 Advances in Bioscience and Biotechnology, 2012, 3, 686-694 ABB http://dx.doi.org/10.4236/abb.2012.326089 Published Online October 2012 (http://www.SciRP.org/journal/abb/) Etoposide-induced apoptosis results in chromosome breaks within the AF9 gene: Its implication in chromosome rearrangement in leukaemia Cynthia Patricia Nicholas, Sai-Peng Sim* Faculty of Medicine and Health Sciences, Universiti Malaysia Sarawak, Kota Samarahan, Malaysia Email: *spsim@fmhs.unimas.my Received 14 August 2012; revised 20 September 2012; accepted 30 September 2012 ABSTRACT Treatment with etoposide (VP-16) has been associ- ated with translocation of the mixed lineage leukae- mia (MLL) gene seen in treatment-related acute mye- loid leukaemia (t-AML). Among the different partner genes, AF9 is the mo st co mmon pa rtne r gene o f MLL. AF9 shares similar structural element with the MLL gene. Various mechanisms of translocation have been proposed for the MLL gene, including apoptosis, par- ticularly the apoptotic nuclease. In the current study, we show that VP-16 induced cleavage of the AF9 gene in both leukaemic cells and cultured normal blood cell. All the breakpoints were mapped within the BCR1 of the AF9 gene. AF9 cleavages in leukaemic cells were abolished by pre-treatment with caspase inhibitor (Z-DEVD-FMK), suggesting the involve- ment of caspase-activated DNase (CAD). The absence of AF9 cleavage in K562 cells further supported the involvement of apoptosis. However, AF9 cleavages in cultured normal blood cell were not eliminated by caspase inhibitor. The possible role of CAD and other apoptotic nucleases/effectors that could be involved in AF9 translocation are discussed. Keywords: Etoposide (VP-16); Apoptosis; AF9; Chromosome Breaks; Leukaemia 1. INTRODUCTION Etoposide (VP-16) is an epipodophyllotoxin that targets DNA topoisomerase II [1]. It stabilises the enzyme-DNA cleavage complex by inhibiting the religation reaction of topoisomerase II [2]. Etoposide exerts its anticancer ef- fects by “poisoning” topoisomerase II, resulting in DNA cleavages [1]. Clinically, etoposide is widely used as an effective anticancer drug. However, about 2% - 12% of the patients eventually developed treatment-related acute myeloid leukaemia (t-AML) [3]. The major chromosome translocation found in this group of patients involves the mixed lineage leukaemia or myeloid lymphoid leukae- mia (MLL) gene at 11q23 [4,5]. Translocation of the MLL gene is not only found in t-AML but in de novo leukaemia as well [6]. In both cases, mapping of the chromosomal breakpoints in MLL revealed that almost all of the breakpoints clustered within an 8.3-kb region known as the breakpoint cluster region (BCR) [6]. Inter- estingly, this region contains a weak- and a high-affinity Scaffold Attachment Region (SAR) [6]. Unlike many other genes, the MLL gene rearranges with more than 66 different partner genes [7]. Among them the AF9 gene located at 9p22 is one of the most common partner genes [8]. The AF9 gene is more than 100 kb in size and contains two patients’ BCR, namely BCR1 at intron 4 and BCR2 that spans introns 7 and 8 [8,9]. There is also a co-localising in vivo DNA topoi- somerase II cleavage site and an in vitro DNase I hyper- sensitive site in intron 7 in BCR2. AF9 also contains two SARs, located centromeric to the topoisomerase II site, bordering the BCR1 and BCR2 [10]. The AF9 BCR was shown to have similar structural elements as the MLL BCR [10]. Various mechanisms have been proposed to mediate the event of chromosome translocation in patients treated with VP-16. These includes Alu repeats [11], V(D)J re- combination [12] and DNA topoisomerase II [13]. How- ever, more recently, the apoptotic nuclease was suggested to play a key role in the initial event of chromosome translocation, that is the breakage of the chromosome [14]. Apoptosis is a naturally occurring programmed cell death process. The apoptotic nuclease, caspase-activated DNase (CAD) is normally inhibited by the inhibitor of CAD (ICAD). During apoptosis induction, caspase cleaves ICAD, releasing the activated CAD. CAD then cleaves the chromosomal DNA resulting in the formation of high molecular weight (HMW) DNA and nucleosomal *Corresponding author. OPEN ACCESS  C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 687 DNA ladder [15]. Although apoptosis is a cell death process, however, cells have the potential to recover upon DNA repair [16]. It is highly possible that during the recovery, different fragments of the chromosomes are joined and resulted in chromosome translocation. This is supported by the observation that etoposide stimulated the formation of MLL fusion product in CAD cDNA- complemented mouse embryonic fibroblasts (MEFs), but not in the CAD knock-out MEF [17]. In the current study, AF9 gene was chosen because it is the most common translocation partner of the MLL gene [8], and AF9 also has similar structure as the MLL gene [10]. We would like to hypothesise that during VP-16-induced apoptosis, chromosome cleavages occur in various parts of the genome including the AF9 gene, which upon erroneous DNA repair, the cells survive with chromosome rearrangement. Thus in the current study we aimed to investigate the role of apoptotic nuclease in VP-16-induced chromosome breaks within the AF9 gene. An understanding of the chromosome break event within this gene is important because this is the initial step of chromosome translocation. 2. MATERIALS AND METHODS 2.1. Cell Lines and Materials CEM leukaemic cell line was purchased from American Type Culture Collection (ATCC). K562 was a generous gift from Prof. Dr. Leroy F. Liu of University of Medi- cine and Dentistry of New Jersey, New Jersey, USA. K562 is a cell line derived from chronic myeloid leu- kaemia (CML) patient and belongs to the granulocytic series of cell [18]. K562 cells exhibits delayed apoptotic features such as cleavage of caspase substrate, cyto- chrome c release, DNA fragmentation and apoptotic morphological changes [19]. Etoposide (VP-16) was purchsed from SIGMA. DNA Polymerase I, Large (Klenow) fragment, T4 DNA Ligase and all the restric- tion enzymes were purchased from New England Biolabs (NEB, England). Phytohaemagglutinin (PHA) was pur- chased from Gibco, USA. PCR primers were obtained from First Base Laboratories. Phusion DNA Polymerase was purchased from Finnzymes, Finland. Caspase-3 in- hibitor II (Z-DEVD-FMK) was purchased from Calbio- chem, USA. Blood and cell culture DNA Mini-Prep kit, and QIAquick Gel Extraction Kit were obtained from QIAGEN, Germany. DYEnamic ET Terminator Cycle Sequencing Kit was purchased from Amersham, UK. 2.2. Cell Culture and Apoptosis Induction Assay Cells were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, 100 g/ml streptomycin, and 2 mM L-gluta- mine. Cells were constantly grown at 37˚C with 5% CO2. CEM and K562 cells at a density of 1 × 106 cells/ml were treated with 50 µM of VP-16 to induce apoptosis. Treatment with 0.1% DMSO as solvent control was in- cluded. All treatments were performed at 37˚C for 5 hours otherwise mentioned. Cells were collected for ge- nomic DNA extraction using QIAGEN Blood and Cell Culture DNA Mini-Prep according to manufacturer’s protocol. The purified DNA was then digested with 100 U of BamH I in a 150 µl reaction at 37˚C for 16 hours. 2.3. Modification of Digested DNA as Template for Inverse Polymerase Chain Reaction (IPCR) Figure 1 shows a summary of DNA modification as well as the IPCR. The BamH I-digested genomic DNA con- sists of a mixture of intact BamH I AF9 fragments and cleaved AF9 fragments. BamH I digestion generates sticky ends while the ends of the cleaved AF9 fragments are believed to be either a 1-base 5’ overhang or a blunt end [20]. Therefore, Klenow fill-in was carried out to generate fragments with blunt ends. Two µg of DNA was used as template for Klenow fill-in following standard protocol [21]. Subsequently ligation/cyclisation with 2000 U of T4 DNA Ligase was performed in a 500 µl reaction at 16˚C for 16 hours. Ethanol precipitation was carried out with 2 volumes of cold absolute ethanol and 0.1 volume of 3 M sodium acetate, pH 5.2. DNA pellet was washed with 70% ethanol and briefly air-dried, then dissolved in TE buffer, pH 8.0. The dissolved DNA was divided into two, one digested with 20 U of Kpn I and the other digested with 20 U of Nde I. Kpn I digestion was performed to linearise the cyclised DNA whereas Nde I digestion was performed to eliminate amplification of the intact BamH I AF9 fragment, thus eliminating its competition with the cleaved AF9 fragment during IPCR. Digested DNA was purified using QIAGEN QIAquick Gel Extraction Kit according to manufacturer’s protocol. 2.4. Nested IPCR and DNA Sequencing Nested IPCR was performed with 200 ng of template DNA (Kpn I- or Nde I-digested), 0.5 mM of each primer, 200 µM of each dNTP, 1× Phusion buffer HF (containing 1.5 mM MgCl2) and 0.4 U of Phusion Polymerase. IPCR cycle condition for the first round was: 30 seconds of 98˚C for 1 cycle, followed by 30 cycles of 98˚C for 10 seconds, 69˚C for 30 seconds, 72˚C for 15 seconds, fol- lowed by a 1 cycle of 72˚C for 10 minutes. Second round IPCR was performed with 2 µl of 5-times diluted first round IPCR product with similar cycle condition, except that the annealing temperature was 57˚C. The primers used during the first round of IPCR were 5’-ATTCTA- GACCCCAAAAAATTCTCAG-3’ and 5’-CTCTTAATG- CCACTGCCATGA-3’, while the primers used in the Copyright © 2012 SciRes. OPEN ACCESS 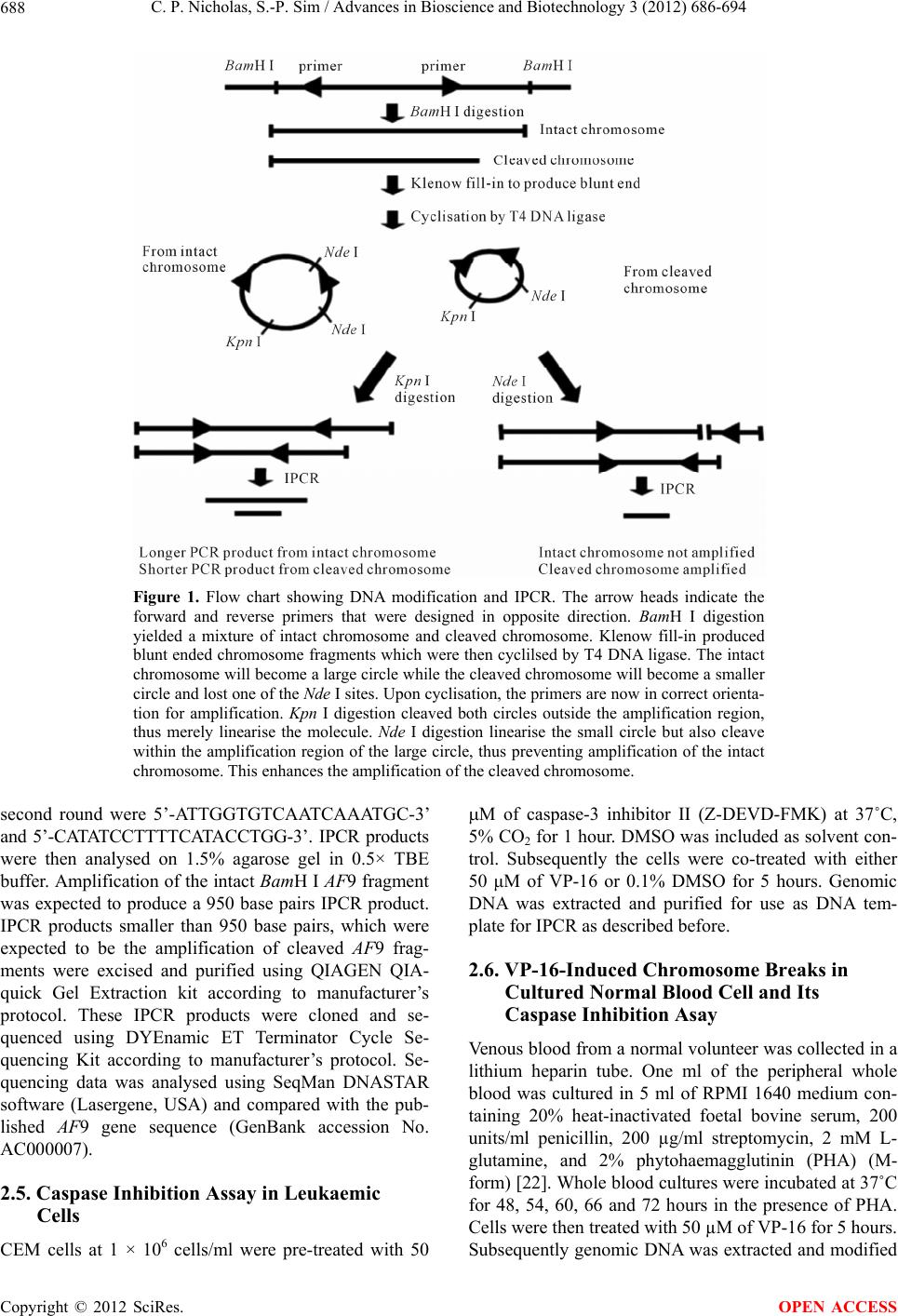 C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 Copyright © 2012 SciRes. 688 Figure 1. Flow chart showing DNA modification and IPCR. The arrow heads indicate the forward and reverse primers that were designed in opposite direction. BamH I digestion yielded a mixture of intact chromosome and cleaved chromosome. Klenow fill-in produced blunt ended chromosome fragments which were then cyclilsed by T4 DNA ligase. The intact chromosome will become a large circle while the cleaved chromosome will become a smaller circle and lost one of the Nde I sites. Upon cyclisation, the primers are now in correct orienta- tion for amplification. Kpn I digestion cleaved both circles outside the amplification region, thus merely linearise the molecule. Nde I digestion linearise the small circle but also cleave within the amplification region of the large circle, thus preventing amplification of the intact chromosome. This enhances the amplification of the cleaved chromosome. second round were 5’-ATTGGTGTCAATCAAATGC-3’ and 5’-CATATCCTTTTCATACCTGG-3’. IPCR products were then analysed on 1.5% agarose gel in 0.5× TBE buffer. Amplification of the intact BamH I AF9 fragment was expected to produce a 950 base pairs IPCR product. IPCR products smaller than 950 base pairs, which were expected to be the amplification of cleaved AF9 frag- ments were excised and purified using QIAGEN QIA- quick Gel Extraction kit according to manufacturer’s protocol. These IPCR products were cloned and se- quenced using DYEnamic ET Terminator Cycle Se- quencing Kit according to manufacturer’s protocol. Se- quencing data was analysed using SeqMan DNASTAR software (Lasergene, USA) and compared with the pub- lished AF9 gene sequence (GenBank accession No. AC000007). OPEN ACCESS 2.5. Caspase Inhibition Assay in Leukaemic Cells CEM cells at 1 × 106 cells/ml were pre-treated with 50 M of caspase-3 inhibitor II (Z-DEVD-FMK) at 37˚C, 5% CO2 for 1 hour. DMSO was included as solvent con- trol. Subsequently the cells were co-treated with either 50 μM of VP-16 or 0.1% DMSO for 5 hours. Genomic DNA was extracted and purified for use as DNA tem- plate for IPCR as described before. 2.6. VP-16-Induced Chromosome Breaks in Cultured Normal Blood Cell and Its Caspase Inhibition Asay Venous blood from a normal volunteer was collected in a lithium heparin tube. One ml of the peripheral whole blood was cultured in 5 ml of RPMI 1640 medium con- taining 20% heat-inactivated foetal bovine serum, 200 units/ml penicillin, 200 µg/ml streptomycin, 2 mM L- glutamine, and 2% phytohaemagglutinin (PHA) (M- form) [22]. Whole blood cultures were incubated at 37˚C for 48, 54, 60, 66 and 72 hours in the presence of PHA. Cells were then treated with 50 µM of VP-16 for 5 hours. Subsequently genomic DNA was extracted and modified  C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 689 for IPCR analysis. For caspase inhibition assay, after the cells have been cultured in the presence of PHA for each time point (60, 66 and 72 hours), cells were pre-treated for 1 hour with caspase inhibitor. Subsequently, they were either co-treated with 50 μM of VP-16 or 0.1% DMSO for 5 hours. Cells were then collected and proc- essed for IPCR. 3. RESULTS 3.1. VP-16 Induces Cleavage of the AF9 BCR CEM cells were treated with VP-16 and the extracted genomic DNA was modified and processed for IPCR analysis. IPCR primers were designed to detect chromo- some breaks within the BCR1 (intron 4 telomeric end) of AF9. As shown in Figure 2, with Kpn I digestion, only the intact AF9 gene was amplified, represented by the 950 base pairs bands (lanes 2-4). IPCR band less than 950 base pairs representing cleaved AF9 was not de- tected. This could be due to the competition between the intact chromosome and the cleaved chromosome (which is in a very small quantity) for the amplification process. Thus, Nde I was used to eliminate the amplification of the intact chromosome while at the same time to linearise the cyclised cleaved fragment as shown in Figure 1. Amplification of the intact AF9 was not eliminated but reduced (Figure 2, lanes 5-7), and at the same time smaller IPCR products of sizes 180 base pairs and 730 base pairs were detected upon VP-16 treatment (Figure 2, lane 7). DNA sequencing shows that the 180 base pairs was an amplification product of an AF9 cleavage frag- ment that carries a breakpoint at coordinate 26805 (GenBank accession No. AC000007), while the 730 base pairs corresponds to amplification of a cleaved AF9 fragment that carries a breakpoint at coordinate 26235 (GenBank accession No. AC000007). 3.2. Caspase Inhibitor Eliminates VP-16-Induced Cleavage of the AF9 BCR VP-16 is known to induce apoptosis [14], thus in order to investigate if this VP-16-induced AF9 cleavage was me- diated by the apoptotic nuclease CAD, caspase inhibition assay was performed. CAD usually exists as a complex with its inhibitor, ICAD. It is only upon apoptosis active- tion, when caspase cleaves ICAD, then CAD will be re- leased and be activated to cleave the genomic DNA [23]. Thus by inhibiting the caspase, CAD will not be acti- vated and would prevent cleavage of the AF9 BCR. As shown in Figure 3(a), in the absence of caspase inhibitor, treatment with VP-16 resulted in cleavage of the AF9 BCR producing a 420 base pairs band (lane 5). Indeed, pre-treatment with caspase inhibitor (Z-DEVD- FMK), and later co-treatment with VP-16 prevented VP-16- induced cleavage (Figure 3(a), lane 8). To ensure that the non-amplification observed in other samples were not due to template DNA degradation, IPCR with Kpn I-digested template DNA was performed, and amplifica- tion of the intact AF9 was observed as a 950 base pairs band (Figure 3(b), lanes 2-8). 3.3. K562 Cells are Resistant to VP-16-Induced Cleavage K562 cells have been shown to be more resistant to apoptosis induction compared with HL-60 cells [19]. Therefore K562 cells were used in this study to investi- gate if VP-16-induced AF9 cleavage would be delayed. As shown in Figure 4, with Kpn I digestion, amplifica- tion of the intact AF9 fragment was detected (lanes 2-8). However, with Nde I digestion, amplification of the in- tact AF9 fragment was eliminated, but there was no de- tection of the cleavage fragment (lanes 10-15). Note that the treatment times for K562 (5, 7 and 24 hours) were much longer as compared with that of CEM (5 hours) as presented before. Even with the extended treatment time, VP-16 failed to induce AF9 cleavage in K562 cells, fur- ther supporting AF9 cleavage was mediated through apoptosis. Figure 2. VP-16 induces AF9 cleavage. CEM cells were treated with 50 M of VP-16 (lanes 4 and 7), 0.1% DMSO (lanes 3 and 6), or left untreated as cell control (lanes 2 and 5). The cells were then lysed, genomic DNA was extracted and modified for IPCR as described in Materials and Methods. The modified DNA was either digested with Kpn I (lanes 2-4) or Nde I (lanes 5-7) prior to IPCR. Lane 1 is a negative control, which does not contain DNA template. The side arrows show the 950 bp, 730 bp, and 180 bp DNA fragments. M1: 1 kb DNA ladder. M2: 100 p DNA ladder. b Copyright © 2012 SciRes. OPEN ACCESS 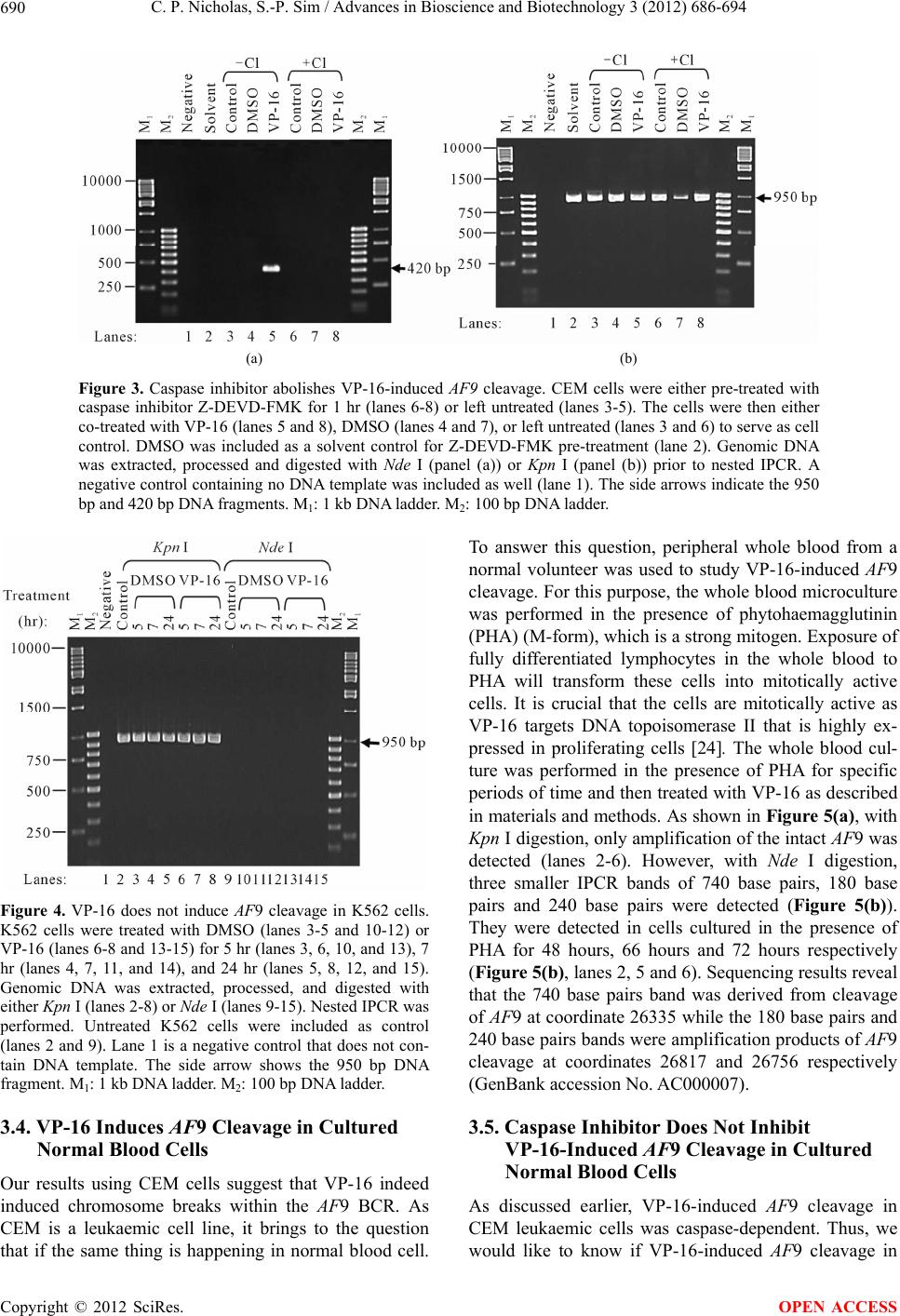 C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 Copyright © 2012 SciRes. 690 (a) (b) Figure 3. Caspase inhibitor abolishes VP-16-induced AF9 cleavage. CEM cells were either pre-treated with caspase inhibitor Z-DEVD-FMK for 1 hr (lanes 6-8) or left untreated (lanes 3-5). The cells were then either co-treated with VP-16 (lanes 5 and 8), DMSO (lanes 4 and 7), or left untreated (lanes 3 and 6) to serve as cell control. DMSO was included as a solvent control for Z-DEVD-FMK pre-treatment (lane 2). Genomic DNA was extracted, processed and digested with Nde I (panel (a)) or Kpn I (panel (b)) prior to nested IPCR. A negative control containing no DNA template was included as well (lane 1). The side arrows indicate the 950 bp and 420 bp DNA fragments. M1: 1 kb DNA ladder. M2: 100 bp DNA ladder. Figure 4. VP-16 does not induce AF9 cleavage in K562 cells. K562 cells were treated with DMSO (lanes 3-5 and 10-12) or VP-16 (lanes 6-8 and 13-15) for 5 hr (lanes 3, 6, 10, and 13), 7 hr (lanes 4, 7, 11, and 14), and 24 hr (lanes 5, 8, 12, and 15). Genomic DNA was extracted, processed, and digested with either Kpn I (lanes 2-8) or Nde I (lanes 9-15). Nested IPCR was performed. Untreated K562 cells were included as control (lanes 2 and 9). Lane 1 is a negative control that does not con- tain DNA template. The side arrow shows the 950 bp DNA fragment. M1: 1 kb DNA ladder. M2: 100 bp DNA ladder. 3.4. VP-16 Induces AF9 Cleavage in Cultured Normal Blood Cells Our results using CEM cells suggest that VP-16 indeed induced chromosome breaks within the AF9 BCR. As CEM is a leukaemic cell line, it brings to the question that if the same thing is happening in normal blood cell. To answer this question, peripheral whole blood from a normal volunteer was used to study VP-16-induced AF9 cleavage. For this purpose, the whole blood microculture was performed in the presence of phytohaemagglutinin (PHA) (M-form), which is a strong mitogen. Exposure of fully differentiated lymphocytes in the whole blood to PHA will transform these cells into mitotically active cells. It is crucial that the cells are mitotically active as VP-16 targets DNA topoisomerase II that is highly ex- pressed in proliferating cells [24]. The whole blood cul- ture was performed in the presence of PHA for specific periods of time and then treated with VP-16 as described in materials and methods. As shown in Figure 5(a), with Kpn I digestion, only amplification of the intact AF9 was detected (lanes 2-6). However, with Nde I digestion, three smaller IPCR bands of 740 base pairs, 180 base pairs and 240 base pairs were detected (Figure 5(b)). They were detected in cells cultured in the presence of PHA for 48 hours, 66 hours and 72 hours respectively (Figure 5(b), lanes 2, 5 and 6). Sequencing results reveal that the 740 base pairs band was derived from cleavage of AF9 at coordinate 26335 while the 180 base pairs and 240 base pairs bands were amplification products of AF9 cleavage at coordinates 26817 and 26756 respectively (GenBank accession No. AC000007). 3.5. Caspase Inhibitor Does Not Inhibit VP-16-Induced AF9 Cleavage in Cultured Normal Blood Cells As discussed earlier, VP-16-induced AF9 cleavage in CEM leukaemic cells was caspase-dependent. Thus, we would like to know if VP-16-induced AF9 cleavage in OPEN ACCESS 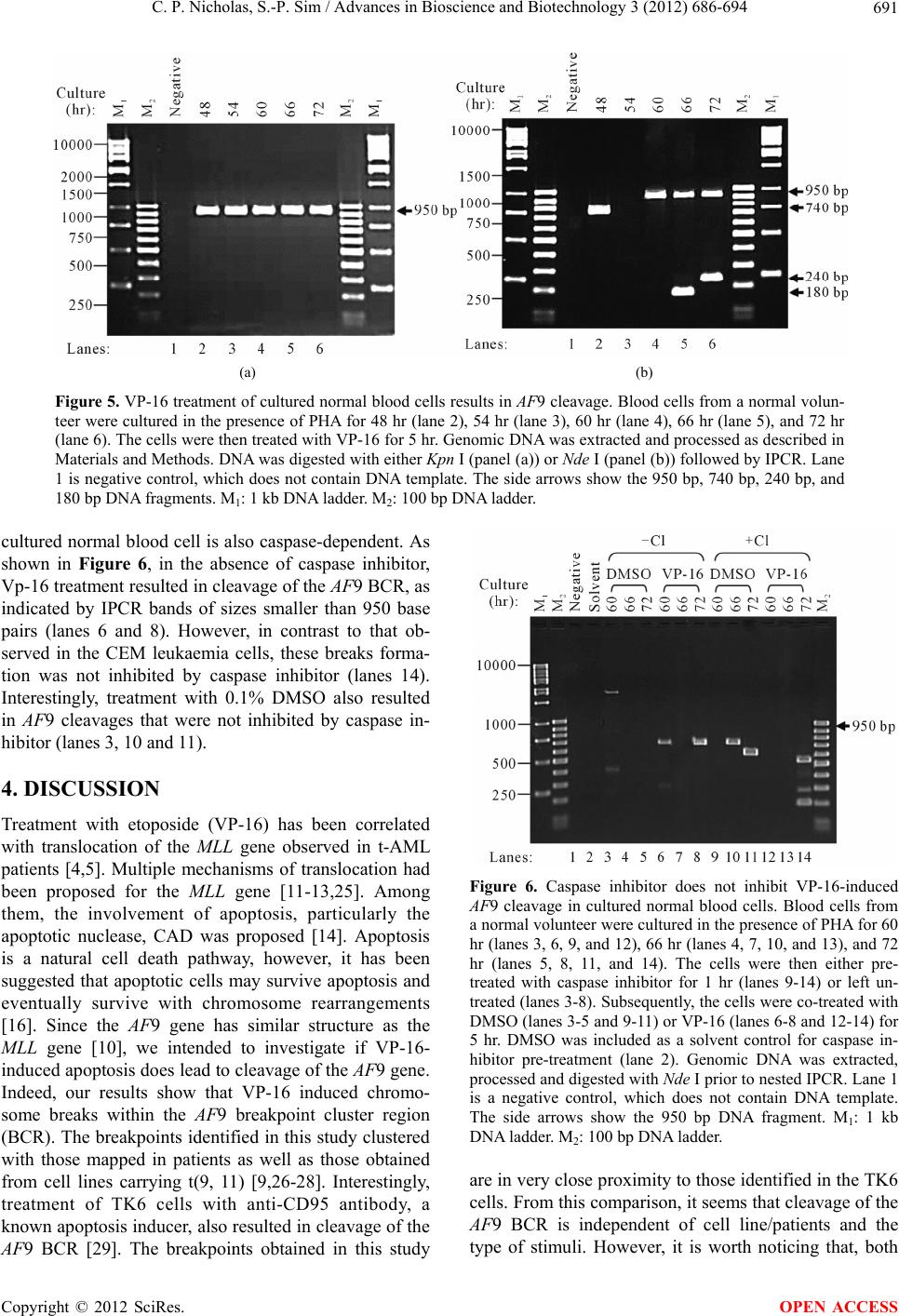 C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 691 (a) (b) Figure 5. VP-16 treatment of cultured normal blood cells results in AF9 cleavage. Blood cells from a normal volun- teer were cultured in the presence of PHA for 48 hr (lane 2), 54 hr (lane 3), 60 hr (lane 4), 66 hr (lane 5), and 72 hr (lane 6). The cells were then treated with VP-16 for 5 hr. Genomic DNA was extracted and processed as described in Materials and Methods. DNA was digested with either Kpn I (panel (a)) or Nde I (panel (b)) followed by IPCR. Lane 1 is negative control, which does not contain DNA template. The side arrows show the 950 bp, 740 bp, 240 bp, and 180 bp DNA fragments. M1: 1 kb DNA ladder. M2: 100 bp DNA ladder. cultured normal blood cell is also caspase-dependent. As shown in Figure 6, in the absence of caspase inhibitor, Vp-16 treatment resulted in cleavage of the AF9 BCR, as indicated by IPCR bands of sizes smaller than 950 base pairs (lanes 6 and 8). However, in contrast to that ob- served in the CEM leukaemia cells, these breaks forma- tion was not inhibited by caspase inhibitor (lanes 14). Interestingly, treatment with 0.1% DMSO also resulted in AF9 cleavages that were not inhibited by caspase in- hibitor (lanes 3, 10 and 11). 4. DISCUSSION Treatment with etoposide (VP-16) has been correlated with translocation of the MLL gene observed in t-AML patients [4,5]. Multiple mechanisms of translocation had been proposed for the MLL gene [11-13,25]. Among them, the involvement of apoptosis, particularly the apoptotic nuclease, CAD was proposed [14]. Apoptosis is a natural cell death pathway, however, it has been suggested that apoptotic cells may survive apoptosis and eventually survive with chromosome rearrangements [16]. Since the AF9 gene has similar structure as the MLL gene [10], we intended to investigate if VP-16- induced apoptosis does lead to cleavage of the AF9 gene. Indeed, our results show that VP-16 induced chromo- some breaks within the AF9 breakpoint cluster region (BCR). The breakpoints identified in this study clustered with those mapped in patients as well as those obtained from cell lines carrying t(9, 11) [9,26-28]. Interestingly, treatment of TK6 cells with anti-CD95 antibody, a known apoptosis inducer, also resulted in cleavage of the AF9 BCR [29]. The breakpoints obtained in this study Figure 6. Caspase inhibitor does not inhibit VP-16-induced AF9 cleavage in cultured normal blood cells. Blood cells from a normal volunteer were cultured in the presence of PHA for 60 hr (lanes 3, 6, 9, and 12), 66 hr (lanes 4, 7, 10, and 13), and 72 hr (lanes 5, 8, 11, and 14). The cells were then either pre- treated with caspase inhibitor for 1 hr (lanes 9-14) or left un- treated (lanes 3-8). Subsequently, the cells were co-treated with DMSO (lanes 3-5 and 9-11) or VP-16 (lanes 6-8 and 12-14) for 5 hr. DMSO was included as a solvent control for caspase in- hibitor pre-treatment (lane 2). Genomic DNA was extracted, processed and digested with Nde I prior to nested IPCR. Lane 1 is a negative control, which does not contain DNA template. The side arrows show the 950 bp DNA fragment. M1: 1 kb DNA ladder. M2: 100 bp DNA ladder. are in very close proximity to those identified in the TK6 cells. From this comparison, it seems that cleavage of the AF9 BCR is independent of cell line/patients and the type of stimuli. However, it is worth noticing that, both Copyright © 2012 SciRes. OPEN ACCESS  C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 692 VP-16 and anti-CD95 are apoptosis stimuli. Similar re- sults were observed for the ML L BCR cleavage, whereby various chemotherapeutic drugs (VP-16, 5-fluorouracil, methotrexate, vinblastine and camptothecin) induced apo- ptosis as well as cleavage of the MLL BCR [14]. VP-16 targets DNA topoisomerase II and results in DNA double stranded breaks [30]. This DNA damage may induce the apoptotic signalling pathway [31], whereby the caspase cascade is activated. CAD is nor- mally inhibited by ICAD. During apoptosis, caspase 3 cleaves ICAD, thus releasing the activated CAD, which eventually cleaves the chromosomal DNA [23,32]. In our study, Z-DEVD-FMK was used to investigate if the VP-16-induced AF9 cleavage was mediated by CAD. Z-DEVD-FMK is a potent, cell-permeable and irreversi- ble inhibitor of caspase 3, 6, 7, 8 and 10. Our results in leukaemic cell line indicated that CAD was most likely responsible for the AF9 cleavage. This is in line with other reports in leukaemic cell line [14] and in naso- pharyngeal carcinoma cell line as well [33]. In apoptotic cells, CAD associates with the nuclear matrix [34], thus allowing it to cleave the base of the chromatin loops at- taching to the nuclear matrix or scaffold, generating high molecular weight (HMW) DNA during early stage apoptosis [15,35]. This is again supported by our results in K562 cells, where AF9 cleavage was absent despite long period of VP-16 treatment. K562 is known to ex- hibit delayed apoptosis induction. This delay resulted from a delay in the signaling cascade upstream of cyto- chrome c release and caspase activation [19]. Taken to- gether, it seems that VP-16-induced cleavages of the AF9 were most likely to be mediated by CAD. Our results showed that normal blood cell also exhib- ited AF9 cleavage upon VP-16 treatment. However, these cleavages were not inhibited by caspase inhibitor. This contradictory observation could be due to a few possi- bilities. Firstly, normal blood cells and leukaemic cells may have different capacity of uptake for the caspase inhibitor. Although the uptake of caspase inhibitor has not been reported, it is known that the uptake capacity of methotrexate is different between leukaemic cells and normal leucocytes [36]. In addition, uptake of nitrogen mustard and choline were shown to be higher in leukae- mic lymphoid cells compared with normal lymphoid cells [37]. Secondly, the normal blood cells might have died via the caspase-independent pathway as demon- strated by others [38,39]. This is seconded by the obser- vation that mouse embryonic fibroblast (MEF) from caspase 3-deficient mice and CAD-deficient chicken DT40 cells were able to generate HMW DNA during apoptosis induction [40,41]. Other than CAD, the other possible candidate is Endonuclease G (Endo G). Endo G is an evolutionary conserved endonuclease that usually localised in the intermembrane space of mitochondria, but released during apoptosis and results in caspase-in- dependent DNA degradation [42,43]. However, based on its cleavage preferences, Endo G most likely does not work alone. It may work with DNase-I-like enzymes together with exonucleases in vivo for apoptotic DNA processing [44]. In addition, apoptosis-inducing factor (AIF) is another enzyme that is released from mitochon- dria during apoptosis and cause chromatin condensation and large scale DNA fragmentation [41]. DNA topoi- somerase II is another possible player. It was proposed to be directly involved in the MLL BCR cleavage [45]. Poisoning of topoisomerase II by VP-16 resulted in chromatin loop excision [46]. This is very logical as to- poisomerase II is one of the major proteins found in the nuclear scaffold [47]. Interestingly, CAD interacts with topoisomerase II and enhances topoisomerase II’s de- catenation activity in vitro [48]. Our sequencing analysis revealed that all the break- points are mapped inside the BCR1 within close prox- imity to one of the MAR/SAR. MAR/SAR sequences are abundantly found at the base of chromatin loop struc- tures [49], and excision of the chromatin loops forming HMW DNA occurs during early apoptosis [35]. Thus our results indicate a possibility that the apoptotic nuclease(s) could be cleaving the base of the chromatin loop struc- ture during early apoptosis. This is a crucial observation, as we hypothesise that apoptotic cells escape the cell death pathway and rescued as cells carrying chromosome rearrangements, thus it is logical that only those at early apoptosis could be rescued. In conclusion, our results suggest that apoptotic nu- clease(s) could be involved in VP-16-induced AF9 cleavage, and thus contributing to chromosome translo- cation in t-AML. 5. ACKNOWLEDGEMENTS We would like to acknowledge Prof. Dr. Leroy F. Liu for the K562 cell line. We are also grateful to the Sarawak General Hospital pathology laboratory staff for their technical advice. This work was supported by the UNIMAS Fundamental Research Grant No. 1(4)/266/02(4) and partly the IRPA Grant No. 09-02-09-1017 from the Ministry of Science, Technology and Innovation. REFERENCES [1] Ross, W., Rowe, T., Glisson, B., Yalowich, J. and Liu, L. (1984) Role of topoisomerase II in mediating epipodo- phyllotoxin-induced DNA cleavage. Cancer Research, 44, 5857-5860. [2] Osheroff, N. (1989) Effect of antineoplastic agents on the DNA cleavage/religation reaction of eukaryotic topoi- somerase II: Inhibition of DNA religation by etoposide. Biochemistry, 28, 6157-6160. doi:10.1021/bi00441a005 Copyright © 2012 SciRes. OPEN ACCESS  C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 693 [3] Felix, C.A. (1998) Secondary leukemias induced by to- poisomerase-targeted drugs. Biochimica et Biophysica Acta, 1400, 233-255. doi:10.1016/S0167-4781(98)00139-0 [4] Felix, C.A., Hosler, M.R., Winick, N.J., Masterson, M., Wilson, A.E. and Lange, B.J. (1995) ALL-1 gene rear- rangements in DNA topoisomerase II inhibitor-related leukemia in children. Blood, 85, 3250-3256. [5] Super, H.J., McCabe, N.R., Thirman, M.J., Larson, R.A., Le Beau, M.M., Pedersen-Bjergaard, J., Philip, P., Diaz, M.O. and Rowley, J.D. (1993) Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA- topoisomerase II. Blood, 82, 3705-3711. [6] Broeker, P.L., Super, H.G., Thirman, M.J., Pomykala, H., Yonebayashi, Y., Tanabe, S., Zeleznik-Le, N. and Row- ley, J.D. (1996) Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leu- kemia and in treatment-related acute myeloid leukemia: Correlation with scaffold attachment regions and topoi- somerase II consensus binding sites. Blood, 87, 1912- 1922. [7] Huret J.L. (2005) MLL(myeloid/lymphoid or mixed line- age leukemia). Atlas of Genetics and Cytogenetics in Oncology and Haematology, 1, 68-69. http://AtlasGeneticsOncology.org/Genes/MLL.html [8] Nakamura, T., Alder, H., Gu, Y., Prasad, R., Canaani, O., Kamada, N., Gale, R.P., Lange, B., Crist, W.M. and Nowell, P.C. (1993) Genes on chromosomes 4, 9, and 19 involved in 11q23 abnormalities in acute leukemia share sequence homology and/or common motifs. Proceedings of the National Academy of Sciences of USA, 90, 4631- 4635. doi:10.1073/pnas.90.10.4631 [9] Super, H.G., Strissel, P.L., Sobulo, O.M., Burian, D., Reshmi, S.C., Roe, B., Zeleznik, L., Diaz, M.O. and Rowley, J.D. (1997) Identification of complex genomic breakpoint junctions in the t(9;11) MLL-AF9 fusion gene in acute leukemia. Genes Chromosomes. Cancer, 20, 185-195. doi:10.1002/(SICI)1098-2264(199710)20:2<185::AID-G CC9>3.0.CO;2-# [10] Strissel, P.L., Strick, R., Tomek, R.J., Roe, B.A., Rowley, J.D. and Zeleznik, L. (2000) DNA structural properties of AF9 are similar to MLL and could act as recombination hot spots resulting in MLL/AF9 translocations and leu- kemogenesis. Human Molecular Genetics, 9, 1671-1679. doi:10.1093/hmg/9.11.1671 [11] Gu, Y., Alder, H., Nakamura, T., Schichman, S.A., Prasad, R., Canaani, O., Saito, H., Croce, C.M. and Ca- naani, E. (1994) Sequence analysis of the breakpoint cluster region in the ALL-1 gene involved in acute leu- kemia. Cancer Research, 54, 2327-2330. [12] Gu, Y., Cimino, G., Alder, H., Nakamura, T., Prasad, R., Canaani, O., Moir, D.T., Jones, C., Nowell, P.C. and Croce, C.M. (1992) The (4;11)(q21;q23) chromosome translocations in acute leukemias involve the VDJ re- combinase. Proceedings of the National Academy of Sci- ences of USA, 89, 10464-10468. doi:10.1073/pnas.89.21.10464 [13] Strissel, P.L., Strick, R., Rowley, J.D. and Zeleznik, L. (1998) An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood, 92, 3793-3803. [14] Sim, S.P. and Liu, L.F. (2001) Nucleolytic cleavage of the mixed lineage leukemia breakpoint cluster region during apoptosis. Journal of Biological Chemistry, 276, 31590-31595. doi:10.1074/jbc.M103962200 [15] Sakahira, H., Enari, M., Ohsawa, Y., Uchiyama, Y. and Nagata, S. (1999) Apoptotic nuclear morphological change without DNA fragmentation. Current Biology, 9, 543-546. doi:10.1016/S0960-9822(99)80240-1 [16] Vaughan, A.T., Betti, C.J. and Villalobos, M.J. (2002) Surviving apoptosis. Apoptosis, 7, 173-177. doi:10.1023/A:1014374717773 [17] Hars, E.S., Lyu, Y.L., Lin, C.P. and Liu, L.F. (2006) Role of apoptotic nuclease caspase-activated DNase in eto- poside-induced treatment-related acute myelogenous leu- kemia. Cancer Research, 66, 8975-8979. doi:10.1158/0008-5472.CAN-06-1724 [18] Klein, E., Ben Bassat, H., Neumann, H., Ralph, P., Zeuthen, J., Polliack, A. and Vanky, F. (1976) Properties of the K562 cell line, derived from a patient with chronic myeloid leukemia. International Journal of Cancer, 18, 421-431. doi:10.1002/ijc.2910180405 [19] Martins, L.M., Mesner, P.W., Kottke, T.J., Basi, G.S., Sinha, S., Tung, J.S., Svingen, P.A., Madden, B.J., Taka- hashi, A., McCormick, D.J., Earnshaw, W.C. and Kauf- mann, S.H. (1997) Comparison of caspase activation and subcellular localization in HL-60 and K562 cells under- going etoposide-induced apoptosis. Blood, 90, 4283- 4296. [20] Widlak, P., Li, P., Wang, X. and Garrard, W.T. (2000) Cleavage preferences of the apoptotic endonuclease DFF40 (caspase-activated DNase or nuclease) on naked DNA and chromatin substrates. Journal of Biological Chemistry, 275, 8226-8232. doi:10.1074/jbc.275.11.8226 [21] Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Mo- lecular cloning: A laboratory manual. 2nd Edition, Cold Spring Harbor Laboratory, Plainview, New York. [22] Watt, J.L. and Stephen, S.G.S. (1986) Lymphocyte cul- ture for chromosome analysis. In: Rooney, D.E. and Czepulkowski, B.H., Eds., Human Cytogenetics: A Prac- tical Approach, Oxford University Press, Oxford, 39-55. [23] Sakahira, H., Enari, M. and Nagata, S. (1998) Cleavage of CAD inhibitor in CAD activation and DNA degrada- tion during apoptosis. Nature, 391, 96-99. doi:10.1038/34214 [24] Hsiang, Y.H., Wu, H.Y. and Liu, L.F. (1988) Prolifera- tion-dependent regulation of DNA topoisomerase II in cultured human cells. Cancer Research, 48, 3230-3235. [25] Tamai, H. and Inokuchi, K. (2010) 11q23/MLL acute leukemia: Update of clinical aspects. Journal of Clinical and Experimental Hematopathology, 50, 91-98. doi:10.3960/jslrt.50.91 [26] Atlas, M., Head, D., Behm, F., Schmidt, E., Zeleznik, L., Roe, B.A., Burian, D. and Domer, P.H. (1998) Cloning and sequence analysis of four t(9;11) therapy-related Copyright © 2012 SciRes. OPEN ACCESS  C. P. Nicholas, S.-P. Sim / Advances in Bioscience and Biotechnology 3 (2012) 686-694 Copyright © 2012 SciRes. 694 OPEN ACCESS leukemia breakpoints. Leukemia, 12, 1895-1902. doi:10.1038/sj.leu.2401223 [27] Odero, M.D., Zeleznik, L., Chinwalla, V. and Rowley, J.D. (2000) Cytogenetic and molecular analysis of the acute monocytic leukemia cell line THP-1 with an MLL- AF9 translocation. Genes Chromosomes. Cancer, 29, 333-338. doi:10.1002/1098-2264(2000)9999:9999<::AID-GCC104 0>3.0.CO;2-Z [28] Super, H.J., Martinez-Climent, J. and Rowley, J.D. (1995) Molecular analysis of the Mono Mac 6 cell line: Detec- tion of an MLL-AF9 fusion transcript. Blood, 85, 855- 856. [29] Betti, C.J., Villalobos, M.J., Diaz, M.O. and Vaughan, A.T. (2003) Apoptotic stimuli initiate MLL-AF9 translo- cations that are transcribed in cells capable of division. Cancer Research, 63, 1377-1381. [30] Yang, L., Rowe, T.C. and Liu, L.F. (1985) Identification of DNA topoisomerase II as an intracellular target of an- titumor epipodophyllotoxins in simian virus 40-infected monkey cells. Cancer Research, 45, 5872-5876. [31] Jackson, S.P. (2002) Sensing and repairing DNA dou- ble-strand breaks. Carcinogenesis, 23, 687-696. doi:10.1093/carcin/23.5.687 [32] Enari, M., Sakahira, H., Yokoyama, H., Okawa, K., Iwamatsu, A. and Nagata, S. (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its in- hibitor ICAD. Nature, 391, 43-50. doi:10.1038/34112 [33] Yee, P.H. and Sim, S.P. (2010) High cell density and latent membrane protein 1 expression induce cleavage of the mixed lineage leukemia gene at 11q23 in naso- pharyngeal carcinoma cell line. Journal of Biomedical Science, 17, 77. doi:10.1186/1423-0127-17-77 [34] Lechardeur, D., Xu, M. and Lukacs, G.L. (2004) Con- trasting nuclear dynamics of the caspase-activated DNase (CAD) in dividing and apoptotic cells. Journal of Cell Biology, 167, 851-862. doi:10.1083/jcb.200404105 [35] Lagarkova, M.A., Iarovaia, O.V. and Razin, S.V. (1995) Large-scale fragmentation of mammalian DNA in the course of apoptosis proceeds via excision of chromoso- mal DNA loops and their oligomers. Journal of Biologi- cal Chemistry, 270, 20239-20241. doi:10.1074/jbc.270.35.20239 [36] Kessel, D., Hall, T.C. and Roberts, D. (1968) Modes of uptake of methotrexate by normal and leukemic human leukocytes in vitro and their relation to drug response. Cancer Research, 28, 564-570. [37] Lyons, R.M. and Goldenberg, G.J. (1972) Active trans- port of nitrogen mustard and choline by normal and leu- kemic human lymphoid cells. Cancer Research, 32, 1679- 1685. [38] Kim, D.K., Cho, E.S. and Um, H.D. (2000) Caspase- dependent and -independent events in apoptosis induced by hydrogen peroxide. Experimental Cell Research, 257, 82-88. doi:10.1006/excr.2000.4868 [39] Kitagawa, K. and Niikura, Y. (2008) Caspase-independent mitotic death (CIMD). Cell Cycle, 7, 1001-1005. doi:10.4161/cc.7.8.5720 [40] Samejima, K., Tone, S. and Earnshaw, W.C. (2001) CAD/DFF40 nuclease is dispensable for high molecular weight DNA cleavage and stage I chromatin condensa- tion in apoptosis. Journal of Biological Chemistry, 276, 45427-45432. doi:10.1074/jbc.M108844200 [41] Susin, S.A., Lorenzo, H.K., Zamzami, N., Marzo, I., Snow, B.E., Brothers, G.M., Mangion, J., Jacotot, E., Co- stantini, P., Loeffler, M., Larochette, N., Goodlett, D.R., Aebersold, R., Siderovski, D.P., Penninger, J.M. and Kroemer, G. (1999) Molecular characterization of mito- chondrial apoptosis-inducing factor. Nature, 397, 441- 446. doi:10.1038/17135 [42] Li, L.Y., Luo, X. and Wang, X. (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature, 412, 95-99. doi:10.1038/35083620 [43] Van Loo, G., Schotte, P., van Gurp, M., Demol, H., Hoorelbeke, B., Gevaert, K., Rodriguez, I., Ruiz-Carrillo, A., Vandekerckhove, J., Declercq, W., Beyaert, R. and Vandenabeele, P. (2001) Endonuclease G: A mitochon- drial protein released in apoptosis and involved in cas- pase-independent DNA degradation. Cell Death & Dif- ferentiation, 8, 1136-1142. doi:10.1038/sj.cdd.4400944 [44] Widlak, P., Li, L.Y., Wang, X. and Garrard, W.T. (2001) Action of recombinant human apoptotic endonuclease G on naked DNA and chromatin substrates: Cooperation with exonuclease and DNase I. Journal of Biological Chemistry, 276, 48404-48409. [45] Strick, R., Strissel, P.L., Borgers, S., Smith, S.L. and Rowley, J.D. (2000) Dietary bioflavonoids induce cleav- age in the MLL gene and may contribute to infant leuke- mia. Proceedings of the National Academy of Sciences of USA, 97, 4790-4795. doi:10.1073/pnas.070061297 [46] Solovyan, V.T., Bezvenyuk, Z.A., Salminen, A., Austin, C.A. and Courtney, M.J. (2002) The role of topoisom- erase II in the excision of DNA loop domains during apoptosis. Journal of Biological Chemistry, 277, 21458- 21467. doi:10.1074/jbc.M110621200 [47] Earnshaw, W.C., Halligan, B., Cooke, C.A., Heck, M.M. and Liu, L.F. (1985) Topoisomerase II is a structural component of mitotic chromosome scaffolds. Journal of Cell Biology, 100, 1706-1715. doi:10.1083/jcb.100.5.1706 [48] Durrieu, F., Samejima, K., Fortune, J.M., Kandels-Lewis, S., Osheroff, N. and Earnshaw, W.C. (2000) DNA topoi- somerase IIalpha interacts with CAD nuclease and is in- volved in chromatin condensation during apoptotic exe- cution. Current Biology, 10, 923-926. doi:10.1016/S0960-9822(00)00620-5 [49] Laemmli, U.K., Kas, E., Poljak, L. and Adachi, Y. (1992) Scaffold-associated regions: Cis-acting determinants of chromatin structural loops and functional domains. Cur- rent Opinion in Genetics & Development, 2, 275-285. doi:10.1016/S0959-437X(05)80285-0.
|