 Advances in Bioscience and Biotechnology, 2012, 3, 720-730 ABB http://dx.doi.org/10.4236/abb.2012.326093 Published Online October 2012 (http://www.SciRP.org/journal/abb/) Modulation of cell death pathways in cancer stem cells: Targeting histone demethylases Ravi Thakur, Durga Prasad Mishra* Cell Death Research Laboratory, Division of Endocrinology, Central Drug Research Institute, Lucknow, India Email: *dpm@cdri.res.in Received 16 August 2012; revised 28 September 2012; accepted 4 October 2012 ABSTRACT Cancer stem cells (CSCs) are tumor initiating cells within the tumor mass; that play a critical role in can- cer pathogenesis. CSCs regulate cancer cell survival, metastatic potential, resistance to conventional radio- chemotherapy, disease relapse and poor prognosis. Recent studies have established that the drug resis- tant cancers and cancer cell lines possess high stem cell like traits compared to their drug sensitive coun- terparts. Histone demethylases are recently been linked to drug induced reversible tolerant state in cancers. Lysine histone demethylases are enzymes those demethylate lysines in histones and can act as transcriptional repressors or activators. Apart from histones other cellular proteins like E2F1, Rb, STAT3 and p53 are also regulated by methylation and de- methylation cycles. In cancer cells these enzymes regulate cell survival, migration, invasion, and pro- liferation. This review summarizes the current pro- gress of research on the role of histone demethylases in support ing drug to lerant canc er stem cell state and their pote ntial as a drug target. Keywords: Cancer Stem Cells; Histone Lysine Demethylases; Cell Death; LSD1; KDM5A 1. INTRODUCTION Cancer stem cells (CSCs) represent a small population of cells existing within the tumor mass and possess many characteristics of normal stem cells [1]. They can divide indefinitely, renew themselves and differentiate into various tumor forming cells. The existence of CSCs has long been debated but now presence of such rare popula- tion of cells is well documented in various cancer types [1,2]. To date, the presence of cancer stem cells has been reported in acute and chronic myeloid leukemia, brain, head and neck, lung, breast, pancreatic, gastrointestinal, colon, prostate and skin cancers [3-15]. Standard radio- therapy and chemotherapy is effective against most of the cancer cells but cancer stem cells are highly resistant to these therapies and remain viable post treatment [15- 17]. Among other cancer cells these cells are present as a very small population though, they are believed to induce tumor relapse, sometimes many years after the “success- ful” treatment of the primary tumors [18-22]. There are many ways by which a cancer stem cell escapes cell death. The differentiation status of a cell is an important intrinsic factor while components of microenvironment such as secreted survival factors, adhesion-mediated apoptosis resistance and hypoxic environment are im- portant extrinsic factors regulating CSC survival [23-29]. CSCs are also responsible for the enhanced migratory and metastatic potential of cancer cells [2]. During me- tastasis cancer cell detaches itself from cancer mass and resists cell death activation after detachment, a process known as anoikis [30-33]. Epithelial to mesenchymal transition (EMT) is an important step in metastasis of various cancers; it is characterized by transition of a cell to a much invasive, elongated mesenchymal form from a less invasive epithelial form [2]. Recent evidences indi- cates that CSC state, chemo-resistance and EMT path- ways are somehow linked and activation of one induces other one and vice-versa [34-39]. The acquired drug tol- erant stem cell state in many cancers was found reverse- ble once drug treatment is discontinued, indicating the role of epigenetic mechanisms in chemo-resistance and disease relapse. Various pharmacological inhibitors of epigenetic modifiers like histone deacetylases (HDACs), DNA methyltransferase (DNMTs), have shown promis- ing results in cancer therapy [40,41]. Recently, a group of epigenetic modifiers known as histone demethylases have been the focus o f in tense investig ation for th eir role in carcinogenesis [42]. Amongst this group, lysine histone demethylases are protein lysine demethylases, which act on histone and non-histone proteins [43,44]. Major function of this group of enzymes is to mediate epigenetic regulation of gene transcription at the chromatin level [44]. Histone de- methylases have important role in various cellular proc- *Corresponding a uthor. OPEN ACCESS 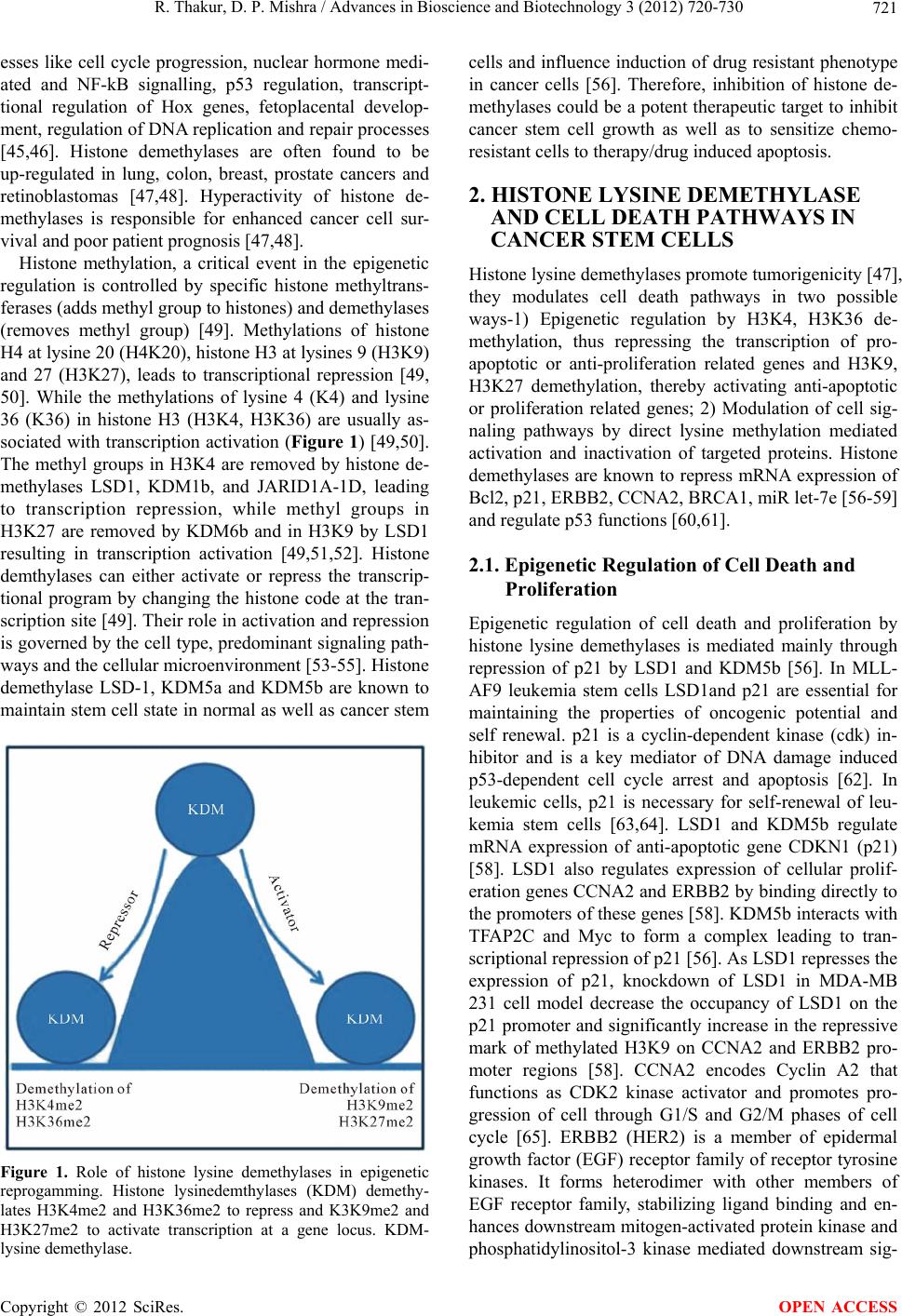 R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 721 esses like cell cycle progression, nuclear hormone medi- ated and NF-kB signalling, p53 regulation, transcript- tional regulation of Hox genes, fetoplacental develop- ment, regulation of DNA replication and repair processes [45,46]. Histone demethylases are often found to be up-regulated in lung, colon, breast, prostate cancers and retinoblastomas [47,48]. Hyperactivity of histone de- methylases is responsible for enhanced cancer cell sur- vival and poor patient prognosis [47,48]. Histone methylation, a critical event in the epigenetic regulation is controlled by specific histone methyltrans- ferases (adds methyl group to histones) and demethylases (removes methyl group) [49]. Methylations of histone H4 at lysine 20 (H4K20), histone H3 at lysines 9 (H3K9) and 27 (H3K27), leads to transcriptional repression [49, 50]. While the methylations of lysine 4 (K4) and lysine 36 (K36) in histone H3 (H3K4, H3K36) are usually as- sociated with transcription activation (Figure 1) [49,50]. The methyl groups in H3K4 are removed by histone de- methylases LSD1, KDM1b, and JARID1A-1D, leading to transcription repression, while methyl groups in H3K27 are removed by KDM6b and in H3K9 by LSD1 resulting in transcription activation [49,51,52]. Histone demthylases can either activate or repress the transcrip- tional program by changing the histone code at the tran- scription site [49]. Their role in activation and rep ression is governed by the cell type, predominant signaling path- ways and the cellular microenviron ment [53-55 ]. Histone demethylase LSD-1, KDM5a and KDM5b are known to maintain stem cell state in normal as well as cancer stem Figure 1. Role of histone lysine demethylases in epigenetic reprogamming. Histone lysinedemthylases (KDM) demethy- lates H3K4me2 and H3K36me2 to repress and K3K9me2 and H3K27me2 to activate transcription at a gene locus. KDM- lysine demethylase. cells and influence inductio n of drug resistant phenotype in cancer cells [56]. Therefore, inhibition of histone de- methylases could be a potent therap eutic target to inhibit cancer stem cell growth as well as to sensitize chemo- resistant cells to therapy/drug induced apoptosis. 2. HISTONE LYSINE DEMETHYLASE AND CELL DEATH PATHWAYS IN CANCER STEM CELLS Histone lysine demethylases promote tumorigenicity [47], they modulates cell death pathways in two possible ways-1) Epigenetic regulation by H3K4, H3K36 de- methylation, thus repressing the transcription of pro- apoptotic or anti-proliferation related genes and H3K9, H3K27 demethylation, thereby activating anti-apoptotic or proliferation related genes; 2) Modulation of cell sig- naling pathways by direct lysine methylation mediated activation and inactivation of targeted proteins. Histone demethylases are known to repress mRNA expression of Bcl2, p21, ERBB2, CCNA2, BRCA1, miR let-7e [56-59] and regulate p53 functions [60,61]. 2.1. Epigenetic Regulation of Cell Death and Proliferation Epigenetic regulation of cell death and proliferation by histone lysine demethylases is mediated mainly through repression of p21 by LSD1 and KDM5b [56]. In MLL- AF9 leukemia stem cells LSD1and p21 are essential for maintaining the properties of oncogenic potential and self renewal. p21 is a cyclin-dependent kinase (cdk) in- hibitor and is a key mediator of DNA damage induced p53-dependent cell cycle arrest and apoptosis [62]. In leukemic cells, p21 is necessary for self-renewal of leu- kemia stem cells [63,64]. LSD1 and KDM5b regulate mRNA expression of anti-apoptotic gene CDKN1 (p21) [58]. LSD1 also regulates expression of cellular prolif- eration genes CCNA2 and ERBB2 by b inding directly to the promoters of these genes [58]. KDM5b interacts with TFAP2C and Myc to form a complex leading to tran- scriptional repression of p21 [56]. As LSD1 represses the expression of p21, knockdown of LSD1 in MDA-MB 231 cell model decrease the occupancy of LSD1 on the p21 promoter and significantly increase in the repressive mark of methylated H3K9 on CCNA2 and ERBB2 pro- moter regions [58]. CCNA2 encodes Cyclin A2 that functions as CDK2 kinase activator and promotes pro- gression of cell through G1/S and G2/M phases of cell cycle [65]. ERBB2 (HER2) is a member of epidermal growth factor (EGF) receptor family of receptor tyrosine kinases. It forms heterodimer with other members of EGF receptor family, stabilizing ligand binding and en- hances downstream mitogen-activated protein kinase and phosphatidylinositol-3 kinase mediated downstream sig- Copyright © 2012 SciRes. OPEN ACCESS  R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 722 naling pathways [66]. Over-expression of cyclin A2 and ERBB2 corresponds to a drug resistant or aggressive phenotype of tumor cells [67,68]. LSD1 can also be linked to the aberrant regulation of Wnt signaling pathways in cancer cells. Wnt signaling is important to maintain cancer stem cell state in various cancers [69]. Treatment of colon cancer cells with LSD1 oligoamine inhibitor SL111144 led to increases in H3K4Me3, restoring expression of secreted frizzled- related proteins 2 (SFRP2) [70]. SFRP2 ia a Wnt signal- ing pathway antagonist and it enhances the expression of the epithelial marker E-cadherin, through inhibition of the expression of SLUG, TWIST and SNAIL [71]. SNAIL, SLUG and Twist are transcription factors involved in the epithelial mesenchymal transition (EMT) program [71]. KDM6b act on H3K27 and is responsible for activation of anti-apoptotic gene Bcl2 transcription in hormone dependent breast cancers [57]. Apart from normal anti- apoptotic functions Bcl2 is thought to be involved in resistance to conventional cancer therapies, suggesting role of decreased apopto sis may play a role in the devel- opment of cancer [72]. KDM5A-mediated H3K4 de- methylase activity plays an importan t role in maintaining the proliferative capacity of breast cancer cells through repression of tumor suppressor genes, including BRCA1 [58]. Another histone lysine demethylase JARID1B leads to repression of let-7e which then increases expression of cyclin D1 [59]. Cyclin D1 is a target gene of mir let-7e mediated gene regulation. JARID1B demethylase con- tributes to tumor cell proliferation throu gh the epigenetic repression of a tumor suppressor miR let-7 that has been reported to be a direct regulator of RAS expression in human cells [59,73]. In lung cancer patient samples, ex- pression of RAS and let-7 showed reciprocal pattern, which has low let-7 and high RAS in cancerous cells, and high let-7 and low RAS in normal cells [74]. Other targets of let-7 are some oncogenes like high mobility group A2 (HMGA2) and MYC [74,75]. Histone lysine demethylase mediated epigenetic gene regulation thus can drive tumorigenisis in cancers and inhibit program- med cell death to support cancer stem cells state. 2.2. Non-Epigenetic Regulation of Cell Death and Proliferation Non-epigenetic regulation by lysine histone demethy- lases is mediated by their potential to demethylate vari- ous cellular proteins [76]. E2F1-p53 axis is the major target of non-epigenetic regulation of cell death and pro- liferation [60,61]. p53 transcriptional activity is neces- sary to inhibit cancer stem cells growth and proliferation [60]. Histone lysine-specific demethylase LSD1 interacts with p53 to repress p53-mediated transcriptional active- tion and to inhibit p53 mediated apoptosis [60,61]. LSD1 removes both mono and di-methylation at K370 of p53 [60]. Mono-methylation K370me1 represses p53 func- tion and prevents interaction of p53 with TP53BP1 (p53- binding protein 1), thus represses p53-mediated tran- scriptional activation [60,61] In p53 negative cells (p53-/-) LSD1 removes methylation mark from E2F1 at lysine- 185 [60,61,77]. Lysine-185 methylation leads to E2F1 accumulation during DNA damage and activation of its pro-apoptotic target genes p73 and Bim (Figure 2) [78]. E2F1 promotes DNA damage-induced apoptosis in p53 dependent as well as p53 independent manner [60,77]. LSD1 mediated demethylation leads to dysregulation of the E2F1 function and pro motes survival in many cancer cells [77]. 2.3. KDM5A (JARID1A) and KDM1A (LSD1) Support Drug Tolerant Cancer Stem Cell State Apart from specific mechanism and targets involved in histone lysine demethylase mediated regulation of cell proliferation and apoptosis, various studies link lysine histone demethylases to cancer drug tolerant stem cell state [56,79-81]. The Settleman lab generated erlotinib resistant versions of PC9 lung cancer cells by exposing these cells to media containing increasing concentrations of erlotinib [79]. This drug tolerant state could be re- versed after withdrawing the drug, suggesting a epige- netic link behind this reversible erlotinib resistance [79]. In the erlotinib resistant population of PC9 cells, K DM5A expression was increased, leading to decreased global levels of H3K4me3 and H3K4me2 (Figure 3) [79]. Figure 2. General role of lysine histone demethylases in main- taining cancer stem cell state. Cancer stem cells over-express histone lysine demethylases and low expression is associated with differentiated state. KDM-lysine demethylase. Copyright © 2012 SciRes. OPEN ACCESS 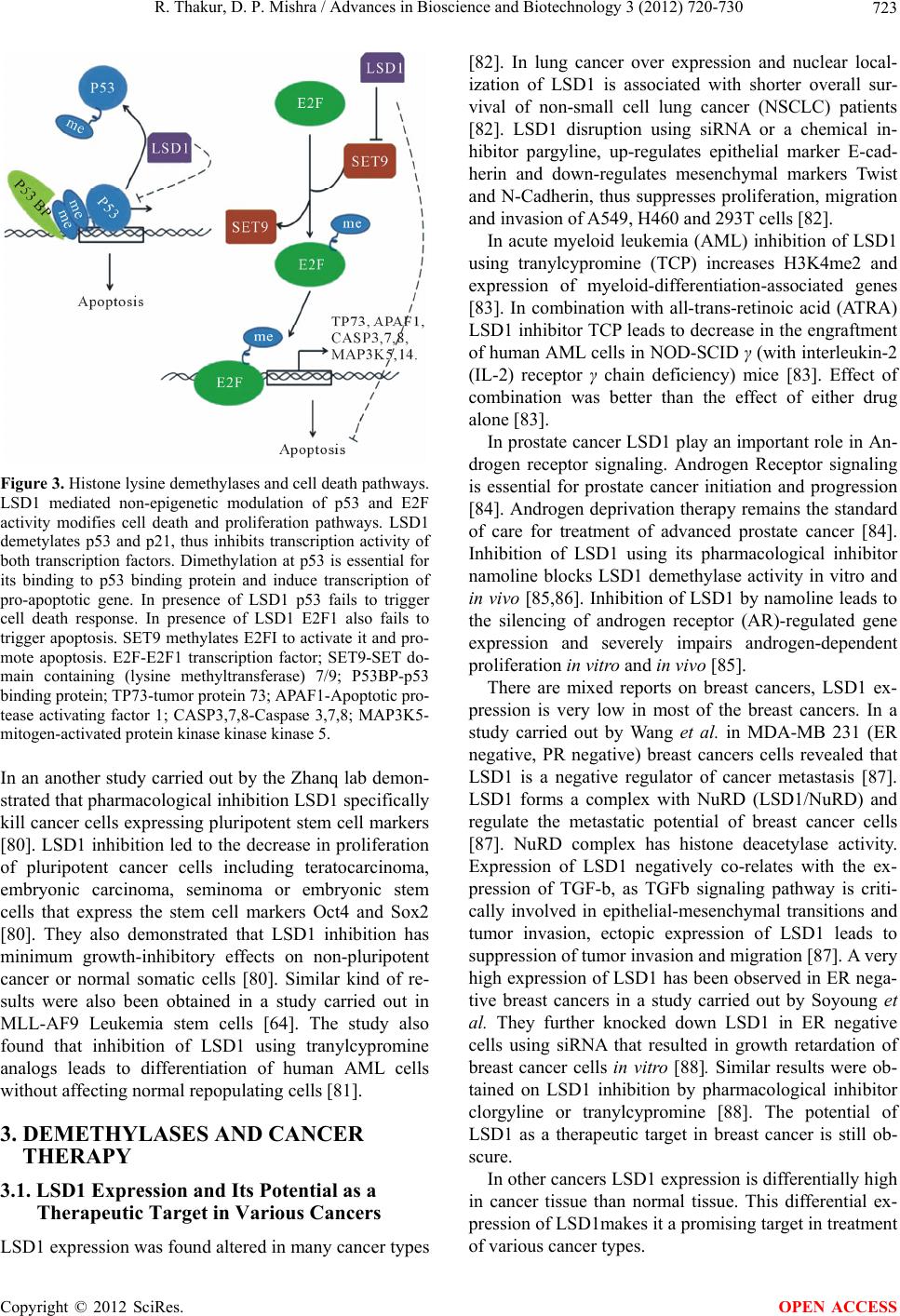 R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 723 Figure 3. Histone lysine demethylases and cell death pathways. LSD1 mediated non-epigenetic modulation of p53 and E2F activity modifies cell death and proliferation pathways. LSD1 demetylates p53 and p21, thus inhibits transcription activity of both transcription factors. Dimethylation at p53 is essential for its binding to p53 binding protein and induce transcription of pro-apoptotic gene. In presence of LSD1 p53 fails to trigger cell death response. In presence of LSD1 E2F1 also fails to trigger apoptosis. SET9 methylates E2FI to activate it and pro- mote apoptosis. E2F-E2F1 transcription factor; SET9-SET do- main containing (lysine methyltransferase) 7/9; P53BP-p53 binding protein; TP73-tumor protein 73; APAF1-Apoptotic pro- tease activating factor 1; CASP3,7,8-Caspase 3,7,8; MAP3K5- mitogen-activated protein kinase kinase kinase 5. In an another study carried out by the Zhanq lab demon- strated that pharmacological inhibition LSD1 specifically kill cancer cells expressing plu ripotent stem cell markers [80]. LSD1 inhibition led to the decrease in p roliferation of pluripotent cancer cells including teratocarcinoma, embryonic carcinoma, seminoma or embryonic stem cells that express the stem cell markers Oct4 and Sox2 [80]. They also demonstrated that LSD1 inhibition has minimum growth-inhibitory effects on non-pluripotent cancer or normal somatic cells [80]. Similar kind of re- sults were also been obtained in a study carried out in MLL-AF9 Leukemia stem cells [64]. The study also found that inhibition of LSD1 using tranylcypromine analogs leads to differentiation of human AML cells without affecting normal repopulating cells [81]. 3. DEMETHYLASES AND CANCER THERAPY 3.1. LSD1 Expression and Its Potential as a Therapeutic Target in Various Cancers LSD1 expression was found altered in many cancer types [82]. In lung cancer over expression and nuclear local- ization of LSD1 is associated with shorter overall sur- vival of non-small cell lung cancer (NSCLC) patients [82]. LSD1 disruption using siRNA or a chemical in- hibitor pargyline, up-regulates epithelial marker E-cad- herin and down-regulates mesenchymal markers Twist and N-Cadherin, thus suppresses proliferation, migration and invasion of A549, H460 and 293T cells [82]. In acute myeloid leukemia (AML) inhibition of LSD1 using tranylcypromine (TCP) increases H3K4me2 and expression of myeloid-differentiation-associated genes [83]. In combination with all-trans-retinoic acid (ATRA) LSD1 inhibitor TCP leads to decrease in the engraftment of human AML cells in NOD-SCID γ (with interleukin-2 (IL-2) receptor γ chain deficiency) mice [83]. Effect of combination was better than the effect of either drug alone [83] . In prostate cancer LSD1 play an important role in An- drogen receptor signaling. Androgen Receptor signaling is essential for prostate cancer initiation and progression [84]. Androgen deprivation therapy remains the standard of care for treatment of advanced prostate cancer [84]. Inhibition of LSD1 using its pharmacological inhibitor namoline blocks LSD1 demethylase activity in vitro and in vivo [85,86]. Inhibition of LSD1 b y namoline leads to the silencing of androgen receptor (AR)-regulated gene expression and severely impairs androgen-dependent proliferation in vitro and in vivo [85]. There are mixed reports on breast cancers, LSD1 ex- pression is very low in most of the breast cancers. In a study carried out by Wang et al. in MDA-MB 231 (ER negative, PR negative) breast cancers cells revealed that LSD1 is a negative regulator of cancer metastasis [87]. LSD1 forms a complex with NuRD (LSD1/NuRD) and regulate the metastatic potential of breast cancer cells [87]. NuRD complex has histone deacetylase activity. Expression of LSD1 negatively co-relates with the ex- pression of TGF-b, as TGFb signaling pathway is criti- cally involved in epithelial-mesenchymal transitions and tumor invasion, ectopic expression of LSD1 leads to suppression of tum or inv asion and m igration [8 7]. A v ery high expression of LSD 1 has been observed in ER nega- tive breast cancers in a study carried out by Soyoung et al. They further knocked down LSD1 in ER negative cells using siRNA that resulted in growth retardation of breast cancer cells in vitro [88]. Similar results were ob- tained on LSD1 inhibition by pharmacological inhibitor clorgyline or tranylcypromine [88]. The potential of LSD1 as a therapeutic target in breast cancer is still ob- scure. In other cancers LSD1 expression is differentially high in cancer tissue than normal tissue. This differential ex- pression of LSD1makes it a promising target in treatment of various cancer types. Copyright © 2012 SciRes. OPEN ACCESS 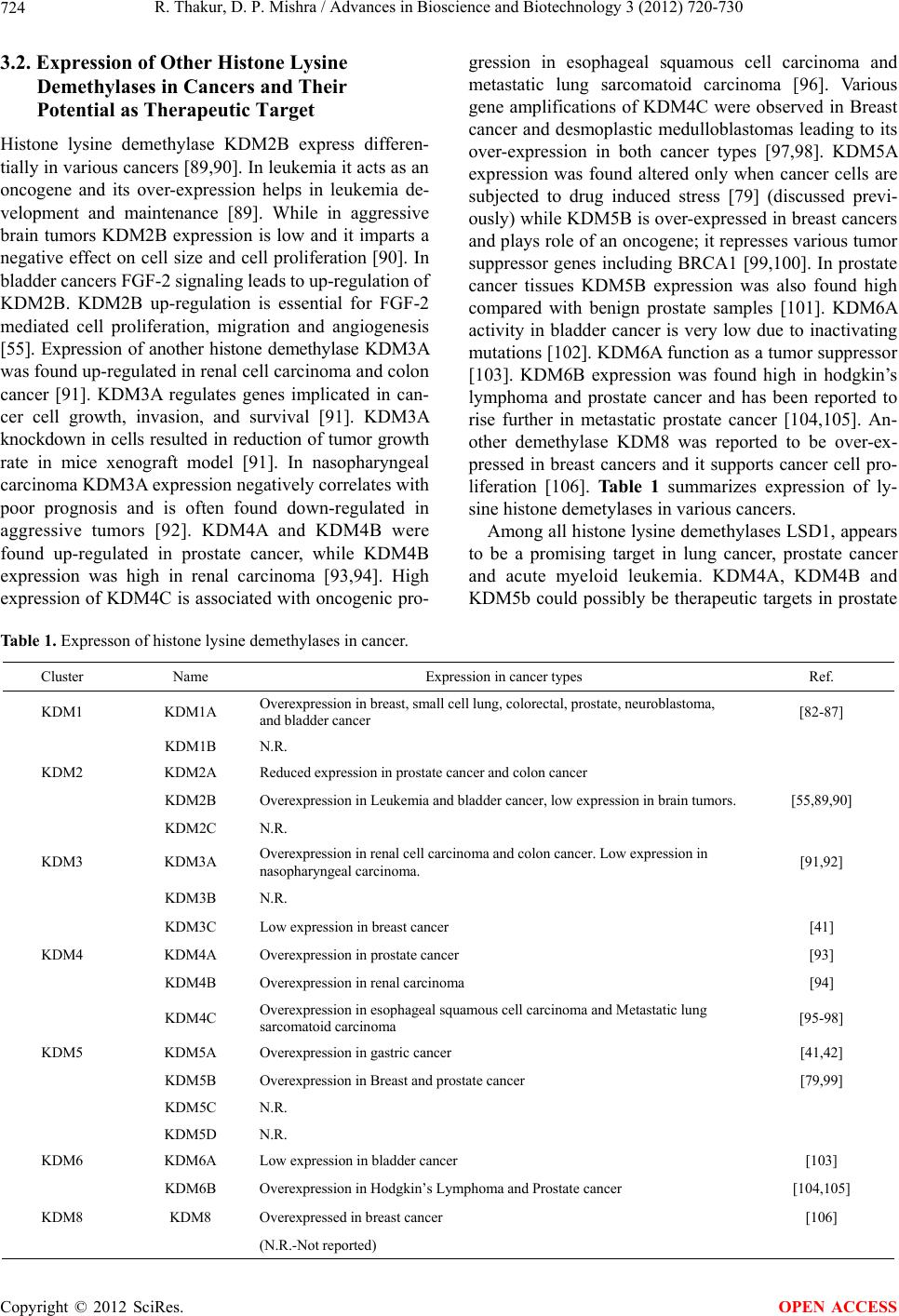 R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 Copyright © 2012 SciRes. 724 OPEN ACCESS 3.2. Expression of Other Histone Lysine Demethylases in Cancers and Their Potential as Therapeutic Target Histone lysine demethylase KDM2B express differen- tially in various cancers [89,90]. In leukemia it acts as an oncogene and its over-expression helps in leukemia de- velopment and maintenance [89]. While in aggressive brain tumors KDM2B expression is low and it imparts a negative effect on cell size and cell proliferation [90]. In bladder cancers FGF-2 signaling leads to up-regulation of KDM2B. KDM2B up-regulation is essential for FGF-2 mediated cell proliferation, migration and angiogenesis [55]. Expression of another histone demethylase KDM3A was found up-regulated in r enal cell carcinoma and colon cancer [91]. KDM3A regulates genes implicated in can- cer cell growth, invasion, and survival [91]. KDM3A knockdown in cells resulted in reduction of tumor growth rate in mice xenograft model [91]. In nasopharyngeal carcinoma KDM3A expression negatively correlates with poor prognosis and is often found down-regulated in aggressive tumors [92]. KDM4A and KDM4B were found up-regulated in prostate cancer, while KDM4B expression was high in renal carcinoma [93,94]. High expression of KDM4C is associated with oncogenic pro- gression in esophageal squamous cell carcinoma and metastatic lung sarcomatoid carcinoma [96]. Various gene amplifications of KDM4C were observed in Breast cancer and desmoplastic medulloblastomas leading to its over-expression in both cancer types [97,98]. KDM5A expression was found altered only when cancer cells are subjected to drug induced stress [79] (discussed previ- ously) while KDM5B is over-expressed in breast cancers and plays role of an oncogene; it represses various tumor suppressor genes including BRCA1 [99,100]. In prostate cancer tissues KDM5B expression was also found high compared with benign prostate samples [101]. KDM6A activity in bladder cancer is very low due to inactivating mutations [102]. KD M6A function as a tumor suppresso r [103]. KDM6B expression was found high in hodgkin’s lymphoma and prostate cancer and has been reported to rise further in metastatic prostate cancer [104,105]. An- other demethylase KDM8 was reported to be over-ex- pressed in breast cancers and it supports cancer cell pro- liferation [106]. Table 1 summarizes expression of ly- sine histone demetylases in various cancers. Among all histone lysine demethylases LSD1, appears to be a promising target in lung cancer, prostate cancer and acute myeloid leukemia. KDM4A, KDM4B and KDM5b could possibly be therapeutic targets in prostate Table 1. Expresson of histone lysine demethylases in cancer. Cluster Name Expression in cancer types Ref. KDM1 KDM1A Overexpression in breast, small cell lung, colorectal, prostate, neuroblastoma, and bladder cancer [82-87] KDM1B N.R. KDM2 KDM2A Reduced expression in prostate cancer and colon cancer KDM2B Overexpression in Leukemia and bladder cancer, low expression in brain tumors. [55,89,90] KDM2C N.R. KDM3 KDM3A Overexpression in renal cell carcinoma and colon cancer. Low expression in nasopharyngeal carcinoma. [91,92] KDM3B N.R. KDM3C Low expression in breast cancer [41] KDM4 KDM4A Overexpression in prostate cancer [93] KDM4B Overexpression in renal carcinoma [94] KDM4C Overexpression in esophageal squamous cell car cinoma and Metastatic lung sarcomatoid carcinoma [95-98] KDM5 KDM5A Overexpression in gastric cancer [41,42] KDM5B Overexpression in Breast and prostate cancer [79,99] KDM5C N.R. KDM5D N.R. KDM6 KDM6A Low expression in bladder cancer [103] KDM6B Overexpression in Hodgkin’s Lymphoma and Prostate cancer [104,105] KDM8 KDM8 Overexpressed in breast cancer [106] (N.R.-Not reported)  R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 725 cancers, while in case of breast cancer KDM8 and KDM4C as possible targets seem promising. 4. LIMITATIONS Histone lysine demethylases were discovered in last decade and their role in cancer and other biological func- tion came into limelight only in the last 5 years. Histone methylation modulates the structure and function of chromatin [49]. The balance between the methylation and demethylation of specific histone residues at gene locus is critical for regulating gene expression. Still much is not known about the targets of these enzymes, also their role in various cancers is yet to be delineated. Current literature limits only to their poten tial role in few malignancies and in some cancer models to drug tolerant cancer stem cell state. Prior to the therapeutic targeting, their precise role in initiation, progression and spread/ metastasis must be d eciphered. Th erefore more thorough insight is required to their biology in cancer and cancer stem cells. 5. FUTURE PROSPECTIVES Histone demethylases work as erasures of histone code and they also regulate function of various proteins by lysine demethylation [43,44]. This group of enzyme is known to regulate various cancer hallmarks; they par- ticipate in self renewal, differentiation, anti-apoptotic, migratory, and angiogenic pathways [45,46]. Histone deacetylases another group of epigenetic enzymes func- tion as erasures of lysine acetyl group at histones [107]. HDACs are also known to modulate various cancer hallmarks and thus are an established group of therapeu- tic targets [107]. Like HDACs, demthylases also modu- late functions and expression o f versatile targets and this makes them a good choice for development of cancer therapeutics. There are various HDAC inhibitors cur- rently under cancer clinical trials [107,108]. HDACs removes acetyl group from histones and make DNA more tightly packed around histones, and thus leads to transcription repression (similar to demethylases) of genes present at target site [108]. Like histone demethy- lases, HDACs also modulate proteins non-epigenetically; p53, E2F1 and STAT3 are common targets [108]. His- tone demethylase inhibitors can be used in combination with the HDAC inhibitors in cancer where both HDAC and demethylase activities are high. HDAC inhibition usually leads to E2F1 and p53 activation and STAT3 inactivation [108]. Therefore combination of both could be an effective way to treat cancers. A recent study using HDAC inhibitor vorinostat in combination with LSD1 inhibitor PCI-24781, reported that the combination in- duced a synergistic apoptotic cell death in glioblastoma multiforme cells [109]. A similar study in breast cancer used HDAC inhibitor in combination with LSD1 and LSD2 inhibitors, and found a synergistic effect only in HDAC-LSD1 inhibitor combination [110]. Few specula- tions can be made out of these studies: 1) HDAC inhibit- tors and Lysine demethylase inhibitors could result in to a synergestic combination; and 2) Probably not all combi- nation will be highly effective. KDM5A inhibitors could also be used in combination with HDAC inhibitors or with standard therapeutic agents to sensitize cancer cells to death. As h istone lysine demethylases LSD1 and KDM5A play an important role in cancer stem cell state and chemo-resistance their in- hibitors could be a promising therapy [79-81]. Histone lysine demethylases inhibitors could be used in combination with inhibitors of the pathways upregu- lated in cancer and responsible for cancer stem cell state and chemoresistance. Inhibitors of various cancer and cancer stem cell related pathways, notch, receptor tyro- sine kinase, hedgehog, wnt-β-catenin and mTOR can be used in combination with each other and also with the inhibitors o f hi st one demethylases [111-114]. 6. CONCLUSION The limited numbers of studies on histone lysine de- methylases so far are suggestive of their potential as an attractive drug target in cancer. Histone lysine demethy- lases in some cases act as oncogenes and support cancer growth and progression, while other cases act as tumor suppressors and inhibit tumor growth. Most important aspect of their function relate to cancer cell quiescence or maintenance of cancer stem cell state and drug resistance. Combination of histone demethylase inhibitors with tra- ditional cancer therapy holds a huge potential for treat- ment of various malignancies. However a thorough mechanistic understanding of the basic biology of his- tone lysine demethylases is imperative in gain ing a better idea about their function in normal cellular processes as well as carcinogenesis. 7. ACKNOWLEDGEMENTS We wish to apologize to all colleagues whose work, because of lack of space, could not be cited. We thank all the members of the DP Mishra laboratory for helpful discussions. This work was supported by the grants from the Defense Research and Development Organization (GAP-0058) to DP Mishra. Ravi Thakur acknowledges the support by the junior research fellowship from the Council of Scientific and In- dustrial Research, New Delhi. The Communication Number of this manuscript is 8325. REFERENCES [1] Nguyen, L.V., Vanner, R., Dirks, P. and Eaves, C.J. (2012) Cancer stem cells: An evolving concept. Nature Copyright © 2012 SciRes. OPEN ACCESS  R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 726 Reviews Cancer, 12, 133-143. [2] Baccelli, I. and Trumpp, A. (2012) The evolving concept of cancer and metastasis stem cells. Journal of Cell Biology, 198, 281-293. doi:10.1083/jcb.201202014 [3] Singh, S.K., Clarke, I.D., Terasaki, M., Bonn, V.E., Hawkins, C., Squire, J. and Dirks, P.B. (2003). Identi- fication of a cancer stem cell in human brain tumors. Cancer Research, 63, 5821-5828 [4] Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., Mor- rison, S.J. and Clarke, M.F. (2003). Prospective identi- fication of tumorigenic breast cancer cells. PNAS, 100, 3983-3988. doi:10.1073/pnas.0530291100 [5] O'Brien, C.A., Pollett, A., Gallinger, S. and Dick, J.E. (2007). A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature, 445, 106-110. doi:10.1038/nature05372 [6] Zhang, S., Balch, C., Chan, M.W., Lai, H.C., Matei, D., Schilder, J.M., Yan, P.S., Huang, T.H. and Nephew, K.P. (2008). Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Research, 68, 4311-4320. doi:10.1158/0008-5472.CAN-08-0364 [7] Li, C., Heidt, D.G., Dalerba, P., Burant, C.F., Zhang, L., Adsay, V., Wicha, M., Clarke, M.F. and Simeone, D.M. (2007). Identification of pancreatic cancer stem cells. Cancer Research, 67, 1030-1037. doi:10.1158/0008-5472.CAN-06-2030 [8] Schatton, T., Murphy, G.F., Frank, N.Y., Yamaura, K., Waaga-Gasser, A.M., Gasser, M., Zhan, Q., Jordan, S., Duncan, L.M., Weishaupt, C., Fuhlbrigge, R.C., Kupper, T.S., Sayegh, M.H. and Frank, M.H. (2008). I de nti fic ati on of cells initiating human melanomas. Nature, 451, 345- 349. doi:10.1038/nature06489 [9] Boiko, A.D., Razorenova, O.V., van de Rijn, M., Swetter, S.M., Johnson, D.L., Ly, D.P., Butler, P.D., Yang, G.P., Joshua, B., Kaplan, M.J., Longaker, M.T. and Weissman, I.L. (2010). Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature, 466, 133-137. doi:10.1038/nature09161 [10] Civenni, G., Walter, A., Kobert, N., Mihic-Probst, D., Zipser, M., Belloni, B., Seifert, B., Moch, H., Dummer, R., van den Broek, M. and Sommer, L. (2011). Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Research, 71, 3098-3109. doi:10.1158/0008-5472.CAN-10-3997 [11] Matsui, W., Huff, C.A., Wang, Q., et al. (2004). Cha- racterization of clonogenic multiple myeloma cells. Blood, 103, 2332-2336. doi:10.1182/blood-2003-09-3064 [12] Singh, S.K., Clarke, I.D., Hide, T. and Dirks, P.B. (2004) Cancer stem cells in nervous system tumors. Oncogene, 23, 7267-7273. doi:10.1038/sj.onc.1207946 [13] Vercauteren, S.M. and Sutherland, H.J. (2001) CD133 (AC133) expression on AML cells and progenitors. Cy- totherapy, 3, 449-459. doi:10.1080/146532401317248054 [14] Matsui, W., Wang, Q., Barber, J.P., et al. (2008). Clono- genic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Research, 68, 190-197. doi:10.1158/0008-5472.CAN-07-3096 [15] Oh, S.Y., Kang, H.J., Kim, Y.S., Kim, H. and Lim, Y.C. (2012) CD44-negative cells in head and neck squamous carcinoma also have stem-cell like traits. European Journal of Cancer. [16] Eyler, C.E. and Rich, J.N. (2008) Survival of the fittest: Cancer stem cells in therapeutic resistance and angio- genesis. Journal of Clinical Oncology, 26, 2839-2845. doi:10.1200/JCO.2007.15.1829 [17] Li, X., Lewi, M.T., Huang, J., Gutierrez, C., Osborne, C.K., Wu, M., Hilsenbeck, S.G., Pavlick, A., Xiaomei Zhang, X., Chamness, G.C., Wong, H., Rosen, J. and Chang, J.C. (2008) Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. Journal of the Natio- nal Cancer Institute, 100, 672-679. doi:10.1093/jnci/djn123 [18] Bao, S., Wu, Q., McLendon, R.E., Hao, Y., Shi, Q., Hjelmeland, A.B., Dewhirst, M.W., Bigner, D.D. and Rich, J.N. (2006) Glioma stem cells promote radiore- sistance by preferential activation of the DNA damage response. Nature, 444, 756-760. doi:10.1038/nature05236 [19] Mitra, A., Conway, C., Walker, C., Cook, M. and Powell, B. (2010) Melanoma sentinel node biopsy and prediction models for relapse and overall survival. British Journal of Cancer, 103, 1229-1236. doi:10.1038/sj.bjc.6605849 [20] Markopoulos, C.J. (2010) minimizing early relapse and maximizing treatment outcomes in hormone-sensitive postmenopausal breast cancer: Efficacy review of AI trials. Cancer and Metastasis Reviews, 29, 581-594. doi:10.1007/s10555-010-9248-x [21] Saphner, T., Tormey, D.C. and Gray, R. (1996) Annual hazard rates of recurrence for breast cancer after primary therapy. Journal of Clinical Oncology, 14, 2738-2746. [22] Mansell, J., Monypenny, I.J., Skene, A.I., Abram, P., Carpenter, R., Gattuso, J.M., et al. (2009) Patterns and predictors of early recurrence in postmenopausal women with estrogen receptor-positive early breast cancer. Breast Cancer Research and Treatment, 117, 91-98. doi:10.1007/s10549-008-0291-z [23] Frisch, S.M. and Francis, H. (1994) Disruption of epi- thelial cell-matrix interactions induces apoptosis, Journal of Cell Biology, 124, 619-626. doi:10.1083/jcb.124.4.619 [24] Meredith, J.E., Fazeli, B. and Schwartz, M.A. (1993) The extracellular matrix as a cell survival factor. Molecular Biology of the Cell, 4, 953-961. [25] Sethi, T., Rintoul, R.C., Moore, S.M., et al. (1999) Extracellular matrix proteins protect small cell lung cancer cells against apoptosis: A mechanism for small cell lung cancer growth and drug resistance in vivo. Nature Medicine, 5, 662-668. doi:10.1038/9511 [26] Konopleva, M.Y. and Jordan, C.T. (2011) Leukemia stem cells and microenvironment: Biology and therapeutic targeting. Journal of Clinical Oncology, 29, 591-599. doi:10.1200/JCO.2010.31.0904 [27] Selvaggi, G., Novello, S., Torri, V., Leonardo, E., De Giuli, P., Borasio, P., Mossetti, C., Ardissone, F., Lausi, P. and Scagliotti, G.V. (2004) Epidermal growth factor Copyright © 2012 SciRes. OPEN ACCESS  R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 727 receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Annals of Oncology, 15, 28-32. doi:10.1093/annonc/mdh011 [28] Boudreau, N., Sympson, C.J., Werb, Z. and Bissell, M.J. (1995) Suppression of ICE and apoptosis in mammary epithelial cells by extracellular matrix. Science, 267, 891-893. doi:10.1126/science.7531366 [29] Qu, Y.T., Li, X.M., Xu, O., Wang, M.X. and Lu, X.Y. (2012) Impacts of hypoxia on the features and chemo- resistance of cancer stem cells in Hep-2 cells and underlying mechanism. Chinese Journal of Otorhinolary- ngology Head and Neck Surgery, 47, 228-233. [30] Frolova, O., Samudio, I., Benito, J.M., Jacamo, R., Kornblau, S.M., Markovic, A., Schober, W., Lu, H., Qiu, Y.H., Buglio, D., McQueen, T., Pierce, S., Shpall, E., Konoplev, S., Thomas, D., Kantarjian, H., Lock, R., Andreeff, M. and Konopleva, M. (2012) Regulation of HIF-1α signaling and chemoresistance in acute lymp- hocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biology & Therapy, 13, 858-870. doi:10.4161/cbt.20838 [31] Frisch, S.M., Vuori, K., Ruoslahti, E. and Chan-Hui, P.Y. (1996) Control of adhesion-dependent cell survival by focal adhesion kinase. Journal of Cell Biology, 134 , 793- 799. doi:10.1083/jcb.134.3.793 [32] Frisch, S.M. and Screaton, R.A. (2001) Anoikis mecha- nisms. Current Opinion in Cell Biology, 13, 555-562. doi:10.1016/S0955-0674(00)00251-9 [33] Kim, Y.N., Koo, K.H., Sung, J.Y., Yun, U.J. and Kim, H. (2012) Anoikis resistance: An essential prerequisite for tumor metastasis. International Journal of Cell Biology, 306879. [34] Ahmed, N., Abubaker, K., Findlay, J. and Quinn, M. (2010) Epithelial mesenchymal transition and cancer stem cell-like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Current Cancer Drug Targets, 10, 268-278. doi:10.2174/156800910791190175 [35] Chuthapisith, S., E remin , J., El-Shee mey, M. and Eremi n, O. (2010) Breast cancer chemoresistance: Emerging importance of cancer stem cells. Surgical Oncology, 19, 27-32. doi:10.1016/j.suronc.2009.01.004 [36] May, C.D., Sphyris, N., Evans, K.W., Werden, S.J., Guo, W. and Mani, S.A. (2011) Epithelial-mesenchymal tran- sition and cancer stem cells: A dangerously dynamic duo in breast cancer progression. Breast Cancer Research, 13, 202. [37] Singh, A. and Settleman, J. (2010) EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene, 29, 4741-4751. doi:10.1038/onc.2010.215 [38] Mani, S.A., Guo, W., Liao, M.J., Eaton, E.N., Ayyanan, A., Zhou, A.Y., Brooks, M., Reinhard, F.,Zhang, C.C., Shi- pitsin, M., Campbell, L.L., Polyak, K., Brisken, C., Yang, J. and Weinberg. R.A. (2008) The epithelial-mesenchy- mal transition generates cells with properties of stem cells. Cell, 133, 704-715. doi:10.1016/j.cell.2008.03.027 [39] Sarrio, D., Franklin, C.K., Mackay, A., Reis-Filho, J.S. and Isacke, C.M. (2012) Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells, 30, 292-303. doi:10.1002/stem.791 [40] Lane, A.A. and Chabner, B.A. (2009) Histone deace- tylase inhibitors in cancer therapy . Journal of Clinical On- cology, 27, 5459-5468. doi:10.1200/JCO.2009.22.1291 [41] Gravina, G.L., Festuccia, C., Marampon, F., Popov, V.M., Pestell, R.G., Zani, B.M. and Tombolini, V. (2010) Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Molecular Cancer, 9, 305. doi:10.1186/1476-4598-9-305 [42] Varier, R.A. and Timmers, H.T. (2011) Histone lysine methylation and demethylation pathways in cancer. Bio- chimica et Biophysica Acta, 1815, 75-89. [43] Tian, X. and Fang, J. (2007) Current perspectives on histone demethylases. Acta Biochimica et Biophysica Sinica, 39, 81-88. d oi :1 0. 1111/ j. 1745-7270.2007.00272.x [44] Trojer, P. and Reinberg, D. (2006) Histone lysine deme- thylases and their impact on epigenetics. Cell, 12 5, 213- 217. doi:10.1016/j.cell.2006.04.003 [45] Cloos, P.A., Christensen, J., Agger, K. and Helin, K. (2008) Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes & Development, 22, 1115-1140. doi:10.1101/gad.1652908 [46] Shi, Y. (2007) Histone lysine demethylases: Emerging roles in development, physiology and disease. Nature Reviews Genetics, 8, 829-833. doi:10.1038/nrg2218 [47] Kampranis, S.C. and Tsichlis, P.N. (2009) Histone demethylases and cancer. Advances in Cancer Research, 102, 103-169. doi:10.1016/S0065-230X(09)02004-1 [48] Lim, S., Metzger, E., Schüle, R., Kirfel, J. and Buettner, R. (2010) Epigenetic regulation of cancer growth by histone demethylases. International Journal of Cancer, 127, 1991-1998. doi:10.1002/ijc.25538 [49] Agger, K., Christensen, J., Cloos, P.A. and Helin, K. (2008) The emerging functions of histone demethylases. Current Opinion in Genetics & Development, 18, 159- 168. doi:10.1016/j.gde.2007.12.003 [50] Klose, R.J. and Zhang, Y. (2007) Regulation of histone methylation by demethylimination and demethylation. Nature Reviews Molecular Cell Biology, 8, 307-318. doi:10.1038/nrm2143 [51] Loh, Y.H., Zhang, W., Chen, X., George, J. and Ng, H.H. (2007) Jmjd1a and Jmjd2c histone H3 Lys 9 demethy- lases regulate self-renewal in embryonic stem cells. Genes & Development, 21, 2545-2557. doi:10.1101/gad.1588207 [52] Christensen, J., Agger, K., Cloos, P.A., Pasini, D., Rose, S., Sennels, L., Rappsilber, J., Hansen, K.H., Salcini, A.E. and Helin, K. (2007) RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell, 128, 1063-1076. doi:10.1016/j.cell.2007.02.003 [53] Osawa, T., Muramatsu, M., Wang, F., Tsuchida, R., Kodama, T., Minami, T. and Shibuya, M. (2011) In- creased expression of histone demethylase JHDM1D under nutrient starvation suppresses tumor growth via Copyright © 2012 SciRes. OPEN ACCESS  R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 728 down-regulating angiogenesis. Proceedings of the National Academy of Sciences of USA, 108, 20725-20729. doi:10.1073/pnas.1108462109 [54] Lu, Y., Chu, A., Turker, M.S. and Glazer, P.M. (2011) Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Molecular and Cellular Biology, 31, 3339-3350. doi:10.1128/MCB.01121-10 [55] Kottakis, F., Polytarchou, C., Foltopoulou, P., Sanidas, I., Kampranis, S.C. and Tsichlis, P.N. (2011) FGF-2 r egula tes cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR -101-EZH2 pathway. Molecular Cell, 43, 285-298. doi:10.1016/j.molcel.2011.06.020 [56] Wong, P.P., Miranda, F., Chan, K.V., Berlato, C., Hurst, H.C. and Scibetta, A.G. (2012) Histone demethylase KDM5B collaborates with TFAP2C and Myc to repress the cell cycle inhibitor p21(cip) (CDKN1A). Molecular and Cellular Biology, 32, 1633-1644. doi:10.1128/MCB.06373-11 [57] Svotelis, A., Bianco, S., Madore, J., Huppé, G., Nordell- Markovits, A., Mes-Masson, A.M. and Gévry, N. (2011) H3K27 demethylation by JMJD3 at a poised enhancer of anti-apoptotic gene BCL2 determines ERα ligand depen- dency. EMBO Journal, 30, 3947-3961. doi:10.1038/emboj.2011.284 [58] Lim, S., Janzer, A., Becker, A., Zimmer, A., Schüle, R., Buettner, R. and Kirfel, J. (2010) Lysine-specific deme- thylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis, 31, 512-520. doi:10.1093/carcin/bgp324 [59] Mitra, D., Das, P.M., Huynh, F.C. and Jones, F.E. (2011) Jumonji/ARID1 B (JARID1B) protein promotes breast tumor cell cycle progression through epigenetic r epre ssio n of microRNA let-7e. Journal of Biological Chemistry, 286, 40531-40535. doi:10.1074/jbc.M111.304865 [60] Huang, J., Sengupta, R., Espejo, A.B., Lee, M.G., Dorsey, J.A., Richter, M., Opravil, S., Shiekhattar, R., Bedford, M.T., Jenuwein, T., Scoumanne, A. and Chen, X. (2007) The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. Journal of Biological Chemistry, 282, 15471- 15475. doi:10.1074/jbc.M701023200 [61] Berger, S.L. (2007) p53 is regulated by the lysine demethylase LSD1. Nature, 449, 105-108. doi:10.1038/nature06092 [62] Gartel, A.L. and Tyner, A.L. (2002) The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mole- cular Cancer Therapeutics, 1, 639-649. [63] Viale, A., De Franco, F., Orleth, A., Cambiaghi, V., Giu- liani, V., Bossi, D., Ronchini, C., Ronzoni, S., Muradore, I., Monestiroli, S., Gobbi, A., Alcalay, M., Minucci, S. and Pelicci, P.G. (2009) Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature, 457, 51-56. doi:10.1038/nature07618 [64] Harris, W.J., Huang, X., Lynch, J.T., Spencer, G.J., Hitchin, J.R., Li, Y., Ciceri, F., Blaser, J.G., Greystoke, B.F., Jordan, A.M., Miller, C.J., Ogilvie, D.J. and Some- rvaille, T.C. (2012) The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell, 21, 473-487. doi:10.1016/j.ccr.2012.03.014 [65] Henglein, B,, Chenivesse, X., Wang, J., Eick, D. and Bréchot, C. (1994) Structure and cell cycle-regulated transcription of the human cyclin A gene. Proceedings of the National Academy of Sciences of the USA, 91, 5490- 5494. doi:10.1073/pnas.91.12.5490 [66] Holbro, T. and Hynes, N.E. (2004) ErbB receptors: Directing key signaling networks throughout life. Annual Review of Pharmacology and Toxicology, 44, 195-217. doi:10.1146/annurev.pharmtox.44.101802.121440 [67] Suzuki, A., Horiuchi, A., Ashida, T., Miyamoto, T., Kash i- ma, H., Nikaido, T., Konishi. I. and Shiozawa, T. (2010) Cyclin A2 confers cisplatin resistance to endometrial car- cinoma cells via up-regulation of an Akt-binding protein, periplakin. Journal of Cellular and Molecular Medicine, 14, 2305-2317. doi :1 0. 1111/ j.15 82 -4934.2009.00839.x [68] Tan, M. and Yu, D. (2007) Molecular mechanisms of erbB2-mediated breast cancer chemoresistance. Advances in Experimental Medicine and Biology, 608, 119-129. doi:10.1007/978-0-387-74039-3_9 [69] Lu, D. and Carson, D.A. (2011) Inhibition of Wnt signaling and cancer stem cells. Oncotarget, 2, 587. [70] Huang, Y., Stewart, T.M., Wu, Y., Baylin, S.B., Marton, L.J., Perkins, B., Jones, R. J., Woster, P. M. an d Case ro Jr., R.A. (2009) Novel oligoamine analogues inhibit lysine- specific demethylase 1 and induce reexpression of epige- netically silenced genes. Clinical Cancer Research, 15, 7217-7228. doi:10.1158/1078-0432.CCR-09-1293 [71] Chung, M.T., Lai, H.C., Sytwu, H.K., Yan, M.D., Shih, Y.L., Chang, C.C., Yu, M.H., Liu, H.S., Chu, D.W. and Lin, Y.W. (2009) SFRP1 and SFRP2 suppress the transformation and invasion abilities of cervical cancer cells through Wnt signal pathway. Gynecologic Oncology, 112, 646-653. doi:10.1016/j.ygyno.2008.10.026 [72] DiPaola, R.S. and Aisner, J. (1999) Overcoming bcl-2- and p53-mediated resistance in prostate cancer. Seminars in Oncology, 26, 112-116. [73] Johnson, S.M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K.L., Brown, D. and Slack, F.J. (2005) RAS is regulated by the let-7 microRNA family. Cell, 120, 635-647. doi:10.1016/j.cell.2005.01.014 [74] Kumar, M.S., Erkeland, S.J., Pester, R.E., Chen, C.Y., Ebert, M.S., Sharp, P.A. and Jacks, T. (2008) Suppression of non-small cell lung tumor development by the let-7 microRNA family . Proceedings of the National Academy of Sciences of the USA, 105, 3903-3908. doi:10.1073/pnas.0712321105 [75] He, X.Y., Chen, J.X. , Zhang, Z., Li, C.L., Peng, Q.L. and Peng, H.M. (2010) The let-7a microRNA protects from growth of lung carcinoma by suppression of k-Ras and c-Myc in nude mice. Journal of Cancer Research and Clinical Oncology, 136, 1023-1028. doi:10.1007/s00432-009-0747-5 [76] Kooistra, S.M. and Helin, K. (2012) Molecular mecha- nisms and potential functions of histone demethylases. Nature Reviews Molecular Cell Biology, 13, 297-311. Copyright © 2012 SciRes. OPEN ACCESS  R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 729 [77] Kontaki, H. and Talianidis, I. (2010) Lysine methylation regulates E2F1-induced cell death. Molecular Cell, 39, 152-160. doi:10.1016/j.molcel.2010.06.006 [78] Stiewe, T. and Pützer, B.M. (2000) Role of the p53- homologue p73 in E2F1-induced apoptosis. Nature Gen- etics, 26, 464-469. doi:10.1038/82617 [79] Sharma, S.V., Lee, D.Y., Li, B., Quinlan, M.P., Takahashi, F., Maheswaran, S., McDermott, U., Azizian, N., Zou, L., Fischbach, M.A., Wong, K.K., Brandstetter, K., Wittner, B., Ramaswamy, S., Classon, M. and Settle- man, J. (2010) A chromatin-mediated reversible drug- tolerant state in cancer cell subpopulations. Cell, 141, 69-80. doi:10.1016/j.cell.2010.02.027 [80] Wang, J., Lu, F., Ren, Q., Sun, H., Xu. Z., Lan, R., Liu, Y., Ward, D., Quan, J., Ye, T. and Zhang, H. (2011) Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Research, 71, 7238-7249. doi:10.1158/0008-5472.CAN-11-0896 [81] Harris, W.J., Huang, X., Lynch, J.T., Spencer, G.J., Hitchin, J.R., Li, Y., Ciceri, F., Blaser, J.G., Greystoke, B.F., Jordan, A.M., Miller, C.J., Ogilvie, D.J. and Some- rvaille, T.C. (2012) The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell, 21, 473-487. doi:10.1016/j.ccr.2012.03.014 [82] Lv, T., Yuan, D., Mia o, X., Lv, Y., Zhan, P., et al. (2012) Over-expression of LSD1 promotes proliferation, mig- ration and invasion in non-small cell lung cancer. PLoS ONE, 7, e35065. doi:10.1371/journal.pone.0035065 [83] Schenk, T., Chen, WC., Göllner, S., Howell, L., Jin, L., Hebestreit, K., Klein, HU., Popescu, A.C., Burnet,t A., Mills, K., Casero Jr., R.A., Marton, L., Woster, P., Minden, M.D., Dugas, M., Wang, J.C., Dick, J.E., Müller-Tidow, C., Pe trie, K. and Zel ent , A. (2012) Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nature Medicine, 18, 605-611. doi:10.1038/nm.2661 [84] Metzger, E., Wissmann, M., Yin, N., Müller, J,M., Schneider, R., Peters, A.H., Gü nther, T., Buettner, R. and Schüle, R. (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcrip- tion. Nature, 437, 436-439. [85] Kahl, P., Gullotti, L., Heukamp, L.C., Wolf, S., Fri ed ri ch s, N., Vorreuther, R., Solleder, G., Bastian, P.J., Ellinger, J., Metzger, E., Schüle, R. and Buettner, R. (2 006) Andr ogen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Research, 66, 11341-11347. doi:10.1158/0008-5472.CAN-06-1570 [86] Willmann, D., Lim, S., Wetzel, S., Metzger, E., Jan- dausch, A., Wilk, W., Jung, M., Forne, I., Imhof, A., Janzer, A., Kirfel, J., Waldmann, H., Schüle, R. and Bue- ttner, R. (2012) Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. International Journal of Cancer, 131, 2704- 2709. doi:10.1002/ijc.27555 [87] Wang, Y., Zhang, H., Chen, Y., Sun, Y., Yang, F., Yu, W., Liang, J., Sun, L., Yang, X., Shi, L., Li, R., Li, Y., Zhang, Y., Li, Q., Yi, X. and Shang, Y. (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell, 13 8, 660-672. doi:10.1016/j.cell.2009.05.050 [88] Lim, S., Janzer, A., Becker, A., Zimmer A, Schüle R, Buettner R and Kirfel J. (2010) Lysine-specific deme- thylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis, 31, 512-520. doi:10.1093/carcin/bgp324 [89] He, J., Nguyen, A.T. and Zhang, Y. (2011) KDM2b/ JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood, 117, 3869-3880. doi:10.1182/blood-2010-10-312736 [90] Frescas, D., Guardavaccaro, D., Bassermann, F., Koya ma- Nasu, R. and Pagano, M. (2007) JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature, 450, 309-313. doi:10.1038/nature06255 [91] Krieg, A.J., Rankin, E.B., Chan, D., Razorenova, O., Fernandez, S. and Giaccia, A.J. (2010) Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Molecular and Cellular Biology, 30, 344- 353. doi:10.1128/MCB.00444-09 [92] Du, Z.M., Hu, L.F., Wang, H.Y., Yan, L.X, Zeng, Y.X., Shao, J.Y. and Ernberg, I. (2011) Upregulation of MiR- 155 in nasopharyngeal carcinoma is partly driven by LMP1 and LMP2A and downregulates a negative pro- gnostic marker JMJD1A. PLoS One, 6, e19137. doi:10.1371/journal.pone.0019137 [93] Lim, S., Metzger, E., Schüle, R., Kirfel, J. and Buettner, R. (2010) Epigenetic regulation of cancer growth by histone demethylases. International Journal of Cancer, 127, 1991-1998. doi:10.1002/ijc.25538 [94] Beyer, S., Kristensen, M.M., Jensen, K.S., Johansen, J.V. and Staller, P. (2008) The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia- inducible factor HIF. Journal of Biological Chemistry, 283, 36542-36552. doi:10.1074/jbc.M804578200 [95] Cloos, P.A., Christensen, J., Agger, K., Maiolica, A., Rappsilber, J., Antal, T., Hansen, K.H. and Helin, K. (2006) The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature, 442, 307-311. doi:10.1038/nature04837 [96] Italiano, A., Attias, R., Aurias, A., Pérot, G., Burel- Vandenbos, F., Otto, J., Venissac, N. and Pedeutour, F. (2006) Molecular cytogenetic characterization of a meta- static lung sarcomatoid carcinoma: 9p23 neocentromere and 9p23-p24 amplification including JAK2 and JMJD2C. Cancer Genetics and Cytogenetics, 167, 122-130. doi:10.1016/j.cancergencyto.2006.01.004 [97] Liu, G., Bollig-Fischer, A., Kreike, B., van de Vijver, M.J., Abrams, J., Ethier, S.P. and Yang, Z.Q. (2009) Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene, 28, 4491-4500. doi:10.1038/onc.2009.297 [98] Ehrbrecht, A., Müller, U., Wolter, M., Hoischen, A., Copyright © 2012 SciRes. OPEN ACCESS  R. Thakur, D. P. Mishra / Advances in Bioscience and Biotechnology 3 (2012) 720-730 Copyright © 2012 SciRes. 730 OPEN ACCESS Koch, A., Radlwimmer, B., Actor, B., Mincheva, A., Pietsch, T., Lichter, P., Reifenberger, G. and Weber, R.G. (2006) Comprehensive genomic analysis of desmoplastic medulloblastomas: Identification of novel amplified genes and separate evaluation of the different histological com- ponents. Journal of Pathology, 208, 554-563. doi:10.1002/path.1925 [99] Yamane, K., Tateishi, K., Klose, R.J., Fang, J., Fabrizio, L.A., Erdjument-Bromage, H., Taylor-Papadimitriou, J., Tempst, P. and Zhang, Y. (2007) PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Molecular Cell, 25, 801- 812. doi:10.1016/j.molcel.2007.03.001 [100] Barrett, A., Madsen, B., Copier, J., Lu, P.J., Cooper, L., Scibetta, A.G., Burchell, J. and Taylor-Papadimitriou, J. (2002) PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: A new cancer/testis antigen? Inter- national Journal of Cancer, 101, 581-588. doi:10.1002/ijc.10644 [101] Xiang, Y., Zhu, Z., Han, G., Ye, X., Xu, B., Pe ng, Z., Ma, Y., Yu, Y., Lin, H., Chen, A.P. and Chen, C.D. (2007) JARID1B is a histone H3 lysine 4 demethylase up- regulated in prostate cancer. Proceedings of the National Academy of Sciences of the USA, 104, 19226-19231. doi:10.1073/pnas.0700735104 [102] Van Haaften, G., et al. (2009) Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nature Genetics, 41, 521-523. doi:10.1038/ng.349 [103] Gui, Y., et al. (2011) Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nature Genetics, 43, 875-878. doi:10.1038/ng.907 [104] Anderton, J.A., Bose, S., Vockerodt, M., Vrzalikova, K., Wei, W., Kuo, M., Helin, K., Christensen, J., Rowe, M., Murray, P.G. and Wo odman, C.B. (2011) The H 3K27 me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene, 30, 2037-2043. doi:10.1038/onc.2010.579 [105] Xiang, Y., Zhu, Z., Han, G., Lin, H., Xu, L. and Chen, C.D. (2007) JMJD3 is a histone H3K27 demethylase. Cell Research, 17, 850-857. doi:10.1038/cr.2007.83 [106] Hsia, D.A., Tepper, C.G., Pochampalli, M.R., Hsia, E.Y., Izumiya, C., Huerta, S.B., Wright, M.E., Chen, H.W., Kung, H.J. and Izumiya, Y. (2010) KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proceedings of the National Academy of Sciences of the USA, 107, 9671-9676. doi:10.1073/pnas.1000401107 [107] Gallinari, P., Di Marco, S., Jones, P., Pallaoro, M. and Steinkühler, C. (2007) HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Research, 17, 195-211. [108] Peng, L. and Seto, E. (2011) Deacetylation of nonhistone proteins by HDACs and the implications in cancer. Handbook of Experimental Pharmacology, 206, 39-56. doi:10.1007/978-3-642-21631-2_3 [109] Singh, M.M., Manton, C.A., Bhat, K.P., Tsai, W.W., Al dape, K., Barto n, M.C . and C han dra, J . (2 011) Inhibition of LSD1 sensitizes glioblastoma cells to histone deace- tylase inhibitors. Neuro-Oncology, 13, 894-903. doi:10.1093/neuonc/nor049 [110] Huang, Y., Vasilatos, S.N., Boric, L., Shaw, P.G. and Davidson, N.E. (2012) Inhibitors of histone demethyla- tion and histone deacetylation cooperate in regulating gene expression and inhibiting growth in human breast cancer cells. Breast Cancer Research and Treatment, 131, 777-789. doi:10.1007/s10549-011-1480-8 [111] Styczynski, J. and Drewa, T. (2007) Leukemic stem cells: From metabolic pathways and signaling to a new concept of drug resistance targeting. Acta Biochimica Polonica, 54, 717-726. [112] Fodde, R. and Brabletz, T. (2007) Wnt/β-catenin signal- ing in cancer stemness and malignant behavior. Current Opinion in Cell Biology, 19, 150-158. doi:10.1016/j.ceb.2007.02.007 [113] Zhou, J., Wulfkuhle, J., Zhang, H., Gu, P., Yang, Y., Deng, J., Margolick, J.B., Liotta, L.A., Petricoin III, E. and Zhang, Y. (2007) Activation of the PTEN/mTOR/ STAT3 pathway in breast cancer stemlike cells is re- quired for viability and maintenance. Proceedings of the National Academy of Sciences of the USA, 104, 16158- 16163. doi:10.1073/pnas.0702596104 [114] Peacock, C.D., Wang, Q., Gesell, G.S., Corcoran- Schwartz, I.M., Jones, E., Kim, J., Devereux, W.L., Rho- des, J.T., Huff, C.A., Beachy, P.A., Watkins, D.N. and Matsui, W. (2007) Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proceedings of the National Academy of Sciences of the USA, 104, 4048-4053.
|