ability has been detected in various species of the Aspergillus genus, inside and outside the Flavi group, Aspergillus flavus and Aspergillus parasiticus remain the most important and representative aflatoxin producers occurring naturally in food commodities [2]. Several mycotoxicoses cases in humans and animals have been reported due to the consumption of mycotoxin contaminated food and feed [3].
Because of the toxic and potent carcinogenic properties of aflatoxins, many developed countries have established very stringent regulations limiting the maximum allowable amount of aflatoxins in food and feed [4]. Classical mycological analysis based on morphological and biochemical characters is time-consuming and not always straight-forward. Molecular techniques have also been widely applied for distinguishing aflatoxigenic and non-aflatoxigenic strains of Aspergillus by targeting one or several genes involved in the aflatoxin biosynthetic pathway with the ability/inability to produce aflatoxins. Polymerase Chain Reaction (PCR) methods have been developed for detection of aflatoxigenic fungi. In this respect, several primers have been designed for several genes in the biosynthetic pathways of aflatoxins; for example, afl, nor, omt, ord, tub, ver [5-9]. It has been demonstrated that most of the 25 identified genes clustered within a specific 70-kb region of the fungal genome are involved in aflatoxin biosynthesis [10]. Among them, nor-1 gene encodes an enzyme that catalyses the ketoreduction of norsolorinic acid (the first stable path-way intermediate) to averantin, plays a major role in aflatoxin biosynthesis [11]. A large number of probe-based methods have already been developed for the detection of various food borne bacterial pathogens and mycotoxigenic fungi [12,13]. In the present study, a novel nonradioactive digoxigenen labelled PCR-DNA probe was developed targeting nor-1 gene to assess the contamination of aflatoxigenic Aspergillus species in contaminated food grain samples. To know the incidence of toxigenic Aspergillus species and aflatoxin concentrations, a survey was conducted among the cereal grain growing regions in Southern India. Developed PCR-DNA probe was evaluated on to an array of fungal species recovered from contaminated food grains. For practical usefulness, the developed PCR-DNA probe was co evaluated with the chemical analysis of aflatoxins by HPLC.
2. Material and Methods
2.1. Fungal Strains, Media and Culture Conditions
The list of Standard strains used in the present study was given in (Table 1). Standard fungal strains were obtained from culture collection centers at the Microbial Type Culture Collection (MTCC), Chandigarh, the National Collection of Industrial Microorganisms (NCIM), Pune and the Indian Type culture Collection (ITCC), New Delhi, India. Aspergillus isolates were recovered from infected food grains (maize, paddy and groundnut) collected from different regions of Southern India including Andhra Pradesh, Kerela, Karnataka and Tamilnadu. A total of 62 maize, 50 paddy and 40 groundnut samples were subjected to standard blotter method (International Seed Testing Association. 1996) to assess the incidence of Aspergillus species and were sub cultured on to potato dextrose agar (PDA; Himedia, Mumbai, India) for 7 days at 25˚C and single spore cultures were lyophilized and maintained at 4˚C.
2.2. Molecular Detection of Aspergillus Species DNA Extraction
The DNA isolation from standard strains and grains was carried out with minor modifications of Ramana et al. [14]. The DNA was dissolved in 50 µL of 2 mM TrisEDTA and stored at –20˚C. DNA concentrations were determined using a NanoDrop-ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, USA).
2.3. Species Specific PCR Assays: Primers and Polymerase Chain Reaction (PCR) Conditions
Species specific PCR assays were carried out by using reported primers PEPO1, PEPO2 and AnF, AnR were used for specific detection of A. flavus and A. niger respectively [28,29]. Primer sequence and references were given in (Table 2).
2.4. Design of Nor-1 Gene Specific Oligonucleotide Primers and PCR Assay
One set of oligonucleotide primers were designed targeting nor-1 gene involved in aflatoxin biosynthesis for developing PCR-DNA probe. The nor-1 gene sequence was obtained from NCBI database (accession No. AY- 510451.1). Gene runner software, version 3.1 (www.generunner.com) was used to design optimal primers: NOR1 F 5’ACCGCTACGCCGGCGCTCTCGGCAC 3’ and NOR 1 R 5’GTTGGCCGCCAGCT TCGACACTCCG 3’. PCR amplification was performed in a 20 μL reaction volume with 0.5 μM each forward and reverse primers, 2 μL of 10 × PCR buffer, 1.5 mM of MgCl2, 80 μM of each dNTP’s, and 1 U of Taq polymerase (MBI Fermentas) in Eppendorf master thermal cycler (Hamburg, Germany).
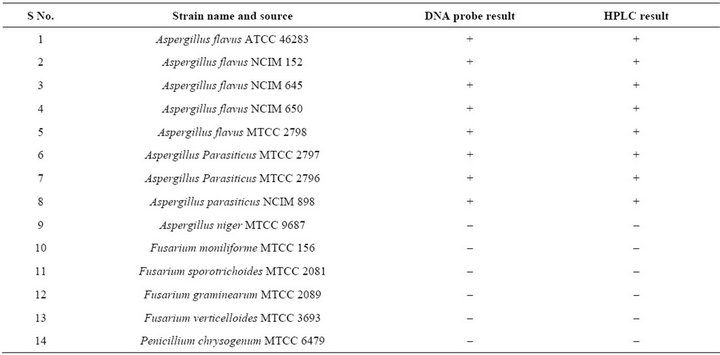
Table 1. Standard isolates used in this study.
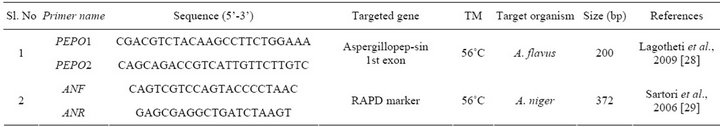
Table 2. Primers used in this study.
A standard protocol was used to perform nor-1 gene PCR. PCR program consisted of an initial denaturation of 4 min at 94˚C followed by 30 cycles denaturation at 94˚C for 40 sec, annealing at 58˚C for 1 min and extension at 72˚C for 90 seconds. A final extension of 72˚C for 5 min was given at the end of the PCR reaction. The amplified product was examined by 1% agarose gel electrophoresis under UV.
2.5. DNA Probe Preparation
The DNA probe was synthesized by PCR amplification of nor-1 gene from the A. flavus. The PCR amplicon was purified using the QIAQuick PCR purification kit (Qiagen, Germany) and labeled by randomly priming with alkali label Digoxigenin-11-dUTP nucleotide following the manufacturer’s instructions (Roche Diagnostics, India). Briefly, a 15 μL volume of purified nor-1 gene PCR amplicon was boiled for 10 min and immediately chilled in ice to obtain completely denatured strands. Two microliters of 10 × hexanucleotide mixes, 2 μL of dNTP labeling mix and 1 μL of klenow enzyme labeling grade, were mixed with the above prepared PCR product and incubated overnight at 37˚C. The reaction was stopped by heating at 65˚C for 10 min.
2.6. Blotting and DNA Hybridization
The extracted DNA of Aspergillus and other fungal strains was boiled with the addition of 0.4 M NaOH and 10 mM EDTA in a volume of 30 μL for 10 min followed by instant cooling on ice to obtain single stranded DNA. These DNA mixtures were neutralized by adding an equal volume of 2 M ammonium acetate and blotted onto positively charged Hybond N+ nylon membrane soaked in 6 × SSC using biodot vacuum manifolds (Biorad laboratories, USA). Cross linking of denatured genomic DNA was carried out using UV cross-linker (UVP). The Dot blots were pre incubated in dig easy Hyb solution with shaking for 30 min at 42˚C. Hybridization was carried out in dig easy hybridization solution (10 mL) with 50 ng/mL of dig-labeled DNA probe at 42˚C in rotating glass tubes, overnight, after overnight hybridization, stringent washes were performed. The membrane was washed twice for 5 min in 50 mL 2 × SSC, 0.1% SDS at ambient temperature (25˚C) under constant agitation. The membranes were then washed in 50 mL 0.5 × SSC, 0.1% SDS (pre-warmed to wash temperature) at 58˚C under constant agitation. After hybridization and stringent washes, the membrane was rinsed briefly (1 - 5 min) in 50 mL washing buffer and incubated for 30 min in 50 mL blocking solution then incubated for another 30 min in 50 mL antibody solution. After incubation membrane was washed twice for 15 min in 50 mL washing buffer and then equilibrated for 2 - 5 min in 50 mL detection buffer. The membrane was incubated in 50 mL freshly prepared colour substrate solution in a dark colored container with a lid in dark cabinet/room. During colour development the container was not shaken. The membranes were exposed to light for short time periods for colour development monitoring. The reaction was stopped when the desired band intensities were achieved, by washing the membrane for 5 min with distilled water and further documented.
2.7. Specificity and Sensitivity of PCR-DNA Probe
The sensitivity of PCR-DNA probe was determined directly onto the DNA prepared from genomic DNA extracted from A. flavus ATCC 46283. 10 ng of DNA was serially diluted and each dilution was analyzed by the optimized DNA hybridization assay. Also sensitivity of the PCR-DNA probe was determined against purified PCR amplicon of nor-1 gene. The highest sensitivity in both the experiments was determined as the lowest concentration of genomic DNA/nor-1 amplicon required to give a visible chromogenic signal on nylon membrane. At the same time, spore suspensions of the organism serially diluted in sterile saline was spiked on to the sterile maize grains. After 48 hrs of incubation DNA was extracted from the maize grains and subjected to dot blot hybridization.
2.8. Chemical Analysis of Aflatoxins by HPLC: Extraction and Clean-Up of Aflatoxins from Food Grains
The extraction of aflatoxins was carried out as per the procedure of Stroka et al. [15]. Immune affinity columns AflaCLEAN (India) was used to clean up the aflatoxins as per the manufacturer instructions. These elutes were used for HPLC analysis. HPLC was carried out by the method described by Ferreira et al. 2004 [16].
3. Results
3.1. Molecular Detection of Aspergillus Species
A total of 152 seed samples of maize, paddy and ground nut collected from different parts of Southern India were screened to recover Aspergillus species. Four different species of Aspergillus were recovered A. flavus (51), A. niger (15), A. oryzae (21) and A. fumigatus (2) (Figure 1). DNA from all A. flavus and A. niger isolates were isolated and subjected to PCR using the species specific PCR assays. When primer pair PEPO1and PEPO2 was used all the A. flavus strains were given single fragment of about 200 bp amplicon (Figure 2(a)), in the case of A. niger, 372 bp amplicon was obtained with the primer set AnF and AnR (Figure 2(b)). Amplification was not observed when these PCR assays were employed on other Aspergillus species viz., A. oryzae, A. fumigatus, A. ochraceous and A. carbonarious. A. oryzae and A. fumigatus were identified by monographs of pitt et al. [17].
3.2. DNA Hybridization Analysis Using Nor-1 Gene DNA Probe
DNA dot blot analysis using the nor-1 gene probe was carried out with the fungal strains listed in (Table 1). The DNA probe hybridized with DNA extracted from standard toxigenic strains of Aspergillus isolates from con-
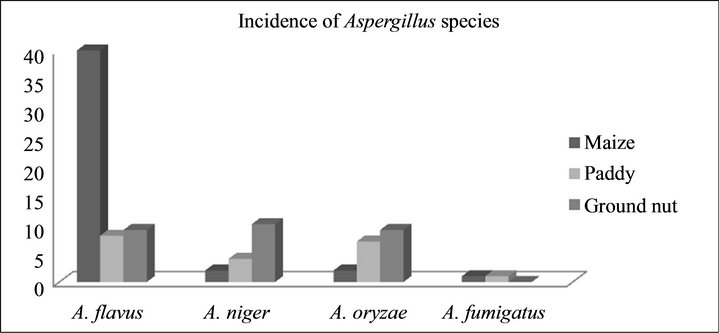
Figure 1. Incidence of Aspergillus species.
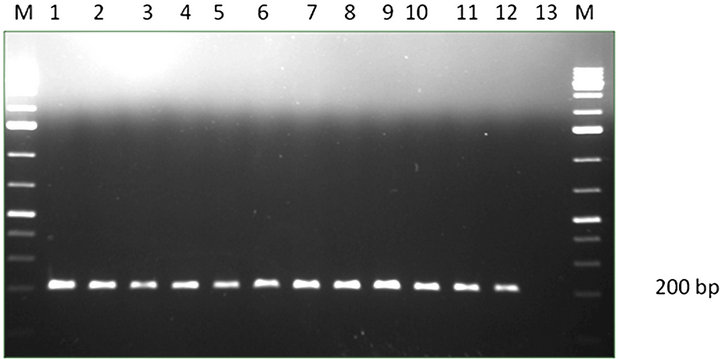 (a) Lane M-1: Kb DNA marker, lane 1-12: A. flavus strains, Lane 13: negative control.
(a) Lane M-1: Kb DNA marker, lane 1-12: A. flavus strains, Lane 13: negative control. 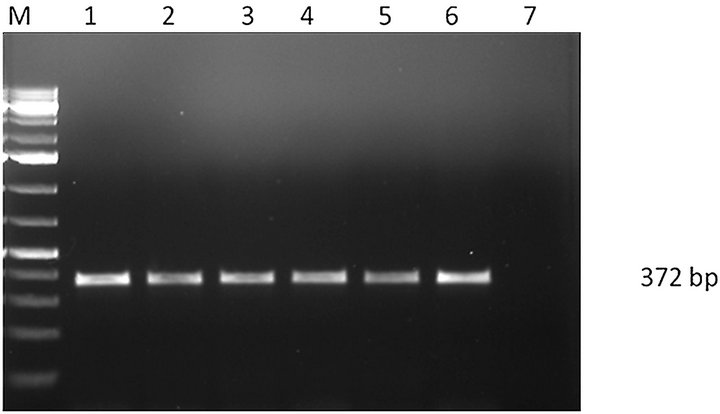 (b) Lane M-1: Kb DNA marker, lane 1-6: A. niger strains, Lane 7: negative control.
(b) Lane M-1: Kb DNA marker, lane 1-6: A. niger strains, Lane 7: negative control.
Figure 2. (a) PCR amplification of A. flavus; (b) PCR amplification of A. niger.
taminated food grains exhibited positive chromogenic signal, whereas no hybridization signal was detected with other non toxigenic Aspergillus strains including A. flavi complex and other fungal strains included in this study (Figure 3). Among the 89 isolates of Aspergillus 60 strains were found to be positive by nor-1 gene probe.
3.3. Detection Sensitivity
The sensitivity of developed PCR-DNA probe was evaluated n to various dilutions of purified PCR product (ranging from 10 ng to 0.01 pg) was achieved as low as 0.01 pg (Figure 4). The detection limit of PCR-DNA probe was estimated onto the DNA purified by conventional protocol. The PCR-DNA probe could detect 10 pg genomic DNA. The sensitivity achieved by PCR-DNA probe was 1 × 106 CFU per gram of the seed sample. The reproducibility was checked by repeating the experiment twice.
3.4. Comparative Analysis of PCR-DNA Probe with HPLC
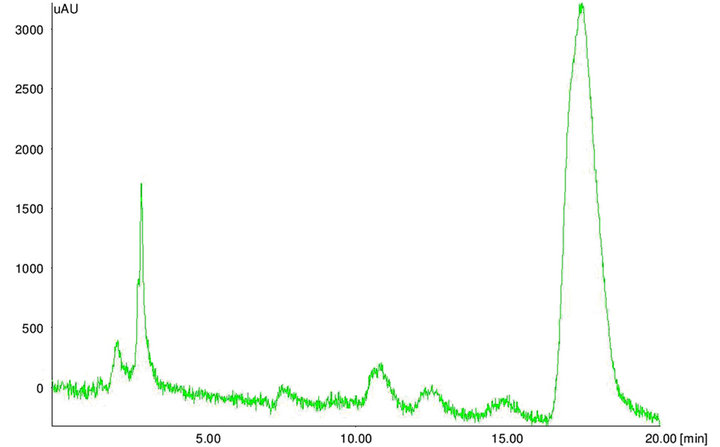
The extracted toxins were analyzed by HPLC along with 10 µg·mL–1 of Aflatoxins standards (B1, B2, G1 and G2). The toxin potentials and quantification was determined by HPLC (Figure 5). When developed PCR-DNA probe was comparatively evaluated with conventional HPLC method, all aflatoxin positive genotypes of Aspergillus stayed positive for chemical analysis by HPLC (Table 3).
4. Discussion
In the present study a non-radiactive nor-1 gene DNA probe was designed for specific detection and differentiation of aflatoxigenic and non-toxigenic Aspergillus species. The nor-1 gene targeted for probe encodes a ketoreductase that regulates the major steps in aflatoxin metabolism [18]. Several experiments were conducted to prove the effective role of this gene in aflatoxin biosynthesis [19,20]. A large number of probe-based methods have already been developed for the detection and enumeration of various food borne bacterial pathogens and also for fumonisin producing Fusarium species [12,13, 21,22]. In Aspergillus DNA probe was characterized for distinguishing strains of A. flavus [23], but so far toxigenic potential of the species has not been targeted.
Molecular detection of aflatoxigenic Aspergillus species from food and clinical samples relies mainly on PCR amplification of specific target genes of interest. PCR methods have been found useful for detection of toxigenic Aspergillus present in contaminated food samples [24]. However, in PCR assays specificity mainly rely on 20 to 25 bases primer and also the stringent practical conditions that require expertise to standardize a reliable and robust PCR prior to evaluation. Dot-blot hybridization and reverse line hybridization assays are more attractive alternatives to overcome these problems. In the case of reverse line dot blot technique, the gene specific oligonucleotide probes are bound to the filter instead of the PCR product and DNA labeled during PCR amplification is used to hybridize to immobilized array of probes and only a single sample can be identified positively for different pathogens simultaneously. In contrast, a dot blot assay can analyze many samples for the presence of a single organism, as the purified DNA/PCR products from the samples are blotted on the membrane and hybridized to a single probe specific for the particular organism. In dot blot hybridization, the specificity is also more reliable as the targeted gene is checked on its entire probe length which would prevent the false positive results [25].
Among the cereal grains examined from Southern parts of India during 2010-2012. Maize had shown highest incidence of toxigenic Aspergillus species (90%) followed by groundnut (86%) and paddy (79%). The previous reports suggested that, maize is the most susceptible crop for aflatoxin contamination in tropical regions like

Figure 3. Dot blot analysis of nor-1 gene.
Lane A1to A5: aflatoxigenic Aspergillus flavus and standard strains of Aspergillus flavus and Aspergillus parasiticus strains; A6: negative control, Lane B1-B6 isolates of Aspergillus, C1-C6: standard strains of Fusarium and Penicillium, D1: Positive control, Lane D2-D6: other fungal species including non toxigenic Aspergillus.
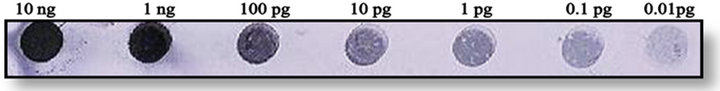
Figure 4. Detection sensitivity.
Sensitivity of PCR-DNA probe on serially diluted nor-1 PCR amplicon ranging from 75 pg to 0.5 pg of DNA (1 pg was considered as detection limit).
 (a)
(a)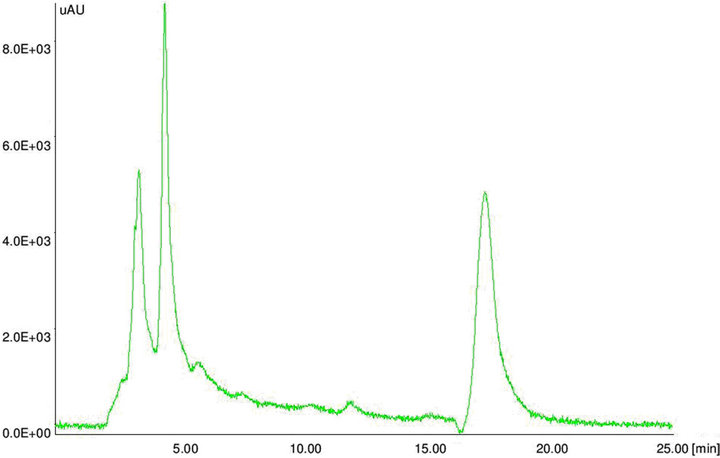 (b)
(b)
Figure 5. HPLC chromatograms. HPLC chromatogram of (a) aflatoxin standard B1 and (b) aflatoxin positive maize sample.
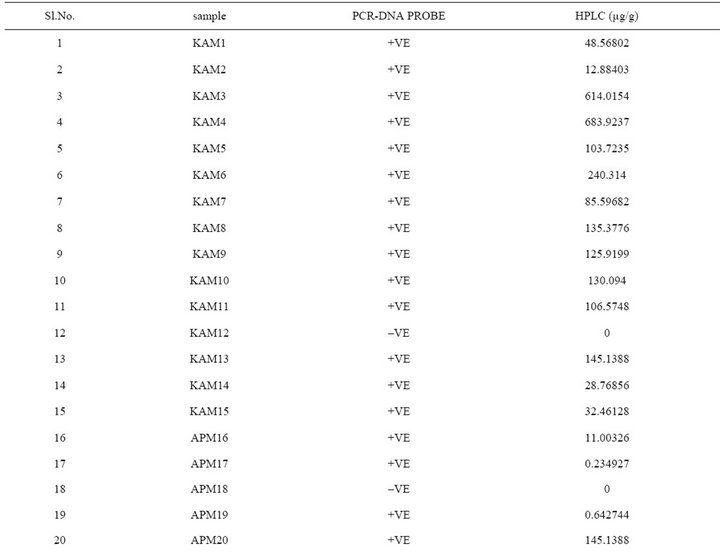

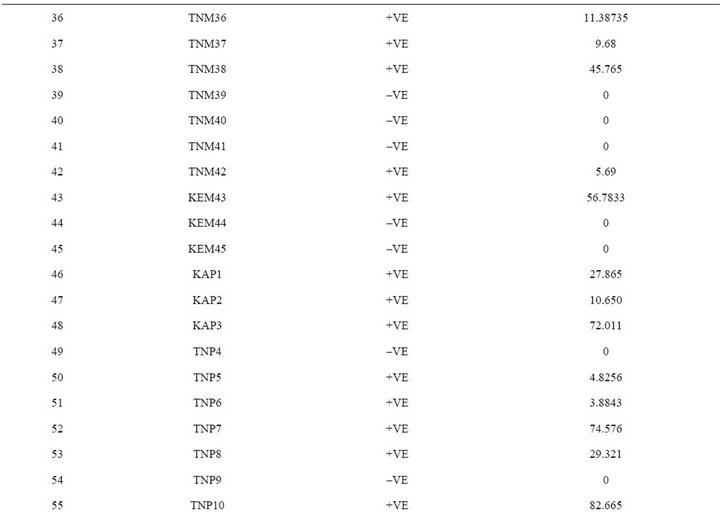

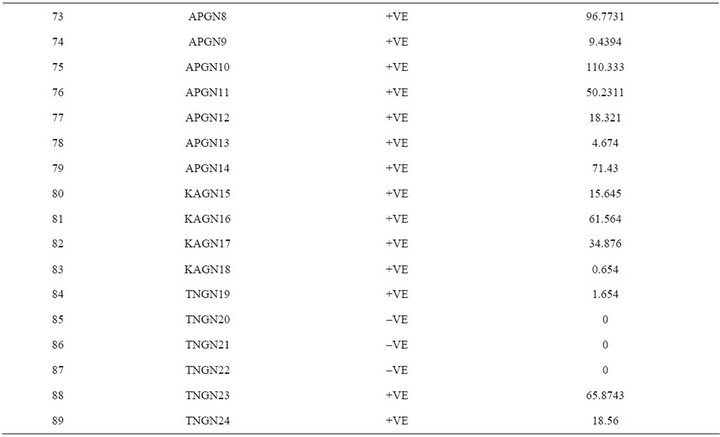
Table 3. Comparative evaluation of PCR-DNA probe with HPLC.
India [26,27]. When developed PCR-DNA probe was evaluated on all the aflatoxin positive samples as well as fungal strains were showed positive for the nor-1 gene. DNA-probe results unequivocally matched with the HPLC analysis.
In conclusion, the high levels of toxigenic strains found in many of the cereal samples warrants the need for better surveillance and monitoring by policy makers or food toxicologists to reduce the exposure of humans and animal life from aflatoxins produced by Aspergillus species. For this purpose, the designed DNA probe, besides having sensitivity and specificity equal to the molecular methods described earlier could score over others in terms of reliability, simplicity, ease of readability of results and safety in terms of avoiding carcinogenic chemicals.
5. Acknowledgements
Authors are thankful to the Director DFRL, Mysore to keen interest in present study.
REFERENCES
- IARC, “Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins,” IARC Monographs on the Evaluation of Carcinogenic Risks to Human, International Agency for Research on Cancer, Lione, Vol. 56, 1993.
- J. W. Cary and K. C. Ehrlich, “Aflatoxigenicity in Aspergillus: Molecular Genetics, Phylogenetic Relationships and Evolutionary Implications,” Mycopathologia, Vol. 162, No. 3, 2006, pp. 167-177.
- M. Peraica and A. M. Domijan, “Mycotoxins in Food and Human Health,” Archives of Industrial Hygiene and Toxicology, Vol. 52, No. 1, 2001, pp. 23-35.
- T. J. Hansen, “Quantitative Testing for Mycotoxins,” American Association Cereal Chemists, Vol. 38, 1993, pp. 346-348.
- R. Geisen, “Multiplex Polymerase Chain Reaction for the Detection of Potential Aflatoxin and Sterigmatocystin Producing Fungi,” Systematic and Applied Microbiology, Vol. 19, 1996, pp. 388-392. doi.10.1016/S0723-2020(96)80067-1
- G. Criseo, A. Bagnara and G. Bisignano, “Differentiation of Aflatoxin Producing and Non-Producing Strains of Aspergillus flavus Group,” Letters in Applied Microbiology, Vol. 33, No. 4, 2001, pp. 291-295. doi.0.1046/j.1472-765X.2001.00998
- P. Rodrigues, A. Venancio, Z. Kozakiewicz and N. Lima, “A Polyphasic Approach to the Identification of Aflatoxigenic and Non-Aflatoxigenic Strains of Aspergillus Section Flavi Isolated from Portuguese Almonds,” International Journal of Food Microbiology, Vol. 129, No. 2, 2009, pp. 187-193.
- P. Rahimi, B. Sharifnabi and M. Bahar, “Detection of Aflatoxin in Aspergillus Species Isolated from Pistachio in Iran,” Journal of Phytopathology, Vol. 156, No. 1, 2008, pp. 15-20.
- B. Scherm, M. Palomba, D. Serra, A. Marcello and Q. Migheli, “Detection of Transcripts of the Aflatoxin Genes Afld, Aflo, and Aflp by Reverse Transcription-Polymerase Chain Reaction Allows Differentiation of Aflatoxin-Producing and Non-Producing Isolates of Aspergillus Flavus and Aspergillus parasiticus,” International Journal of FoodMicrobiology, Vol. 98, No. 2, 2005, pp. 201-210.
- C. A. Townsend, “Progress towards a Biosynthetic Rationale of the Aflatoxin Pathway,” Pure and Applied Chemistry, Vol. 58, No. 2, 1997, pp. 227-238.
- F. Trail, N. Mahanti, M. Rarick, M. Mehigh, S. H. Liang, R. Zhou and J. E. Linz, “Physical and Transcriptional Map of an Aflatoxin Gene Cluster in Aspergillus parasiticus and Functional Disruption of a Gene Involved Early in the Aflatoxin Pathway,” Applied and Environmental Microbiology, Vol. 61, No. 7, 1995, pp. 2665- 2673.
- A. R. Datta, B. A. Wentz, D. Shook and M. W. Trucksess, “Synthetic Oligodeoxyribonucleotide Probes for Detection of Listeria monocytogenes,” Applied and Environmental Microbiology, Vol. 54, 1988, pp. 2933-2937.
- M. V. Ramana, S. Chandrayanaka, K. Balakrishna, H. S. Murali and H. V. Batra, “A Novel PCR-DNA Probe for the Detection of Fumonisin Producing Fusarium Species from Major Food Crops Grown in Ssouthern India” Mycology: An International Journal on Fungal Biology, Vol. 3, 2012, pp. 167-220.
- M. V. Ramana, K. Balakrishna, H. S. Murali and H. V. Batra, “Multiplex PCR-Based Strategy to Detect Contamination with Mycotoxigenic Fusarium Species in Rice and Fingermillet Collected from Southern INDIA,” Journal of the Science of Food and Agriculture, Vol. 91, No. 9, 2011, pp. 1666-1673.
- J. Stroka, E. Anklam, U. Jorissen and J. Gilbert, “Immuno-Affinity Column Clean-Up with Liquid Chromatography Post Column Bromination for Determination of Aflatoxins in Peanut Butter, Pistachio Paste, Fig Paste and Paprika Powder: Collaborative Study,” Journal of Association of Analytical Communication and Interaction, Vol. 83, No. 2, 2000, pp. 320-340.
- I. Ferreira, E. Mendes and M. Oliveira, “Quantification of Aflatoxins B1, B2, G1, and G2 in Pepper by HPLC/Fluorescence,” Journal of Liquid of Chromatography, Vol. 27, No. 2, 2004, pp. 325-334.
- J. I. Pitt, R. A. Samson and J. C. Frisvad “List of Accepted Species and Their Teleomorphs in the Family Trichocomaceae,” In: R. A. Samson and J. I. Pitt, Eds., Integration of Modern Taxonomic Methods for Penicillium and Aspergillus, Harwood Academic Publishers, Amsterdam, 2000, pp. 9-47.
- G. A. Payne and M. P. Brown, “Genetics and Physiology of Aflatoxin Biosynthesis,” Annual Review of Phytopathology, Vol. 36, 1998, pp. 329-362.
- J. Yu, J. W. Cary, D. Bhatnagar, T. E. Cleveland, N. P. Keller and F. S. Chu, “Cloning and Characterization of a cDNA from Aspergillus parasiticus Encoding an OMethyl-Transferase Involved in Aflatoxin Biosynthesis,” Applied and Environmental Microbiology, Vol. 59, No. 11, 1993, pp. 564-571.
- F. Trail, N. Mahanti, M. Rarick, M. Mehigh, S. H. Liang, R. Zhou and J. E. Linz, “Physical and Transcriptional Map of an Aflatoxin Gene Cluster in Aspergillus parasiticus and Functional Disruption of a Gene Involved Early in the Aflatoxin Pathway,” Applied and Environmental Microbiology, Vol. 61, No. 7, 1995, pp. 2665- 2673.
- G. S. Sandhu, B. Kline, L. Stockman and G. D. Roberts, “Molecular Probes for Diagnosis of Fungal Infections,” Journal of Clinical Microbiology, Vol. 33, No. 11, 1995, pp. 2913-2919.
- J. L. Jones, “DNA Probes: Application in the Food Industry, ” Trends in Food Science and Technology, Vol. 2, 1991, pp. 28-32.
- C. E. McAlpin and B. Mannarelli, “Construction and Characterization of a DNA Probe for Distinguishing Strains of Aspergillus flavus,” Applied and Environmental Microbiology, Vol. 61, No. 3, 1995, pp. 1068-1072.
- M. Z. Abdin, M. Malik and A. J. Saleem, “Advances in Molecular Detection of Aspergillus: An Update,” Archives of Microbiology, Vol. 192, No. 6, 2010, pp. 409- 425.
- C. A. Lévesque, C. E. Harlton and A. W. A. M. De-Cock, “Identification of Some Oomycetes by Reverse Dotblot Hybridization,” Phytopathology, Vol. 88, No. 9, 1998, pp. 213-222.
- G. R. Janardhana, K. A. Raveesha and H. S. Shetty, “Mycotoxin Contamination of Maize Grains Grown in Karnataka (India),” Food and Chemical Toxicology, Vol. 37, No. 8, 1999, pp. 863-868. doi.:10.1016/S0278-6915(99)00067-8
- S. V. Reddy, D. K. Mayi, M. U. Reddy, K. ThirumalaDevi and D. V. R. Reddy, “Aflatoxins B1 in Different Grades of Chilies (Capsicum annum L.) in India as Determined by Indirect Competitive ELISA,” Food Additives and Contaminants, Vol. 18, No. 6, 2001, pp. 553- 558.
- M. Logotheti, A. Kotsovili-Tseleni, G. Arsenis and N. I. Legakis, “Multiplex PCR for the Discrimination of A. fumigatus, A. flavus, A. niger and A. terreus,” Journal of Microbiological Methods, Vol. 76, 2009, pp. 209-211.
- D. Sartori, M. C. Furlaneto, M. K. Martins, M. R. F. Paula, A. Pizzirani-Kleiner, M. H. Taniwaki and M. H. P. Fungaro, “PCR Method for the Detection of Potential Ochratoxin-Producing Aspergillus Species in Coffee Beans,” Research in Microbiology, Vol. 157, No. 4, 2006, pp. 350-354. doi.org/10.1016/j.resmic.2005.09.008
NOTES
*Authors do not having any conflict of interest in present study.
#Corresponding author.