A. M. TAROLA, R. PRETI
844
(500 mg, 6 ml) were purchased from Sigma Aldrich. Ul-
trapure water was obtained from a Milli-Q water system
(Millipore, Bedford, MA, USA). An extraction manifold
from Alltech (Alltech Associates, Deerfield, USA) was
used for the SPE analysis.
2.3. Instrumentation
GC-MS Separation and Determination
All analysis were performed with Finnigan Trace GC
ultra gas chromatograph coupled to a Finnigan Polaris Q
mass spectrometer (Thermo Electron Co., Austin, Texas).
Separations were conducted on a DB-5 ms fused-silica
column, 30 m × 0.25 mm × 0.25 µm film thickness
(J&W Scientific, Folsom, CA), with helium as carrier gas,
at a flow of 1.0 ml/min.
The column was held at 70˚C ramped 15˚C/min to
150˚C, then up to 200˚C at 3˚C/min and finally ramped at
8˚C/min to 300˚C and held for 5 minutes. A volume of 2
µl of sample extract was injected manually on a PTV
injector operating in splitless mode. The injector tem-
perature was set at 60˚C. The mass spectrometer operated
in the EI mode. The parameters were set at the following
values: an electron energy of 70 eV and a filament emis-
sion current of 200 µA. The interface and ion source tem-
peratures were maintained at 250˚C and 200˚C, respec-
tively. The scan mode was used between m/z 40 and 350.
2.4. Procedure
Samples of mineral water naturally carbonated were de-
gassed in an ultrasonic bath for 5 min.
Spe Procedure
The procedure followed the guidelines EPA Method n
525 [8]. SPE C18 cartridges were conditioned with 5 ml
of ethyl acetate, followed by 5 ml of methanol and 10 ml
of bidistillated water, without allowing the cartridge to
dry out. Then, 2.5 ml of methanol were added to 500 ml
of water sample that was passed through the conditioned
cartridges at a flow rate of approximately 8 ml/min under
vacuum. The cartridges were dried for 10 min under
vacuum and afterwards the analytes were eluted from the
solid phase with 5 ml of ethyl acetate, traces of water
were removed with anhydrous sodium sulphate. The elu-
ate was evaporated to dryness under a stream of nitrogen
and the residue was dissolved in 0.5 ml of n-hexane.
3. Results
The quality of water for human consumption has always
been and still is one of the most serious challenges. Since
the late decades, concern about the contamination of wa-
ter sources has risen due to the increasing number of pes-
ticides detected. Regulations for drinking water are re-
quired in order to limit human risks and environmental
pollution. These regulations are well defined in Europe,
setting at 0.05 µg/l of each pesticide concentration limit
in mineral water samples. Consequently, it becomes nec-
essary to provide control laboratories with analytical
methods allowing the monitoring of pesticide residues at
this trace level, with basic performance data in agreement
with the drinking water EC Directives 98/83 and 2003/40
requirements [9,10].
Pesticides used in agricultural practices are several,
therefore to ensure an effective quality control it is nec-
essary to develop a list of priority substances to be mon-
itored by the producers themselves who are obliged to
carry out annual controls by Italian law. The methodol-
ogy developed to generate the list of priority substances
is based on relevant factors, including for example sale
data, the target, their degradation, the environmental dis-
tribution, which results from many chemical characteris-
tics such as molecular weight, vapour pressure, solubility
in water and octanol/water partition coefficient (Kow).
The priority list, that includes the pesticides considered
in this study, is developed by the Italian Environmental
Protection Agency.
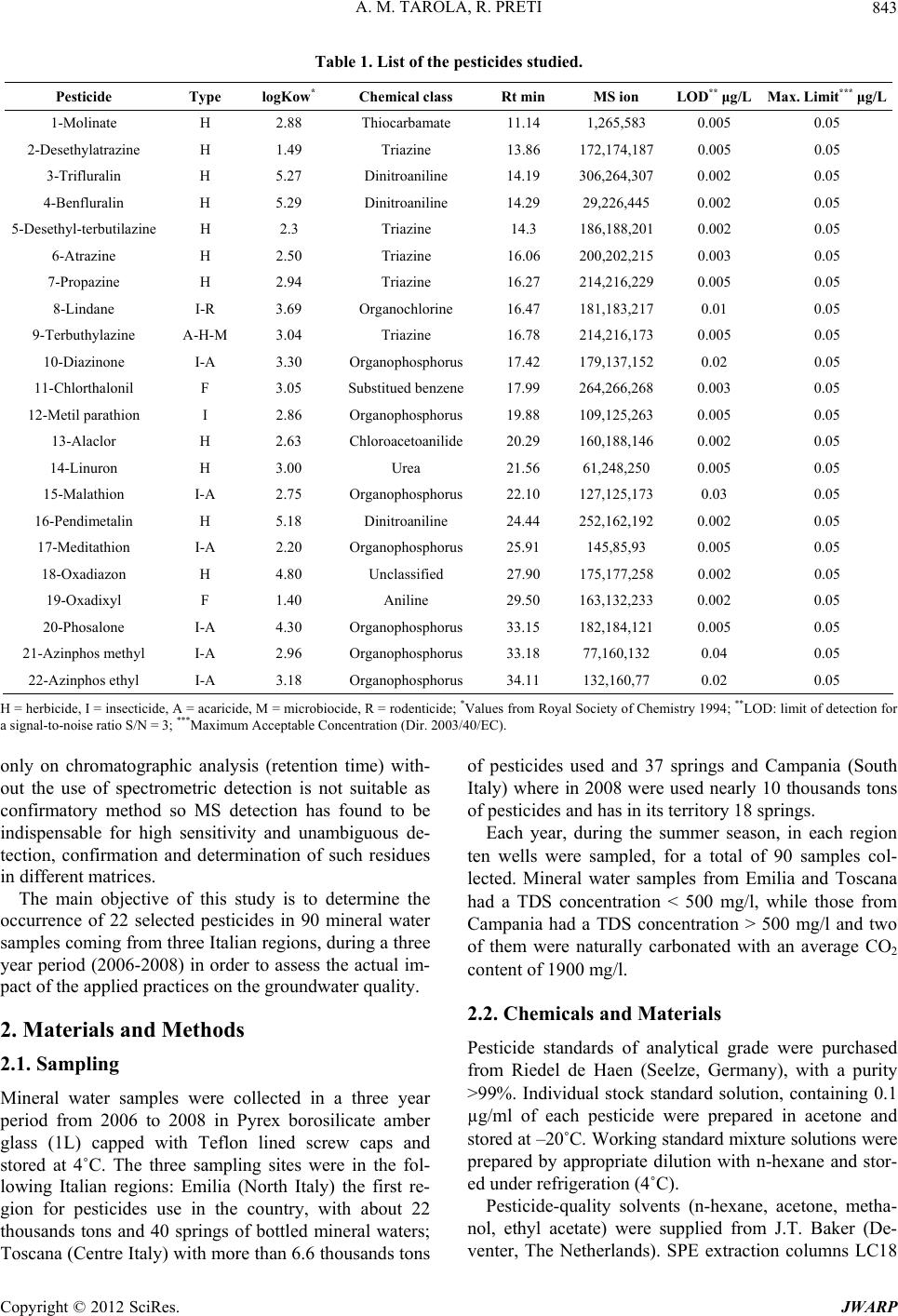
In this study a multiresidue method based on SPE and
GC separation with MS detection were utilised to assess
the presence of 22 selected pesticides residues in 30 Ital-
ian mineral water wells for three years. The method sho-
wed to be suitable to the analysis of these compounds
since they were detected at low concentrations, according
to European Union maximum admissible concentration
(Table 2). The LODs were calculated multiplying by
three the average value of the noise sampled at the reten-
tion time of each analyte. Repeatability and reproducibil-
ity studies yielded Relative Standard Deviations (RSDs)
lower than 7% in all the cases, with recoveries ranging
from 86% to 105% evaluated at 0.05 µg/l spiked level.
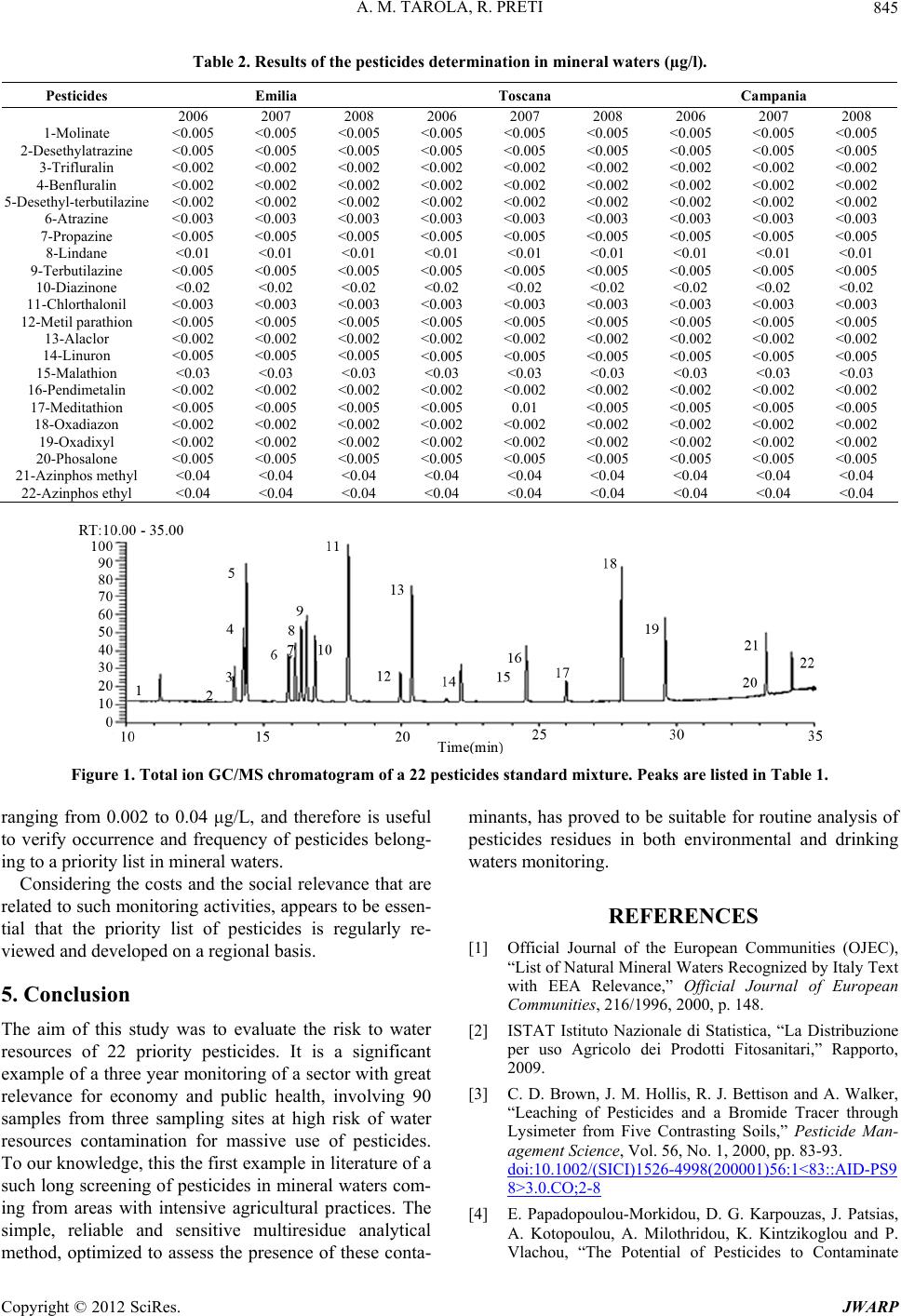
All measurements were performed in triplicate. The ty-
pical chromatogram is showed in Figure 1. During the
screening only in 2007 one sample belonging to an Emilia
well had a level of meditathion of 0.01 mg/l which is
over our detection limit but under the European Union
Maximum Acceptable Concentration (MAC) (Table 2).
4. Discussion
Italy is the nation with highest production and consump-
tion of mineral waters in the world. The results of this
three-year study on pesticide residues in 90 mineral wa-
ter samples coming from three regions with intensive use
of pesticides, are reassuring for the quality of Italian
mineral waters, being residues detected in just one sam-
ple and at a concentration lower than the European Union
Maximum Acceptable Concentration (MAC).
The multiresidue analytical method used has proved to
be sensitive and reliable. The method does fulfill the de-
tection limits required by the EC Directive, with LOD
Copyright © 2012 SciRes. JWARP