 Advances in Bioscience and Biotechnology, 2012, 3, 731-739 ABB http://dx.doi.org/10.4236/abb.2012.326094 Published Online October 2012 (http://www.SciRP.org/journal/abb/) Pentoxifylline suppressed LPS-induced inflammatory and apoptotic signaling in neuronal cells Sameer K. Muchhala, Kenza E. Benzeroual* Division of Pharmaceutical Sciences, Arnold & Marie Schwartz College of Pharmacy & Health Sciences, Long Island University, Brooklyn, USA Email: *kenza.benzeroual@liu.edu Received 16 August 2012; revised 23 September 2012; accepted 3 October 2012 ABSTRACT Several studies have reported a relation between in- creased pro-inflammatory mediators such as TNF-α and apoptosis in neurodegenerative diseases such as Alzheimer’s disease (AD). It is known that lipopoly- saccharide (LPS) treatment induces neuroinflamma- tion and memory deterioration, and it has been re- ported that LPS induces apoptosis mostly through the production of TNF-α. Pentoxifylline (PTX), is a vas- cular protective agent and a potent TNF-alpha in- hibitor. However, the molecular neuroprotective me- chanisms of PTX against LPS-induced neurotoxicity have not been well studied. In this study, we inves- tigated the direct protective effect of PTX against LPS-induced toxicity in Rat pheochromocytoma (PC12) cell line. The results showed that a pretreatment with PTX prior exposure to LPS significantly decreased LPS-induced cell death. Mechanisms study showed that PTX has the potential to inhibit pro-inflamma- tory and pro-apoptotic pathways via the suppression of TNF-α and a caspase-dependent pathway in neu- ronal PC12 cells. This is the first study to report the anti-inflammatory and anti-apoptotic effects of PTX via inhibition of TNF-α and a caspase-dependent pathway in neuronal PC12 cells. Altogether, these observations indicate that PTX is capable of promot- ing neuroprotective effects, meanwhile also present some insights into the potential signaling pathways that are involved. Thus, these findings support the potential for PTX to be investigated as a potential agent fo r the treat ment of neurod egenerativ e diseases such as AD. Keywords: Pentoxifylline; Lipopolysaccharide (LPS); Neuronal Cells; Apoptosis; Inflammatory Cytokines; Neurodegeneration; TNF-Alpha; Alzheimer’s Disease 1. INTRODUCTION Alzheimer’s disease (AD) is an irreversible and progres- sive disorder affecting regions of the brain that control memory and cognitive functions. The disease gradually results in memory loss, and the ability to communicate and carry out daily activities [1]. The three main struc- tural changes in the AD brain include extracellular de- posits of amyloid beta (Aβ) peptides known as senile plaques, intracellular deposits of hyper-phosphorylated tau proteins termed neurofibrillary tangles (NFT), and diffuse neuronal loss [2]. In addition, inflammation has been reported to occur in pathologically vulnerable re- gions of the AD brain [3]. In line with this, pro-inflame- matory mediators such as tumor necrosis factor-alpha (TNF-α) has been reported to play an important role in apoptosis and neuronal cell death [4-6], and apoptosis has been documented in AD brains [2,7] and in models of neuronal cell death utilizing various neurotoxins in- cluding lipopolysaccharide (LPS) [8,9]. LPS is an endotoxin constituent of the outer mem- brane of gram-negative bacteria, and is widely distrib- uted in the environment including in house dust [10] and various agricultural settings [11]. Studies have reported that a direct injection of LPS to the brain leads to neu- ronal death in the hippocampus [12]. This may be due to the presence of inflammatory factors as LPS administra- tion to the brain in rodents induces chronic neuroin- flammation associated with impaired hippocampal-de- pendent spatial cognitive function [13-15]. In line with this, LPS administration to mice showed altered expres- sions of genes associated with learning and memory [16]. Moreover, behavioral and biochemical studies have also shown that spatial memory [17], object recognition and long-term potentiation (LTP) [18,19], and neuronal net- work activity [15] are all impaired following acute LPS injections to models that mimic AD pathology. At the cellular level, LPS binding to the toll-like receptor (TLR4) on host cells can trigger a signaling cascade that induces the expression of inflammatory cytokines such *Corresponding author. OPEN ACCESS  S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 732 as TNF-α, and other signaling pathways such as caspases that subsequently contribute to apoptosis and neuronal death [20]. In line with this, TNF-α may directly induce neuronal apoptosis and injury and its elevated levels in AD have been documented [21,22]. In the present study, we used LPS-induced pheochromocytoma (PC12) cells, as an in vitro model of neurodegeneration to screen for neuroprotective agents. Since the neuropathology of several neurodegenerative diseases, especially AD, has been linked to mitochon- drial dysfunctions, and inflammation-induced apoptosis [21,22] there has been intense interest in screening for neuroprotective agents with anti-inflammatory and anti- apoptotic effects. Pentoxifylline (PTX), a methylated xanthine derivative, is a competitive nonselective phos- phodiesterase inhibitor used as a vascular-protecting agent to improve blood blood circulation in the arms and legs in the case of intermittent claudication, and possibly to improve cerebral blood flow in the case of vascular dementia [23]. In addition to its ability to improve microcirculation, PTX has potent anti-TNF-α action [24] which have prompted investigations into its potential efficacy in disease states such as AD, where TNF-α lev- els are elevated and may intimately be associated with neuronal death [21,22]. Evidence from clinical practices and experimental animal studies indicate that treatment with PTX improves learning, memory and brain metabo- lism [25]. Others reported that PTX inhibited kainic acid- induced cognitive impairment in mice [26]. Only few studies have shown that PTX inhibited apoptosis in non- neuronal cells [27-29]. However, its anti-inflammatory and anti-apoptotic mechanisms in LPS-induced PC12 cells, a model of phenotypic neuronal cells, have not been studied. PTX, if able to protect against inflamma- tory mediators and apoptosis, may play a protective role in neurodegenerative diseases, especially vascular di- mentia of AD. Hence, in the present study we investigated the possi- ble protective mechanisms of PTX in LPS-induced neu- rotoxicity in PC12 cells. Our results showed that PTX significantly and dose-dependently reduced cell death caused by LPS. Furthermore, PTX significantly and dose-dependently suppressed LPS-induced TNF-α ex- pression, and prevented the apoptotic mechanisms. The present result showed that PTX exerts its neuroprotection probably via the inhibition of TNF-α expression and a caspase-dependent apoptotic pathway. 2. MATERIAL AND METHODS 2.1. Materials Rat pheochromocytoma (PC12) cells, Fetal Bovine Se- rum, Horse Serum, Penicillin-Streptomycin, and Tryp- sin-EDTA were obtained from American Type Culture Collection (ATCC, USA). Mouse monoclonal primary antibodies (TNF α, β-actin, Bad, and Bcl2) and Anti- mouse IgG-HRP conjugated secondary antibody, were purchased from Santa Cruz biotechnology. Pentoxifyl- line, LPS, Dulbeccos’s Modified Eagle Medium (DMEM), PBS, DMSO, SDS, Glycine, TBE Buffer, Tris base, β- mercaptoethanol, ammonium persulfate and other chemi- cals were obtained from Sigma-Aldrich (St. Louis, MO). Caspase-3 colorimetric assay kit was obtained from Invi- trogen. 2.2. Cell Culture and Treatments Rat pheochromocytoma (PC12) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, Aldrich), supplemented with 10% horse serum, 5% fetal bovine serum and 1% Penicillin/Streptomycin in an in- cubator with a constant supply of 5% CO2 and 95% oxygen at 37˚C, and medium was changed every 2 days. Cells were separated by trypsin-EDTA, when confluent. In all experiments, cells were seeded in 12 or 96-well plates and grown to a confluence of 75% - 85%. PC12 cells were treated as follows: For MTT assay, vehicle (control cells were treated with 0.1% DMSO for 24 hr, 48 hr, and 72 hr), LPS (cells were pretreated with 0.1% DMSO for 2 hr followed with LPS treatment for 24 hr, 48 hr, and 72 hr), Pentoxifylline (PTX) (cells were treated with 1.0 mM of PTX for 24 hr, 48 hr, and 72 hr). For all other conditions, cells were pretreated with varying concentrations of PTX (0.1 mM, 0.5 mM, and 1 mM) for 2 hr prior explosure to LPS (4 g/ml) for 24 hr, 48 hr, and 72 hr. For all other experiments, cells were treated with 0.1% DMSO (vehicle for 48 hr) or PTX only (1 mM for 48 hrs). Pretreatment consisted of 0.1% DMSO (LPS condi- tion) or PTX (at varying concentrations) for 2 hr, prior exposure to LPS (4 g/ml) for 48 hr. PTX was dissolved in DMSO at a final concentration of 0.1%, and final DMSO concentration in the media was below 0.1%. 2.3. MTT Assay and Cell Viability Cell viability was determined using 3-(4,5-dimethylthi- azol-2-yl)-2,5-diph enyltetrazolium bromide (MTT) re- duction assay according to the following method. In 96 well plates, 1 × 105 PC12 cells/well were seeded and treated as described previously. MTT solution (0.5 mg/ mL) was then added in each well, and the plates were kept at 37˚C in a 5% CO2 and 95% air humidified envi- ronment for 4 hours. The medium was discarded and 100 µL of Dimethyl sulphoxide (DMSO) was added to each cell, and the plate was kept on a shaker for 15 min to dissolve the formazan crystals formed in intact cells. Micro plate reader (Bio-Rad) was used to measure the Copyright © 2012 SciRes. OPEN ACCESS 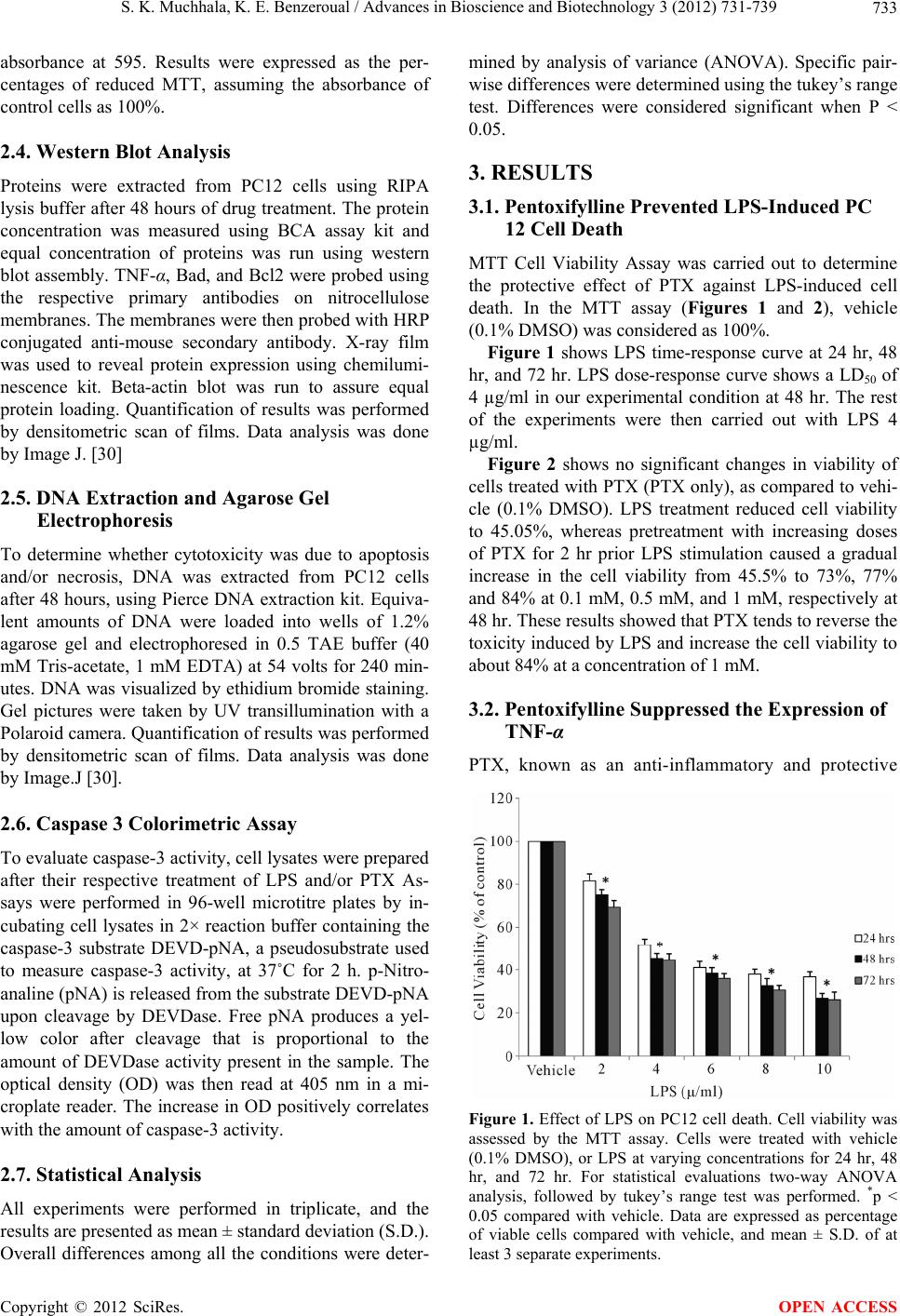 S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 733 absorbance at 595. Results were expressed as the per- centages of reduced MTT, assuming the absorbance of control cells as 100%. 2.4. Western Blot Analysis Proteins were extracted from PC12 cells using RIPA lysis buffer after 48 hours of drug treatment. The protein concentration was measured using BCA assay kit and equal concentration of proteins was run using western blot assembly. TNF-α, Bad, and Bcl2 were probed using the respective primary antibodies on nitrocellulose membranes. The membranes were then probed with HRP conjugated anti-mouse secondary antibody. X-ray film was used to reveal protein expression using chemilumi- nescence kit. Beta-actin blot was run to assure equal protein loading. Quantification of results was performed by densitometric scan of films. Data analysis was done by Image J. [30] 2.5. DNA Extraction and Agarose Gel Electrophoresis To determine whether cytotoxicity was due to apoptosis and/or necrosis, DNA was extracted from PC12 cells after 48 hours, using Pierce DNA extraction kit. Equiva- lent amounts of DNA were loaded into wells of 1.2% agarose gel and electrophoresed in 0.5 TAE buffer (40 mM Tris-acetate, 1 mM EDTA) at 54 volts for 240 min- utes. DNA was visualized by ethidium bromide staining. Gel pictures were taken by UV transillumination with a Polaroid camera. Quantification of results was performed by densitometric scan of films. Data analysis was done by Image.J [30]. 2.6. Caspase 3 Colorimetric Assay To evaluate caspase-3 activity, cell lysates were prepared after their respective treatment of LPS and/or PTX As- says were performed in 96-well microtitre plates by in- cubating cell lysates in 2× reaction buffer containing the caspase-3 substrate DEVD-pNA, a pseudosubstrate used to measure caspase-3 activity, at 37˚C for 2 h. p-Nitro- analine (pNA) is released from the substrate DEVD-pNA upon cleavage by DEVDase. Free pNA produces a yel- low color after cleavage that is proportional to the amount of DEVDase activity present in the sample. The optical density (OD) was then read at 405 nm in a mi- croplate reader. The increase in OD positively correlates with the amount of caspase-3 activity. 2.7. Statistical Analysis All experiments were performed in triplicate, and the results are presented as mean ± standard deviation (S.D.). Overall differences among all the conditions were deter- mined by analysis of variance (ANOVA). Specific pair- wise differences were determined using the tukey’s range test. Differences were considered significant when P < 0.05. 3. RESULTS 3.1. Pentoxifylline Prevented LPS-Induced PC 12 Cell Death MTT Cell Viability Assay was carried out to determine the protective effect of PTX against LPS-induced cell death. In the MTT assay (Figures 1 and 2), vehicle (0.1% DMSO) was considered as 100%. Figure 1 shows LPS time-response curve at 24 hr, 48 hr, and 72 hr. LPS dose-response curve shows a LD50 of 4 µg/ml in our experimental condition at 48 hr. The rest of the experiments were then carried out with LPS 4 µg/ml. Figure 2 shows no significant changes in viability of cells treated with PTX (PTX only), as compared to vehi- cle (0.1% DMSO). LPS treatment reduced cell viability to 45.05%, whereas pretreatment with increasing doses of PTX for 2 hr prior LPS stimulation caused a gradual increase in the cell viability from 45.5% to 73%, 77% and 84% at 0.1 mM, 0.5 mM, and 1 mM, respectively at 48 hr. These results showed that PTX tends to reverse the toxicity induced by LPS and increase the cell viability to about 84% at a concentration of 1 mM. 3.2. Pentoxifylline Suppressed the Expression of TNF-α PTX, known as an anti-inflammatory and protective Figure 1. Effect of LPS on PC12 cell death. Cell viability was assessed by the MTT assay. Cells were treated with vehicle (0.1% DMSO), or LPS at varying concentrations for 24 hr, 48 hr, and 72 hr. For statistical evaluations two-way ANOVA analysis, followed by tukey’s range test was performed. *p < 0.05 compared with vehicle. Data are expressed as percentage of viable cells compared with vehicle, and mean ± S.D. of at least 3 separate experiments. Copyright © 2012 SciRes. OPEN ACCESS  S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 734 Figure 2. Effect of Pentoxifylline on cell viability. Cell viabil- ity was assessed by the MTT assay. Cells were treated with vehicle (0.1% DMSO), or PTX (1 mM). Pretreatment consisted of 0.1% DMSO (LPS condition) or PTX at different concentra- tions (0.1 mM, 0.5 mM, and 1 mM) for 2 hrs prior to stimula- tion with LPS (4 g/ml) for 24 hr, 48 hr, and 72 hr. For sta- tistical evaluations two-way ANOVA analysis, followed by tukey’s range test was performed. *p < 0.05 compared with vehicle; **p < 0.05 compared with LPS treated cells only. Data are expressed as percentage viable cells compared with vehicle, and mean ± S.D. of at least 3 separate experiments. agent in a variety of cells, has been used in this study to confirm this property in neuronal cells. To determine the effect of PTX on LPS-induced cytokine expression, western blot assay of TNF-α expression, as an inflame- matory marker, was carried out as described under mate- rial and methods. Cells treated with LPS showed 8.3-fold increase in the expression of TNF-α as compared to ve- hicle. PTX only treated cells did not show any significant increase in the expression of TNF-α suggesting that no toxicity was imparted by the drug itself on neuronal cells. Pretreatment of cells with 0.1 mM, 0.5 mM, 1 mM of PTX, significantly reduced the LPS-induced expression of TNF-α to 4.8, 2.3, and 1.1 fold, respectively, as com- pared to LPS-treated cells (Figure 3). Over expression of TNF-α in the presence of LPS was reduced dose-depen- dently, and was even suppressed at the highest concen- tration (1 mM) of PTX. The lack of significant differ- ences in the corresponding beta-actin immunonoreactiv- ity among the groups suggests that differences in the expression of TNF-α are not attributed to loading differ- ent amount of proteins per lane. 3.3. Pentoxifylline Inhibited LPS-Induced DNA Fragmentation To examine whether apoptotic mechanism were involved in LPS-induced TNF-α mediated cell death, DNA frag- mentation method was performed as described under Section 2.5. As depicted in Figure 4, LPS induced a lad- der-like pattern on agarose gel electrophoresis, free of β-actin TNFα PTX (mM) prior LPS stimulation Vehicle LPS PTX 0.1 0.5 1 Figure 3. Effect of Pentoxifylline on the expression of TNF-α. Cells were treated with vehicle, or PTX only for 48 hr. Pre- treatment consisted of 0.1% DMSO (LPS condition) or PTX at different concentrations (0.1 mM, 0.5 mM, and 1 mM) for 2 hr prior to stimulation with LPS (4 g/ml) for 48 hrs. Quantitative data for the expression of TNF-α in all groups was performed. For each treatment the corresponding beta-actin immunoreac- tivity is reported to show equal protein loading of the lanes. For statistical evaluations two-way ANOVA analysis, followed by tukey’s range test was performed. Data are expressed as rela- tive intensity of protein expression as compare with vehicle, and mean ± S.D. of at least 3 separate experiments.*p < 0.05 compared with vehicle; **p < 0.05 compared with LPS treated cells only. Figure 4. Effect of Pentoxifylline on DNA fragmentation. Representative agarose gel electrophoresis showing DNA lad- dering of cells treated as described in Figure 3 legends. MW represents the 100-base pair DNA ladder marker. smear like pattern. This result shows that neurotoxicity induced by LPS is in the form of apoptosis. Vehicle and PTX treated cells show intact DNA which rules out drug- Copyright © 2012 SciRes. OPEN ACCESS  S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 Copyright © 2012 SciRes. 735 induced apoptosis. PTX pretreatment at a concentration of 0.1 and 0.5 mM show a decrease in DNA fragmenta- tion while PTX at a concentration of 1 mM shows an intact DNA as compared to vehicle, suggesting that the drug may exerts its protective effect through the inhibit- tion of apoptotic pathways. OPEN ACCESS highest dose used (1 mM), and increased Bcl2 levels significantly (Figure 5(b)), as compared to LPS-treated cells. These results show that pretreatment of PC 12 cells with PTX inhibited LPS-induced expression of the pro- apoptotic bad protein (Figure 5(a)) and increased the expression of the anti-apoptotic Bcl-2 protein (Figure 5(b)). PTX only treated cells did not show any signifi- cant increase in the expression of Bad Protein suggesting that PTX by itself, did not induce apoptosis in neuronal cells. Thus, PTX mediated its protective effect on PC 12 cells through its anti-apoptotic mechanism. 3.4. Pentoxifylline Reversed LPS-Induced Apoptotic Pathways Apoptosis, also known as programmed cell death, plays an important role in neurodegeneration [4]. One pro- posed mechanism is the upregulation of the pro-apoptotic Bad protein. The over-expression of Bad protein affects the permeability of mitochondrial membranes and leads to the activation of caspases which proteolyses cellular components and leads to cell death. In contrast, anti- apoptotic genes such as Bcl-2 are known to down regu- late Bad, and therefore, has the potency to inhibit the intrinsic mitochondrial pathway of apoptosis [31]. As shown in Figures 5(a) and (b), in the presence of LPS, apoptosis went along with a significant increase of in the expression of Bad protein as compared to vehicle, while the expression of Bcl-2 protein was not changed signifi- cantly. This confirms results reported by other studies investigating LPS effects on PC12 cells [32]. However, pretreatment of cells with increasing doses of PTX de- creased Bad expression and even suppressed it at the Caspases also play an important role in the induction, and amplification of apoptotic mechanisms [9,33], and previous studies reported that caspase inhibitors pre- vented LPS-induced apoptosis and cell death in PC12 cells [34]. To further confirm the protective effect of PTX on LPS-induced cell death, we measured caspase-3 levels by caspase-3 colorimetric assay. A fold increase in the absorbance at 405 nm reflects an increase in the cas- pase-3 activity that may indicate ongoing apoptosis. A decrease would reflect inhibition of apoptosis and rever- sal of cell death. Figure 6 shows that LPS treated cells exhibited a significant increase of 11 fold in absorbance as compared to vehicle and PTX only treated cells. This suggests that LPS increased caspase-3 activity in cells leading to ongoing apoptosis. Pretreatment of cells with 0.1 mM, 0.5 mM, and 1 mM of PTX dose-dependently (a) (b) Figure 5. Effect of Pentoxifylline on the expression of Bad (5A) and Bcl2 (5B) proteins. PC12 cells were pretreated with 0.1% DMSO (LPS condition) or 0.1 mM, 0.5 mM, and 1 mM of PTX for 2 hr and then exposed to LPS for 48 hr. Proteins were separated on SDS-PAGE, Western blotted, probed with anti-Bad and/or anti-Bcl-2 antibodies, and reprobed with anti-β-actin antibody (One representative Western blot was shown; n = 3). The relative intensity of corresponding bands was measured and the median of three ndependent experiments is shown. *p < 0.05 compared with vehicle; **p < 0.05 compared with LPS treated cells only. i 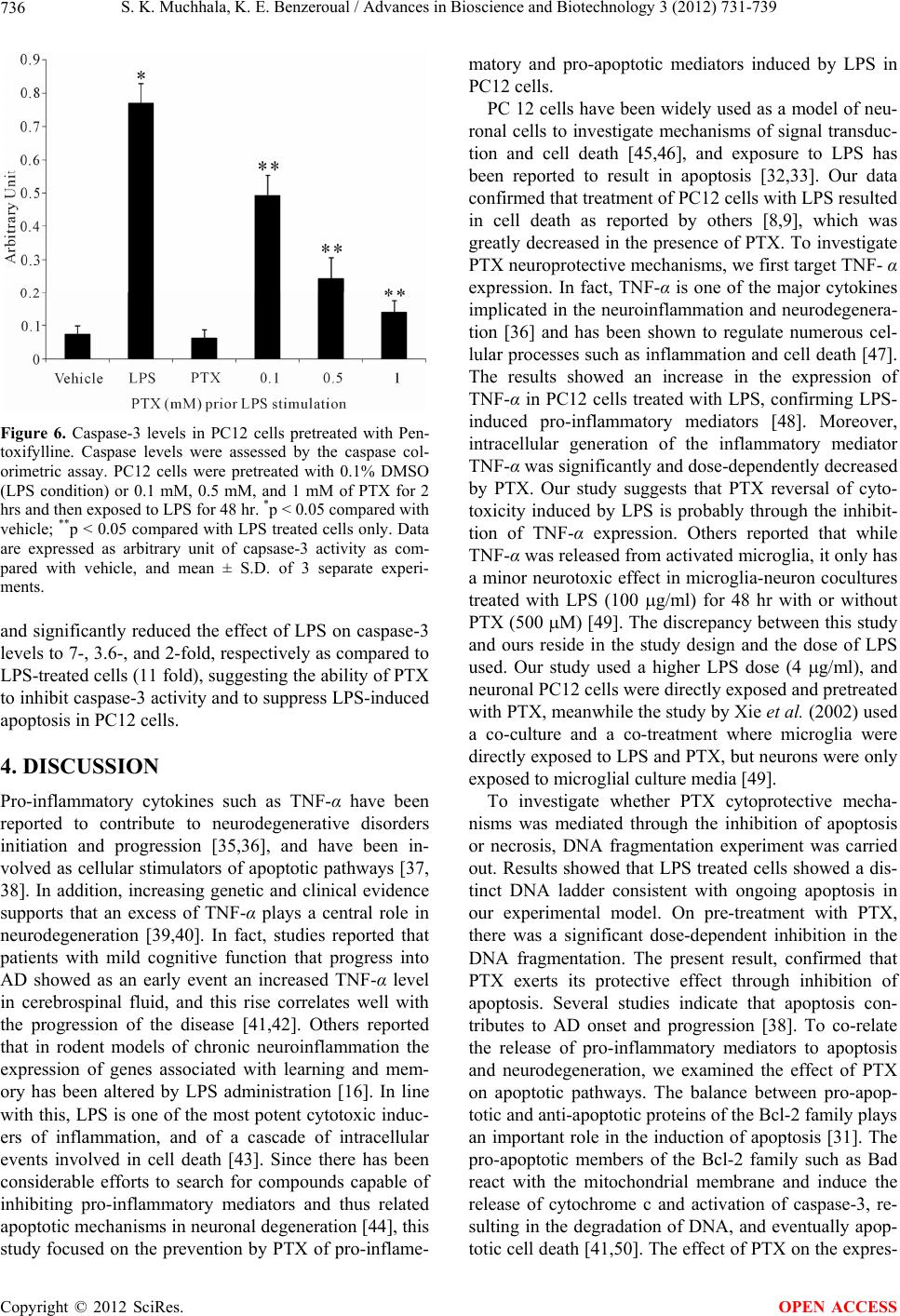 S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 736 Figure 6. Caspase-3 levels in PC12 cells pretreated with Pen- toxifylline. Caspase levels were assessed by the caspase col- orimetric assay. PC12 cells were pretreated with 0.1% DMSO (LPS condition) or 0.1 mM, 0.5 mM, and 1 mM of PTX for 2 hrs and then exposed to LPS for 48 hr. *p < 0.05 compared with vehicle; **p < 0.05 compared with LPS treated cells only. Data are expressed as arbitrary unit of capsase-3 activity as com- pared with vehicle, and mean ± S.D. of 3 separate experi- ments. and significantly reduced the effect of LPS on caspase-3 levels to 7-, 3.6-, and 2-fold, respectively as compared to LPS-treated cells (11 fold), suggesting the ability of PTX to inhibit caspase-3 activity and to suppress LPS-induced apoptosis in PC12 cells. 4. DISCUSSION Pro-inflammatory cytokines such as TNF-α have been reported to contribute to neurodegenerative disorders initiation and progression [35,36], and have been in- volved as cellular stimulators of apoptotic pathways [37, 38]. In addition, increasing genetic and clinical evidence supports that an excess of TNF-α plays a central role in neurodegeneration [39,40]. In fact, studies reported that patients with mild cognitive function that progress into AD showed as an early event an increased TNF-α level in cerebrospinal fluid, and this rise correlates well with the progression of the disease [41,42]. Others reported that in rodent models of chronic neuroinflammation the expression of genes associated with learning and mem- ory has been altered by LPS administration [16]. In line with this, LPS is one of the most potent cytotoxic induc- ers of inflammation, and of a cascade of intracellular events involved in cell death [43]. Since there has been considerable efforts to search for compounds capable of inhibiting pro-inflammatory mediators and thus related apoptotic mechanisms in neuronal degeneration [44], this study focused on the prevention by PTX of pro-inflame- matory and pro-apoptotic mediators induced by LPS in PC12 cells. PC 12 cells have been widely used as a model of neu- ronal cells to investigate mechanisms of signal transduc- tion and cell death [45,46], and exposure to LPS has been reported to result in apoptosis [32,33]. Our data confirmed that treatment of PC12 cells with LPS resulted in cell death as reported by others [8,9], which was greatly decreased in the presence of PTX. To investigate PTX neuroprotective mechanisms, we first target TNF- α expression. In fact, TNF-α is one of the major cytokines implicated in the neuroinflammation and neurodegenera- tion [36] and has been shown to regulate numerous cel- lular processes such as inflammation and cell death [47]. The results showed an increase in the expression of TNF-α in PC12 cells treated with LPS, confirming LPS- induced pro-inflammatory mediators [48]. Moreover, intracellular generation of the inflammatory mediator TNF-α was significantly and dose-dependently decreased by PTX. Our study suggests that PTX reversal of cyto- toxicity induced by LPS is probably through the inhibit- tion of TNF-α expression. Others reported that while TNF-α was released from activated microglia, it only has a minor neurotoxic effect in microglia-neuron cocultures treated with LPS (100 g/ml) for 48 hr with or without PTX (500 M) [49]. The discrepancy between this study and ours reside in the study design and the dose of LPS used. Our study used a higher LPS dose (4 g/ml), and neuronal PC12 cells were directly exposed and pretreated with PTX, meanwhile the study by Xie et al. (2002) used a co-culture and a co-treatment where microglia were directly exposed to LPS and PTX, but neurons were only exposed to microglial culture media [49]. To investigate whether PTX cytoprotective mecha- nisms was mediated through the inhibition of apoptosis or necrosis, DNA fragmentation experiment was carried out. Results showed that LPS treated cells showed a dis- tinct DNA ladder consistent with ongoing apoptosis in our experimental model. On pre-treatment with PTX, there was a significant dose-dependent inhibition in the DNA fragmentation. The present result, confirmed that PTX exerts its protective effect through inhibition of apoptosis. Several studies indicate that apoptosis con- tributes to AD onset and progression [38]. To co-relate the release of pro-inflammatory mediators to apoptosis and neurodegeneration, we examined the effect of PTX on apoptotic pathways. The balance between pro-apop- totic and anti-apoptotic proteins of the Bcl-2 family plays an important role in the induction of apoptosis [31]. The pro-apoptotic members of the Bcl-2 family such as Bad react with the mitochondrial membrane and induce the release of cytochrome c and activation of caspase-3, re- sulting in the degradation of DNA, and eventually apop- totic cell death [41,50]. The effect of PTX on the expres- Copyright © 2012 SciRes. OPEN ACCESS  S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 737 sion of pro-apoptotic protein Bad and anti-apoptotic pro- tein Bcl2 were investigated in this study. Our results showed an increase in the pro-apoptotic Bad expression on LPS treatment, indicating the ongoing apoptosis, and a significant suppression of Bad levels when treated with the highest concentration of PTX. On the other hand, PTX treated PC12 cells showed an increase in the anti- apoptotic Bcl-2 protein expression, thereby supporting an anti-apoptotic role of the drug. To confirm our anti- apoptotic hypothesis, caspase-3 colorimetric assay showed a significant increase in the caspase-3 activity in the LPS-induced cells as reported by others in neuronal PC12 cells [9], and a gradual decrease in PTX pretreated cells confirming inhibition of apoptosis and hence a neuroprotective mechanism of the drug. Considering that caspase-3 in particular, promote apoptotic cell death, it is possible that neuroprotection by PTX is achieved, par- tially by a reduction in a caspase-dependent pathway. These effects on cell death may also be partly due to the attenuating effects of PTX on TNF-α expression. In ac- cordance with our results, others have reported that PTX may induce protection against ischemia injury in the spinal cord in rabbits by preventing both necrosis and apoptosis. The study also reported a significant decrease in serum TNF-α and spinal cord caspase-3 activity in the presence of PTX [52]. Thus, confirming a protective mechanism of the drug. PTX is a vascular protective drug and also exhibits anti-inflammatory and potent anti-TNF- action [24]. In addition, the association between hemorheological changes and alterations of TNF-α has been reported and indicates a potential immunorheologic mechanism associated with cerebrovascular damages in dementia of AD, suggesting the use of vascular protective drugs such as PTX to sup- port the main pharmacological and non-pharmacological therapy of AD [53]. While PTX have been reported to produce a significant improvement in vascular dementia [54] others reached the conclusions that PTX have shown some, but partly limited benefits in vascular di- mentia patients [55]. The study reported that the possible causes of the negative results of many randomized clini- cal trials in vascular dimentia include enrollment of pa- tients with heterogeneous subtypes of vascular dimentia, the small sample size, and the use of endpoints and cog- nitive tests inadequate for vascular dimentia setting be- cause derived from previous experience in the field of AD [55]. Thus, PTX may need to be reevaluated for its neuroprotective effects in vascular dimentia of AD. In summary, our study reports a neuroprotective effect of PTX against LPS-induced PC12 cell death. PTX has been shown to exert a cytoprotecitve effect through the suppression of the expression of the pro-inflammatory mediator TNF-α, and the increase in cell viability. It also inhibited apoptosis pathways induced by LPS, by abol- ishing the pro-apoptotic Bad protein expression and in- creasing the anti-apoptotic Bcl2 protein expression. To the best of our knowledge, this is the first study to dem- onstrate the direct anti-inflammatory mechanisms of PTX in neuronal PC12 cells through inhibition of TNF-α expression and a caspase-3-dependent apoptotic pathway, suggesting potential protective effects against inflamma- tion-mediated neurodegeneration. Future works along this line will lead to a potential therapeutic use of the compound for the treatment of neurodegenerative disor- ders, especially AD. 5. ACKNOWLEDGEMENTS This work was supported by Science Research Funds from Arnold and Marie Schwartz College of Pharmacy of Long Island University. REFERENCES [1] Forlenza, O.V., Diniz, B.S. and Gattaz, W.F. (2010) Di- agnosis and biomarkers of predementia in Alzheimer’s disease. BMC Medicine, 8, 89. doi:10.1186/1741-7015-8-89 [2] Rohn, T.T., Head, E., Su, J.H., Anderson, A.J., Bahr, B.A., Cotman, C.W. and Cribbs, D.H. (2001) Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer’s disease. American Journal of Pathology, 158, 189-198. doi:10.1016/S0002-9440(10)63957-0 [3] Rubio-Perez, J.M. and Morillas-Ruiz, J.M. (2012) A re- view: Inflammatory process in Alzheimer’s disease, role of cytokines. Scientific World Journal, 756357 [4] Shimohama, S. (2000) Apoptosis in Alzheimer’s dis- ease—An update. Apoptosis, 5, 9-16. doi:10.1023/A:1009625323388 [5] McGeer, P.L. and McGeer, EG. (2001) Inflammation, autotoxicity and Alzheimer disease. Neurobiology of Aging, 22, 799-809. doi:10.1016/S0197-4580(01)00289-5 [6] Frankola, K.A., Greig. N.H., Luo. W. and Tweedie. D. (2011) Targeting TNF-α to elucidate and ameliorate neuroinflammation in neurodegenerative diseases. CNS & Neurological Disorders—Drug Targets, 10, 391-403. doi:10.2174/187152711794653751 [7] Jellinger, K.A. (2006) Challenges in neuronal apoptosis. Current Alzheimer Research, 3, 377-391. doi:10.2174/156720506778249434 [8] Huang, Y.H., Chang, A.Y., Huang, C.M., Huang, S.W. and Chan, S.H. (2002) Proteomic analysis of lipopoly- saccharide-induced apoptosis in PC12 cells. Proteomics, 2, 1220-1228. doi:10.1002/1615-9861(200209)2:9<1220::AID-PROT12 20>3.0.CO;2-3 [9] Ansari, N., Khodagholi, F., Amini, M. and Shaerzadeh, F. (2011) Attenuation of LPS-induced apoptosis in NGF- differentiated PC12 cells via NF-κB pathway and regula- tion of cellular redox status by an oxazine derivative. Biochimie, 93, 899-908. doi:10.1016/j.biochi.2011.01.012 Copyright © 2012 SciRes. OPEN ACCESS  S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 738 [10] Michel, O., Kips, J., Duchateau, J., Vertongen, F., Robert, L., Collet, H., Pauwels, R. and Sergysels, R. (1996). Se- verity of asthma is related to endotoxin in house dust. American Journal of Respiratory and Critical Care Medicine, 154, 1641-1646. [11] Kullman, G.J., Thorne, P.S., Waldron, P.F., Marx, J.J., Ault, B., Lewis, D.M., Siegel, P.D., Olenchock, S.A. and Merchant, J.A. (1998). Organic dust exposures from work in dairy barns. American Industrial Hygiene Association Journal, 59, 403-413. doi:10.1080/15428119891010668 [12] Choi, S.H., Langenbach, R. and Bosetti, F. (2007) Ge- netic deletion or pharmacological inhibition of cyclooxy- genase-1 attenuate lipopolysaccharide-induced inflam- matory response and brain injury. FASEB Journal, 22, 1491-501. doi:10.1096/fj.07-9411com [13] Rosi, S., Ramirez-Amaya, V., Vazdarjanova, A., Worley, P.F., Barnes, C.A. and Wenk, G.L. (2005) Neuroinflam- mation alters the hippocampal pattern of behaviorally in- duced Arc expression. Journal of Neuroscience, 25, 723- 731. doi:10.1523/JNEUROSCI.4469-04.2005 [14] Rosi, S., Vazdarjanova, A., Ramirez-Amaya, V., Worley, P.F., Barnes, C.A. and Wenk, G.L. (2006) Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience, 142, 1303-1315. doi:10.1016/j.neuroscience.2006.08.017 [15] Rosi, S., Ramirez-Amaya, V., Vazdarjanova, A., Esparza, E.E., Larkin, P.B., Fike, J.R., Wenk, G.L. and Barnes, C.A. (2009) Accuracy of hippocampal network activity is disrupted by neuroinflammation: Rescue by memantine. Brain, 132, 2464-2477. doi:10.1093/brain/awp148 [16] Bonow, R.H., Aïd, S., Zhang, Y., Becker, K.G. and Bosetti, F. (2009) The brain expression of genes involved in inflammatory response, the ribosome, and learning and memory is altered by centrally injected lipopolysaccha- ride in mice. Pharmacogenomics Journal, 9, 116-126. doi:10.1038/tpj.2008.15 [17] Shaw, K.N., Commins, S. and O’Mara, S.M. (2001) Lipopolysaccharide causes deficits in spatial learning in the watermaze but not in BDNF expression in the rat dentate gyrus. Behavioural Brain Research, 124, 47-54. doi:10.1016/S0166-4328(01)00232-7 [18] Hennigan, A., Trotter, C. and Kelly, A.M. (2007) Lipo- polysaccharide impairs long-term potentiation and recog- nition memory and increases p75NTR expression in the rat dentate gyrus. Brain Research, 1130, 158-166. doi:10.1016/j.brainres.2006.10.066 [19] Hauss-Wegrzyniak, B., Lynch, M.A., Vraniak, P.D. and Wenk, G.L. (2002) Chronic brain inflammation results in cell loss in the entorhinal cortex and impaired LTP in perforant path-granule cell synapses. Experimental Neu- rology, 176, 336-341. doi:10.1006/exnr.2002.7966 [20] Lee, J.W., Lee, Y.K., Yuk, D.Y., Choi, D.Y., Ban, S.B., Oh, K.W. and Hong, J.T. (2008) Neuroinflammation in- duced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. Jour- nal of Neuroinflammation, 5, 37. doi:10.1186/1742-2094-5-37 [21] Fillit, H., Ding, W.H., Buee, L., Kalman, J., Altstiel, L., Lawlor, B. and Wolf-Klein, G. (1991) Elevated circulat- ing tumor necrosis factor levels in Alzheimer’s disease. Neuroscience Letters, 129, 318-320. doi:10.1016/0304-3940(91)90490-K [22] Perry, R.T., Collins, J.S., Wiener, H., Acton, R. and Go, R.C.P. (2001) The role of TNF and its receptors in Alz- heimer’s disease. Neurobiology of Aging, 22, 87-83. doi:10.1016/S0197-4580(01)00291-3 [23] Essayan, D.M (2001) Cyclic nucleotide phosphodies- terases. Journal of Allergy and Clinical Immunology, 108, 671-680. doi:10.1067/mai.2001.119555 [24] Deree, J., Martins, J.O., Melbostad, H., Loomis, W.H. and Coimbra, R. (2008) Insights into the regulation of TNF-α production in human mononuclear cells: The ef- fects of non-specific phosphodiesterase inhibition. Clinics, 63, 321-328. doi:10.1590/S1807-59322008000300006 [25] Ukraintseva, S.V., Arbeev, K.G., Michalsky, A.I. and Yashin, A.I. (2004) Antiaging treatments have been le- gally prescribed for approximately thirty years. Annals of the New York Academy of Sciences, 1019, 64-69. doi:10.1196/annals.1297.014 [26] Bluthé, R.M., Frenois, F., Kelley, K.W. and Dantzer, R. (2005) Pentoxifylline and insulin-like growth factor-I (IGF-I) abrogate kainic acid-induced cognitive impair- ment in mice. Journal of Neuroimmunology, 169, 50-58. doi:10.1016/j.jneuroim.2005.07.017 [27] Stüber, E., Büschenfeld, A., von Freier, A., Arendt, T. and Fölsch, U.R. (1999) Intestinal crypt cell apoptosis in murine acute graft versus host disease is mediated by tu- mour necrosis factor α and not by the FasL-Fas interac- tion: Effect of pentoxifylline on the development of mu- cosal atrophy. Gut, 45, 229-235. doi:10.1136/gut.45.2.229 [28] Belloc, F., Jaloustre, C., Dumain, P., Lacombe, F., Leno- ble, M. and Boisseau, M.R. (1995) Effect of pentoxifyl- line on apoptosis of cultured cells. Journal of Cardiovas- cular Pharmacology, 25, S71-S74. doi:10.1097/00005344-199500252-00015 [29] Rüdiger, H.A. and Clavien, P.A. (2002) Tumor necrosis factor alpha, but not Fas, mediates hepatocellular apop- tosis in the murine ischemic liver. Gastroenterology, 122, 202-210. doi:10.1053/gast.2002.30304 [30] Girish, V. and Vijayalakshmi, A. (2004). Affordable im- age analysis using NIH Image/ImageJ. Indian Journal of Cancer, 41, 47. [31] Korsmeyer, S.J., Shutter, J.R., Veis, D.J., Merry, D.E., and Oltvai, Z.N. (1993) Bcl-2/Bax: A rheostat that regu- lates an anti-oxidant pathway and cell death. Seminars in Cancer Biology, 4, 327-332. [32] Sharifi, A.M., Hoda. F.E. and Noor, A.M. (2010) Study- ing the effect of LPS on cytotoxicity and apoptosis in PC12 neuronal cells: Role of Bax, Bcl-2, and Caspase-3 protein expression. Toxicology Mechanisms and Methods, 20, 316-320. doi:10.3109/15376516.2010.486420 [33] Dholakiya, S. and Benzeroual, K.E. (2011) Protective effect of Diosmin on LPS-induced apoptosis in PC12 cells and inhibition of TNF- expression. Toxicology in Vitro, 25, 1039-1044. doi:10.1016/j.tiv.2011.04.003 [34] Miller, D.K. (1997) The role of caspase family of cys- Copyright © 2012 SciRes. OPEN ACCESS  S. K. Muchhala, K. E. Benzeroual / Advances in Bioscience and Biotechnology 3 (2012) 731-739 Copyright © 2012 SciRes. 739 OPEN ACCESS teine proteases in apoptosis. Seminars in Immunology, 9, 35-49. doi:10.1006/smim.1996.0058 [35] Holmes, C., Cunningham, C., Zotova, E., Woolford, J., Dean, C., Kerr, S., Culliford, D. and Perry, V.H. (2009) Systemic inflammation and disease progression in Alz- heimer disease. Neurology, 73, 768-774. doi:10.1212/WNL.0b013e3181b6bb95 [36] Tweedie, D., Ferguson, R.A., Fishman, K., Frankola, K.A., Van Praag, H., Holloway, H.W., Luo, W., Li, Y., Caracciolo, L., Russo, I., Barlati, S., Ray, B., Lahiri, D.K., Bosetti, F., Greig, N.H. and Rosi, S. (2012) Tumor ne- crosis factor-α synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroin- flammation and Alzheimer’s disease. Journal of Neuroin- flammation, 9,106. doi:10.1186/1742-2094-9-106 [37] Uberti, D., Ferrari-Toninelli, G., Bonini, S.A., Sarnico, I., Benarese, M., Pizzi, M., Benussi, L., Ghidoni, R., Binetti, G., Spano, P., Facchetti, F. and Memo, M. (2007) Block- ade of the tumor necrosis factor-related apoptosis induc- ing ligand death receptor DR5 prevents beta-amyloid neurotoxicity. Neuropsychopharmacology, 32, 872-880. doi:10.1038/sj.npp.1301185 [38] Calissano, P., Matrone, C. and Amadoro, G. (2009) Apoptosis and in vitro Alzheimer disease neuronal mod- els. Communicative & Integrative Biology, 2, 163-169. [39] Tarkowski, E., Liljeroth, A.M., Minthon, L., Tardowski, A., Wallin, A. and Blennow, K. (2003) Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Research Bulletin, 61, 255-260. doi:10.1016/S0361-9230(03)00088-1 [40] Culpan, D., Cornish, A., Love, S., Kehoe, P.G. and Wil- cock, G.K. (2007) Protein and gene expression of tumor necrosis factor receptors I and II and their promoter gene polymorphisms in Alzheimer’s disease. Experimental Gerontology, 42, 538-544. doi:10.1016/j.exger.2006.12.001 [41] Tarkowski, E., Andreasen, N., Tarkowski, A. and Blen- now, K. (2003) Intrathecal inflammation precedes devel- opment of Alzheimer’s disease. Journal of Neurology, Neurosurgery & Psychiatry, 74, 1200-1205. doi:10.1136/jnnp.74.9.1200 [42] Alvarez, A., Cacabelos, R., Sanpedro, C., García-Fantini, M. and Aleixandre, M. (2007) Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiology of Aging, 28, 533-536. doi:10.1016/j.neurobiolaging.2006.02.012 [43] Wong, P.M.C., Chung, S.W. and Sultzer, B.M. (2000) Genes, receptors, signals and responses to lipopolysac- charide endotoxin. Scandinavian Journal of Immunology, 51, 123-127. doi:10.1046/j.1365-3083.2000.00689.x [44] Tweedie, D., Sambamurti, K. and Greig, N.H. (2007) TNF-alpha inhibition as a treatment strategy for neu- rodegenerative disorders: New drug candidates and tar- gets. Current Alzheimer Research, 4, 378-385. doi:10.2174/156720507781788873 [45] Greene, L.A. and Tischler A. (1976) Establishment of a noradrenergic cell line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proceedings of the National Academy of Sciences, 73, 2424-2428. doi:10.1073/pnas.73.7.2424 [46] Farinelli, S.E., Park D.S. and Greene L.A. (1996) Nitric oxide delays the death of trophic factor deprived PC 12 cells and sympathetic neurons by a cGMP-mediated mechanism. Journal of Neuroscience, 16, 2325-2334. [47] Park, K.M. and Bowers, W.J. (2010) Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal, 22, 977-983. doi:10.1016/j.cellsig.2010.01.010 [48] Wang, M., Chen, Y., Zhang, Y., Zhang, L., Lu, X. and Chen, Z. (2011) Mannan-binding lectin directly interacts with Toll-like receptor 4 and suppresses lipopolysaccha- ride-induced inflammatory cytokine secretion from THP- 1 cells. Cellular & Molecular Immunology, 8, 265-275. doi:10.1038/cmi.2011.1 [49] Xie, Z., Wei. M., Morgan, T.E., Fabrizio, P., Han, D., Finch, C.E. and Longo, V.D. (2002) Peroxynitrite medi- ates neurotoxicity of amyloid beta-peptide1-42-and lipo- polysaccharide-activated microglia. Journal of Neurosci- ence, 22, 3484- 3492. [50] Liu, X., Zou, H., Slaughter, C. and Wang, X. (1997). DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell, 89, 175-184. doi:10.1016/S0092-8674(00)80197-X [51] Lavrik, I.N., Golks, A. and Krammer, P.H. (2005) Cas- pases: Pharmacological manipulation of cell death. Jour- nal of Clinical Investigation, 15, 2665-2672. doi:10.1172/JCI26252 [52] Zhu, D.J., Xia, B., Bi, Q., Zhang, S.J., Qiu, B.S. and Zhao, C. (2008) Functional protection of pentoxifylline against spinal cord ischemia/reperfusion injury in rabbits: Necro- sis and apoptosis effects. Chinese Medical Journal, 121, 2444-2449. [53] Solerte, S.B., Ceresini, G., Ferrari, E. and Fioravanti, M. (2000) Hemorheological changes and overproduction of cytokines from immune cells in mild to moderate demen- tia of the Alzheimer’s type: Adverse effects on cere- bromicrovascular system. Neurobiology of Aging, 21, 271-281. doi:10.1016/S0197-4580(00)00105-6 [54] Romàn, G. (2000) Perspectives in the treatment of vascu- lar dementia. Drugs Today, 36, 641-653. [55] Pantoni, L. (2004) Treatment of vascular dementia: Evi- dence from trials with non-cholinergic drugs. Journal of the Neurological Sciences, 226, 67-70. doi:10.1016/j.jns.2004.09.014
|