 Advances in Bioscience and Biotechnology, 2012, 3, 657-664 ABB http://dx.doi.org/10.4236/abb.2012.326085 Published Online October 2012 (http://www.SciRP.org/journal/abb/) Increased survivin expression contributes to apoptosis-resistance in IPF fibroblasts Thomas H. Sisson1, Toby M. Maher2, Iyabode O. Ajayi1, Jessie E. King1, Peter D. R. Higgins3, Adam J. Booth1, Rommel L. Sagana1, Steven K. Huang1, Eric S. White1, Bethany B. Moore1, Jeffrey C. Horowitz1* 1Division of Pulmonary and Critical Care Medicine, Department of Medicine, University of Michigan Medical Center, Ann Arbor, USA 2Interstitial Lung Disease Unit, Royal Brompton Hospital, London, UK 3Division of Gastroenterology, Department of Medicine, University of Michigan Medical Center, Ann Arbor, USA Email: *jchorow@umich.edu Received 30 July 2012; revised 6 September 2012; accepted 16 September 2012 ABSTRACT Fibroblasts perform critical functions during the normal host response to tissue injury, but the inap- propriate accumulation and persistent activation of these cells results in the development of tissue fibrosis. The mechanisms accounting for the aberrant accu- mulation of fibroblasts during fibrotic repair are poorly understood, although evidence supports a role for fibroblast resistance to apoptosis as a contributing factor. We have shown that TGF-β1 and endothelin-1 (ET-1), soluble mediators implicated in fibrogenesis, promote fibroblast resistance to apoptosis. Moreover, we recently found that ET-1 induced apoptosis resis- tance in normal lung fibroblasts through the upregu- lation of survivin, a member of the Inhibitor of Apoptosis (IAP) protein family. In the current study, we sought to determine the role of survivin in the apoptosis resistance of primary fibroblasts isolated from the lungs of patients with Idiopathic Pulmonary Fibrosis (IPF), a fibrotic lung disease of unclear eti- ology for which there is no definitive therapy. First, we examined survivin expression in lung tissue from patients with IPF and found that there is robust ex- pression in the fibroblasts residing within fibroblastic foci (the “active” lesions in IPF which correlate with mortality). Next, we show that survivin expression is increased in fibroblasts isolated from IPF lung tissue compared to cells from normal lung t issue. Co nsist ent with a role in fibrogenesis, we demonstrate that TGF- β1 increases survivin expression in normal lung fi- broblasts. Finally, we show that inhibition of sur- vivin enhances susceptibility of a subset of IPF fibro- blasts to apoptosis. Collectively, these findings suggest that increased survivin expression represents one mechanism contributing to an apoptosis-resistant phe - notype in IPF fibroblasts. Keywords: Myofibroblast; Idiopathic Pulmonary Fibrosis; Inhibitor of Apoptosis; Lung Fibrosis; Fas 1. INTRODUCTION Idiopathic Pulmonary Fibrosis (IPF) is a chronic lung disease characterized by the progressive development of scar tissue which can ultimately lead to respiratory fail- ure and death. The incidence of IPF is increasing and, with an average survival of 2 - 3 years following the dia- gnosis, the overall prognosis is poor [1-3]. Although several randomized clinical trials have evaluated a num- ber of therapeutic targets in IPF over the last decade, there continues to be a lack of clearly effective pharma- cologic therapy [4]. Despite the generally poor prognosis, a well-recognized clinical feature of IPF is the markedly heterogeneous clinical course, with some patients main- taining stable lung function for years while others ex- perience a rapid deterioration culminating in respiratory failure and death within months of the diagnosis [5]. It is critical to better understand the mechanisms of biologic heterogeneity that underlie the variable clinical course of IPF in order to identify novel targets for intervention that can be applied in a personalized manner [6]. Alveolar scarring in IPF is hypothesized to result from dysfunctional wound repair which is characterized by the accumulation of mesenchymal cells (including fibro- blasts and their differentiated phenotype, myofibroblasts) within pathologic lesions called fibroblastic foci [7,8]. During normal reparative responses following injury, mesenchymal cells perform critical functions which pro- vide healing wounds with tensile strength and facilitate the homeostatic restoration of epithelial integrity, in- cluding the synthesis, secretion, organization and con- *Corresponding author. OPEN ACCESS  T. H. Sisson et al. / Advances in Bioscience and Biotechnology 3 (2012) 657-664 658 traction of new extracellular matrix (ECM), [1,9-11]. Resolution of the normal repair process requires clear- ance of fibroblasts via apoptosis [12,13]. However, in the dysfunctional repair that is evident in IPF and other dis- eases characterized by fibrosis, the persistence of repara- tive fibroblasts leads to excessive deposition and organi- zation of ECM [14]. Several recent studies demonstrate a paucity of apoptosis within the mesenchymal cells that comprise the fibroblastic foci in IPF, highlighting one mechanism that contributes to the persistence of these cells [15-18]. Accumulating studies also show that fi- brotic lung fibroblasts are resistant to a variety of apop- totic stimuli, further supporting a role for impaired fibro- blast apoptosis in the pathobiology of IPF [18-25]. How- ever, the mechanisms regulating resistance to apoptosis in lung fibroblasts have not been fully elucidated [14]. Recent studies suggest that Inhibitor of Apoptosis (IAP) family proteins can regulate susceptibility to apop- tosis in normal lung fibroblasts [18,20,24]. The IAP genes encode at least eight different proteins with diverse functions, including the direct and indirect regulation of apoptosis. The protein family is characterized by the presence of at least one baculoviral IAP repeat (BIR) domain which is critical for the ability of these proteins to inhibit caspase activation in vitro [26,27]. The small- est member of the IAP family, survivin (also known as BIRC5) is a 16.3 kD protein with one BIR domain that is predominantly expressed as a homodimer. Physiologi- cally, survivin serves a critical function in cell-cycle progression and is essential during embryonic develop- ment. In adult cells, however, it is typically expressed only in actively dividing cells [28]. As with other IAP family members, survivin inhibits activation of caspase 3, 7 and 9 in-vitro. Moreover, a number of studies demon- strate regulation of cell susceptibility to apoptosis by survivin, although the precise in vivo mechanisms re- main unclear [27]. In accord with its crucial function in cell division and apoptosis regulation, inappropriate sur- vivin expression is associated with cancers involving multiple organs including lung, colon, breast, brain and skin [27,29,30]. We have recently shown that induction of survivin by the soluble fibrogenic mediator endo- thelin-1 (ET-1) reduces the apoptotic susceptibility of normal fetal lung fibroblasts and that the anti-fibrotic mediator prostaglandin E2 enhances fibroblast suscepti- bility to apoptosis while suppressing survivin expression [20,24]. The goal of this study was to examine the ex- pression of survivin in IPF lung fibroblasts and to deter- mine if survivin regulates IPF fibroblast susceptibility to Fas-mediated apoptosis. 2. MATERIALS AND METHODS 2.1. Cells and Cell Culture Normal human adult lung fibroblasts (CCL-210) were obtained from ATCC (Manassas, VA). Cells between passages 8 and 16 were cultured as previously described and growth arrested for 24 hours in serum-free media prior to experiments [20]. Patient-derived primary human lung fibroblasts were cultured from the lungs of patients with IPF or from normal lung tissue from patients un- dergoing thoracic surgery for non-fibrotic disease as we have described previously [31]. Written informed con- sent was obtained from all subjects in accordance with the University of Michigan Institutional Review Board. 2.2. Reagents Porcine TGF-β1 was from R&D Systems (Minneapolis, MN). The activating anti-Fas antibody (clone CH11) was from Millipore (Billerica, MA). The mouse monoclonal antibody to survivin used for Western blotting was from Abnova. Antibodies to glyceraldehyde-3-phosphate de- hydrogenase (GAPDH) were from Cell Signaling (Dan- vers, MA). The Cell Death Detection ELISA Kit detect- ing histone-associated DNA fragments was from Roche Applied Science (Indianapolis, IN). Horseradish peroxi- dase conjugated secondary antibodies were from Pierce (Rockford, IL). The survivin inhibitor CAY10625, which disrupts the interaction between survivin and Smac/ Diablo (second mitochondrial activator of caspases/direct IAP binding protein with low isoelectric point), thereby allowing Smac/Diablo to carry out its pro-apoptotic func- tion, was from Cayman Chemical Company (Ann Arbor, MI) [32]. YM155, a functionally distinct inhibitor which inhibits survivin transcription [33], was from Selleck Chemicals (Houston, TX). 2.3. Immunohistochemistry 4 µm sections on polylysiene coated slides (VWR, Leicestershire, UK) were de-waxed and rehydrated by immersion in xylene followed by decreasing concentra- tions of ethanol. Sections were washed with tris-buffered saline (TBS, pH 7.6), and endogenous peroxidise activity was blocked by immersion in 3% H2O2 (v/v) for 30 min- utes at room temperature. Sections were washed twice in TBS and treated with a 1 in 6 solution of goat serum containing 4 drops/ml of Avidin block (Vector Laborato- ries, Peterborough, UK) for 20 minutes and incubated overnight with rabbit anti-survivin polyclonal antibodies (4 µg/ml; Santa Cruz Biotechnology) or an isotype con- trol in TBS with 1% bovine serum albumin and 4 drops/ ml of Biotin block. Following another wash, samples were treated with a 1:200 dilution of biotinylated second- dary antibody (Dako, Cambridshire, UK), washed again and immersed in a 1:200 dilution of Streptavadin HRP (Dako) for 30 minutes at room temperature. After wash- ing, DAB peroxidise substrate (Vector Laboratories) was added for ten minutes. Samples were rinsed in H2O, Copyright © 2012 SciRes. OPEN ACCESS  T. H. Sisson et al. / Advances in Bioscience and Biotechnology 3 (2012) 657-664 659 counterstained with Gill’s Haematoxylin, dehydrated in Xylene and mounted using a Sakura Coveraid system. 2.4. Quantitative Reverse Transcription Real-Time PCR Quantitative reverse transcription real time-PCR was performed on an Applied Biosystems (Foster City, CA) 7300 real time PCR machine as we have reported previ- ously [31]. Relative quantitation was based on the ΔΔCT method and used predesigned primer-probe sets obtained from Applied Biosystems TaqMan Gene Expression As- says for human Survivin (BIRC5; product number Hs00153353_m1). 2.5. SDS Page Electrophoresis and Western Blotting Whole cell lysates were collected and subjected to SDS- PAGE electrophoresis and Western blotting as previ- ously described [20]. All Western blots were stripped and re-probed for GAPDH. 2.6. Densitometry Western blots were analyzed using the public domain NIH ImageJ program available at http://rsbweb.nih.gov/ij/ as previously described [20]. The relative expression of survivin was determined by dividing the survivin band density (adjusted for the background) by the density of the corresponding GAPDH. This ratio of survivin to GAPDH was then normalized by dividing these values by the average ratio in the “normal” fibroblasts, such that the average of all normal (or untreated) lung fibroblasts is equal to 1.0 and the IPF samples (or the TGF-β1 treated samples) represent the fold change. 2.7. Apoptosis Apoptosis was assessed by ELISA-based detection of histone-associated DNA fragments using the Cell Death Detection ELISA Kit (Roche Applied Science, Indian- apolis, IN) as previously described [20]. To allow com- parisons across experiments using different IPF fibro- blast cell lines, the apoptosis data is represented as the percentage of the absorbance of the positive control sample (provided by the manufacturer) that was run on each ELISA plate. Each IPF sample was run in triplicate. 2.8. Statistical Analyses Statistical analyses were done using ANOVA with Tukey post-test for experiments involving multiple comparisons (apoptosis assays). An unpaired T-test with Welch’s cor- rection was used for comparisons between normal and IPF samples (Graphpad Prism software version 5.01 for Windows). The relationship between apoptosis induced by Fas-activating ligand combined with YM155 or CAY10625 was determined using a Spearman correlation (r). 3. RESULTS 3.1. Survivin Is Expressed in IPF Lung Tissue To determine if survivin was increased in fibroblasts residing in areas of active fibrosis, we used immunohis- tochemistry to examine survivin expression in normal and IPF lung biopsies (Figure 1). In normal lung tissue, survivin staining was observed in subsets of alveolar epithelial cells (Figure 1(a), arrows) and in the normal bronchial epithelium (data not shown). In IPF tissue, survivin expression was identified in interstitial cells within thickened alveolar septae (Figure 1(b), arrowhead) and in the alveolar epithelium (Figure 1(b), arrow). Marked survivin expression was also observed in mes- enchymal cells residing within the IPF fibroblastic foci and in the epithelium overlying the fibroblastic foci (Figure 1(c)). 3.2. Survivin Expression Is Increased in IPF Lung Fibroblasts Having shown expression of survivin within fibroblastic foci of IPF tissue, we next sought to compare survivin expression in primary lung fibroblasts from patients with normal lung architecture to fibroblasts isolated from IPF lung tissue. Using quantitative real-time PCR of banked RNA from 12 patient-derived normal lung fibroblast cell lines and 21 IPF fibroblast cell lines, we found a signifi- cant (2-fold) increase in survivin expression in the IPF fibroblasts (Figure 2). While the overall difference was statistically significant, the distribution of survivin ex- pression in IPF fibroblasts was heterogeneous and ap- peared to segregate into two distinct groups, with ap- proximately half (12/21) of the IPF cell lines showing survivin expression that was no different than the aver- age expression seen in the normal lung fibroblasts (Fig- ure 2, white diamonds). In contrast, 9/21 IPF fibroblast cell lines had a more pronounced (average 3.5-fold) in- crease in survivin expression compared to the normal lung fibroblasts (Figure 2, black diamonds). We next assessed the relative survivin protein levels in 12 IPF fibroblast cell lines compared to 5 normal lung fibroblast cell lines (Figure 3). Consistent with the RNA findings, survivin expression was significantly (approxi- mately 2.5-fold) higher in the IPF fibroblasts. Survivin protein expression in IPF fibroblasts was also heteroge- neous, with some cell lines indistinguishable from the average expression in normal lung fibroblasts (white dia- monds), and others demonstrating more than a 3-fold Copyright © 2012 SciRes. OPEN ACCESS 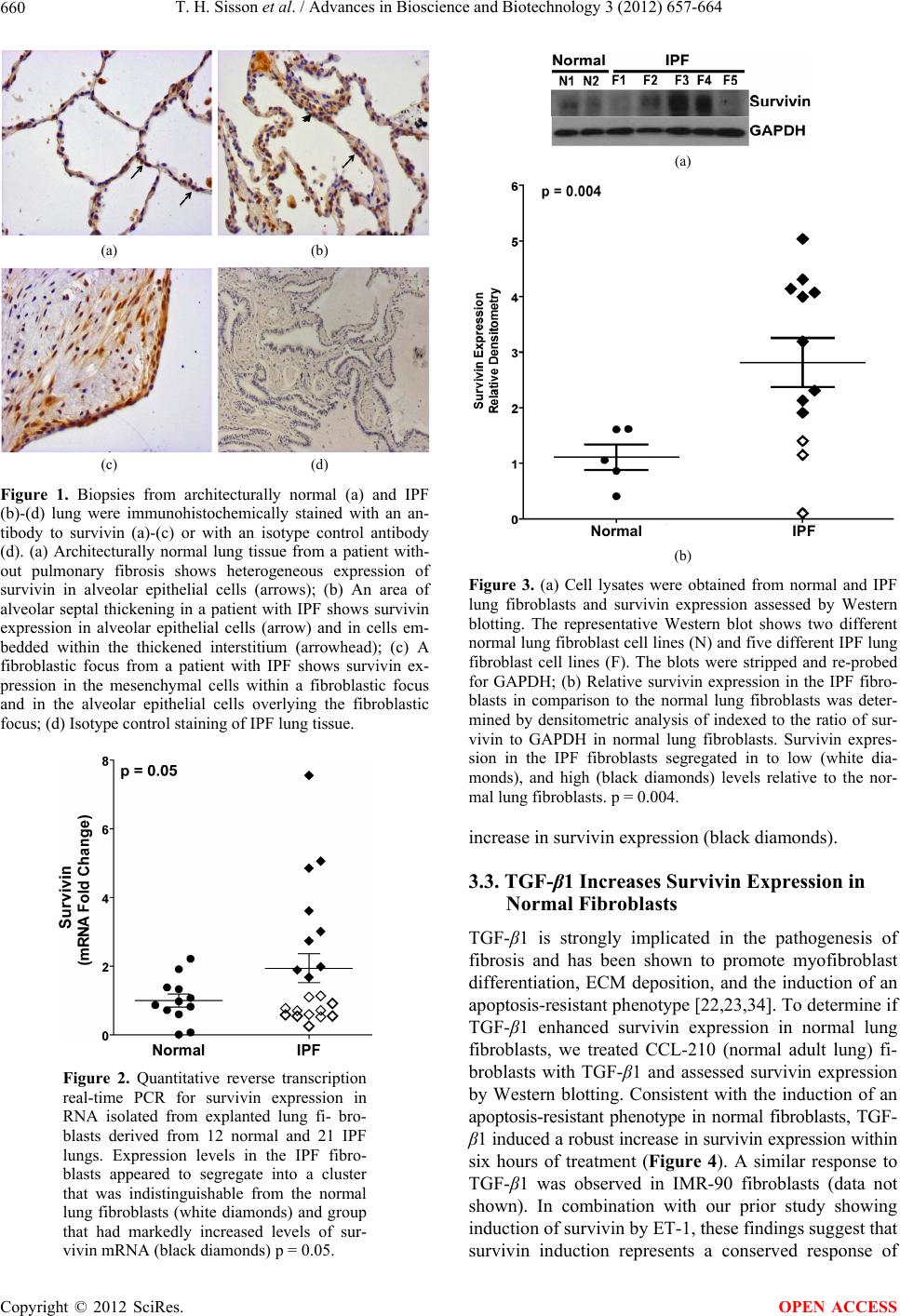 T. H. Sisson et al. / Advances in Bioscience and Biotechnology 3 (2012) 657-664 660 (a) (b) (c) (d) Figure 1. Biopsies from architecturally normal (a) and IPF (b)-(d) lung were immunohistochemically stained with an an- tibody to survivin (a)-(c) or with an isotype control antibody (d). (a) Architecturally normal lung tissue from a patient with- out pulmonary fibrosis shows heterogeneous expression of survivin in alveolar epithelial cells (arrows); (b) An area of alveolar septal thickening in a patient with IPF shows survivin expression in alveolar epithelial cells (arrow) and in cells em- bedded within the thickened interstitium (arrowhead); (c) A fibroblastic focus from a patient with IPF shows survivin ex- pression in the mesenchymal cells within a fibroblastic focus and in the alveolar epithelial cells overlying the fibroblastic focus; (d) Isotype control staining of IPF lung tissue. Normal IPF Figure 2. Quantitative reverse transcription real-time PCR for survivin expression in RNA isolated from explanted lung fi- bro- blasts derived from 12 normal and 21 IPF lungs. Expression levels in the IPF fibro- blasts appeared to segregate into a cluster that was indistinguishable from the normal lung fibroblasts (white diamonds) and group that had markedly increased levels of sur- vivin mRNA (black diamonds) p = 0.05. (a) Normal IPF (b) Figure 3. (a) Cell lysates were obtained from normal and IPF lung fibroblasts and survivin expression assessed by Western blotting. The representative Western blot shows two different normal lung fibroblast cell lines (N) and five different IPF lung fibroblast cell lines (F). The blots were stripped and re-probed for GAPDH; (b) Relative survivin expression in the IPF fibro- blasts in comparison to the normal lung fibroblasts was deter- mined by densitometric analysis of indexed to the ratio of sur- vivin to GAPDH in normal lung fibroblasts. Survivin expres- sion in the IPF fibroblasts segregated in to low (white dia- monds), and high (black diamonds) levels relative to the nor- mal lung fibroblasts. p = 0.004. increase in survivin expression (black diamonds). 3.3. TGF-β1 Increases Survivin Expression in Normal Fibroblasts TGF-β1 is strongly implicated in the pathogenesis of fibrosis and has been shown to promote myofibroblast differentiation, ECM deposition, and the induction of an apoptosis-resistant phenotype [22,23,34]. To determine if TGF-β1 enhanced survivin expression in normal lung fibroblasts, we treated CCL-210 (normal adult lung) fi- broblasts with TGF-β1 and assessed survivin expression by Western blotting. Consistent with the induction of an apoptosis-resistant phenotype in normal fibroblasts, TGF- β1 induced a robust increase in survivin expression within six hours of treatment (Figure 4). A similar response to TGF-β1 was observed in IMR-90 fibroblasts (data not shown). In combination with our prior study showing induction of survivin by ET-1, these findings suggest that survivin induction represents a conserved response of Copyright © 2012 SciRes. OPEN ACCESS  T. H. Sisson et al. / Advances in Bioscience and Biotechnology 3 (2012) 657-664 661 (a) TGF-β1 (2 ng/ml) (b) Figure 4. Normal adult lung fibroblasts (CCL-210) were cultured to 60% confluence in DMEM supple- mented with 5% fetal bovine serum and growth ar- rested in serum-free DMEM for 24 hours prior to treatment with TGF-β1 (2 ng/ml) for 6 hours. (a) Sur- vivin expression was assessed by Western blotting. The blot was stripped and re-probed for GAPDH, and is representative of three independent experi- ments; (b) Densitometric analysis of three independ- ent replicates of this experiment, indexed to the ratio of survivin to GAPDH in untreated CCL-210 cells. normal lung fibroblasts to soluble pro-fibrotic mediators [20]. 3.4. Survivin Inhibition Enhances IPF Fibroblast Susceptibility to Apoptosis In normal lung fibroblasts, enhanced survivin expression by ET-1 is associated with decreased susceptibility to apoptosis [20]. In contrast, the suppression of survivin in response to treatment with PGE2 is associated with en- hanced susceptibility to apoptosis [20,24]. Finding that survivin is expressed within the fibroblastic foci in IPF tissue, that IPF fibroblasts demonstrate increased levels of survivin, and that TGF-β1 induced survivin expression in normal lung fibroblasts, we sought to determine if survivin inhibition could modulate susceptibility to apoptosis in IPF fibroblasts. Eight patient derived IPF fibroblast cell lines were treated with/without Fas-acti- vating antibody in the presence/absence of two mecha- nistically distinct inhibitors of survivin (CAY10625 or YM155) and apoptosis was assessed at 16 hours (Figure 5). Consistent with prior reports, treatment with Fas- activating ligand alone did not significantly increase IPF lung fibroblast apoptosis [18,34,35]. However, treatment with the Fas-activating antibodies in the presence of ei- ther survivin inhibitor was associated with a statistically significant increase in IPF fibroblast apoptosis. As with (a) (b) Figure 5. IPF fibroblasts from 8 different patients were cul- tured to 60% confluence in DMEM supplemented with 10% fetal bovine serum and growth-arrested for 24 hours in serum- free DMEM prior to treatment for 16 hours with/without the Fas-activating antibody CH11 (FasL; 250 ng/ml) in the pres- ence/absence of the survivin inhibitors CAY10625 (5 µM) or YM155 (10.0 µM), or with the survivin inhibitors alone. (a) Apoptosis was assessed using ELISA-based detection of his- tone associated DNA fragments. To allow relative comparisons of apoptosis across experiments, the data for each cell line is expressed as the percentage of the assay positive control that was included on each ELISA plate. The scatter plot shows the distribution of responses in individual cell lines along with the mean ± standard error for each treatment group. p = 0.038 overall (ANOVA). p = 0.027 for FasL compared to FasL/ CAY10625, and p = 0.046 for FasL compared to FasL/YM155; (b) The correlation of apoptosis between IPF fibroblasts treated with FasL/CAY10625 and FasL/YM155. Each point represents a different IPF cell line. The Spearman correlation (r) = 0.95. survivin expression levels, the IPF fibroblast cell lines demonstrated heterogeneity in their apoptotic responses to Fas-activation in the presence of the survivin inhibit- tors, with enhanced apoptosis observed in only half of the IPF fibroblast cell lines. Neither inhibitor signifi- cantly increased apoptosis in the absence of Fas-activate- Copyright © 2012 SciRes. OPEN ACCESS  T. H. Sisson et al. / Advances in Bioscience and Biotechnology 3 (2012) 657-664 662 ing antibodies. Finally, there was strong correlation be- tween apoptosis induced by Fas-activation with YM155 and Fas-activation with CAY1062 (Figure 5(b)), sug- gesting that the increased susceptibility to apoptosis was specifically related to survivin inhibition and not the re- sult of an off-target effect of the inhibitors used. 4. DISCUSSION In the current study, we examined the role of survivin in the regulation of IPF lung fibroblast apoptosis. We show that survivin expression is increased in the mesenchymal cells within the fibroblastic foci of IPF lungs, supporting a role for this IAP in the apoptosis-resistant phenotype of these cells. Further, we demonstrate increased survivin expression in IPF lung fibroblasts compared to normal lung fibroblasts, although the expression of survivin in these IPF fibroblasts is heterogeneous. Additionally, we show that TGF-β1, like ET-1, induces survivin expres- sion in normal lung fibroblasts [20]. Finally, we demon- strate that pharmacologic inhibition of survivin enhances the apoptotic susceptibility of a subset of IPF lung fibro- blasts. Collectively, these findings indicate that increased survivin expression represents one mechanism by which IPF fibroblasts acquire resistance to apoptosis. IPF lung tissue is characterized by the heterogeneous distribution of fibroblastic foci composed of fibroblasts and myofibroblasts which, in contrast to the overlying epithelium, lack evidence of active apoptosis [2,14,15, 17,18]. The mechanisms leading to the lack of apoptosis in IPF fibroblasts have not been fully elucidated. Given the heterogeneity seen in IPF, it is possible that different stimuli converge on overlapping signaling pathways and effector mechanisms to promote pro-fibrotic fibroblast phenotypes in different patients. Supporting this concept of mechanistic heterogeneity with phenotypic conver- gence in complex disease states such as fibrosis, we have shown that TGF-β1 and ET-1, two soluble mediators implicated in fibrosis in-vivo, independently promote fibroblast resistance to apoptosis through activation of the pro-survival tyrosine kinases, focal adhesion kinase (FAK) and phosphatidylinositol 3-hydroxy kinase/AKT [23,34]. We recently demonstrated a mechanistic role for sur- vivin in the regulation of normal lung fibroblast apop- tosis, showing that FAK and PI3K/AKT activation are required for the upregulation of survivin by ET-1 and that survivin inhibition attenuates ET-1 mediated resis- tance to apoptosis in these cells [20]. In this study, we extend these findings into primary fibrotic lung fibro- blasts, showing that a significant subset of IPF fibroblast cell lines have markedly increased levels of survivin. Moreover, while all of the IPF lung fibroblast cell lines studied were resistant to Fas-mediated apoptosis, sur- vivin inhibition enhanced apoptosis in only one-half of the cell lines. The implication of this observation is that alternative mechanisms that are not dependent on sur- vivin mediate apoptosis resistance in the other half of the IPF fibroblast cell lines. Such segregation of IPF fibro- blasts into similar apoptosis-resistant phenotypes via he- terogeneous mechanisms is reminiscent of a prior study demonstrating that diminished expression of the EP2 receptor accounted for the lack of IPF fibroblast respon- siveness to the anti-fibrotic mediator PGE2 in 4/9 cell lines while the remaining IPF fibroblasts had decreased responsiveness to PGE2 despite normal levels of the EP2 receptor [36]. Interestingly, we have also shown that PGE2 suppresses survivin expression in normal lung fi- broblasts, suggesting a potential mechanistic link be- tween PGE2 resistance and increased survivin expression in the IPF fibroblasts. Although the mechanisms by which survivin regulates cell apoptosis are controversial, there is abundant evi- dence demonstrating that survivin inhibition can enhance the susceptibility of cancer cells to apoptotic stimuli, making survivin an attractive target for anti-cancer treat- ments [27,30,33,37-39]. We employed two mechanisti- cally distinct inhibitors to assess the role of survivin in IPF fibroblast resistance to Fas-mediated apoptosis [32, 33,40]. Similar to the distribution of survivin expression in IPF fibroblasts, inhibition of survivin significantly enhanced Fas-mediated apoptosis in approximately half of the IPF fibroblasts studied. While all pharmacologic inhibitors can have off target effects, we found a high correlation between the effects of YM155 and CAY10625 on cell susceptibility to apoptosis (r = 0.95, p = 0.002). This high correlation between the effects of two mecha- nistically distinct inhibitors, combined with the lack of any significant effect on apoptosis when cells were treated with the inhibitors alone, suggests that the en- hanced apoptosis was specific to the inhibition of sur- vivin and did not represent an off-target effect of either inhibitor. Studies exploring a relationship between survivin and fibrosis are lacking. Our previous in vitro studies support a link between survivin and a pro-fibrotic fibroblast phenotype [20,36]. Additionally, several studies have linked survivin expression with PI3K/AKT activation, findings consistent with the established roles of PI3K/ AKT signaling in pulmonary fibrosis [18,41-44]. In vivo, only one prior study has shown a link between survivin and fibrosis [45]. In that study, which employed a murine model of reversible liver fibrosis, survivin expression was significantly increased during the establishment of fibrosis. Moreover, the resolution phase of this fibrosis model, which is dependent on hepatic stellate cell (myo- fibroblast) apoptosis, coincided with the reduction of survivin expression to pre-fibrosis levels. However, a Copyright © 2012 SciRes. OPEN ACCESS  T. H. Sisson et al. / Advances in Bioscience and Biotechnology 3 (2012) 657-664 663 mechanistic link between survivin and fibrosis was not examined, and no reports have shown that survivin is required for the establishment of fibrosis or that suppres- sion of survivin is necessary for the reversal of fibrosis. Together with the current demonstration that inhibition of survivin can restore susceptibility of IPF fibroblasts to Fas-mediated apoptosis, these findings suggest that sur- vivin may represent a potential target for anti-fibrotic therapies. In conclusion, this study is the first to show that sur- vivin is expressed in the fibroblastic foci of IPF tissue. Moreover, it is the first study to examine survivin ex- pression in IPF fibroblasts and to assess the impact of survivin inhibition on fibroblast susceptibility to apop- tosis. Our findings demonstrate that survivin expression is increased in a subset of IPF fibroblasts and that it is possible to enhance the susceptibility of these cells to Fas-mediated apoptosis by inhibiting survivin. The het- erogeneity of survivin expression in IPF fibroblasts, and the heterogeneous responses to apoptotic stimuli seen in the presence of survivin inhibitors, may be a reflection of the biologic and clinical heterogeneity observed in pa- tients with IPF. Further studies are required to elucidate the mechanisms by which survivin regulates apoptosis susceptibility in these cells, and to determine if survivin inhibition can modulate fibrosis in vivo. 5. ACKNOWLEDGEMENTS Funding for these studies was provided by NIH/NHLBI HL105489 (JCH), HL R01085083 (ESW), and HL078871 (THS). REFERENCES [1] Collard, H.R., et al. (2012) Burden of illness in idiopathic pulmonary fibrosis. Journal of Medical Economics, 15, 829-835. doi:10.3111/13696998.2012.680553 [2] Raghu, G., et al. (2011) An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. American Journal of Respiratory and Critical Care Medicine, 183, 788-824. doi:10.1164/rccm.2009-040GL [3] Navaratnam, V., et al. (2011) The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax, 66, 462-467. doi:10.1136/thx.2010.148031 [4] Maher, T.M. (2012) Idiopathic pulmonary fibrosis: Pa- thobiology of novel approaches to treatment. Clinics in Chest Medicine, 33, 69-83. doi:10.1016/j.ccm.2011.11.002 [5] Ley, B., Collard, H.R. and King Jr., T.E. (2011) Clinical course and prediction of survival in idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine, 183, 431-440. doi:10.1164/rccm.201006-0894CI [6] Ding, Q., et al. (2011) New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis. Drugs, 71, 981-1001. doi:10.2165/11591490-000000000-00000 [7] King Jr., T.E., Pardo, A. and Selman, M. (2011) Idio- pathic pulmonary fibrosis. Lancet, 378, 1949-1961. doi:10.1016/S0140-6736(11)60052-4 [8] Horowitz, J.C. and Thannickal, V.J. (2006) Idiopathic pulmonary fibrosis: New concepts in pathogenesis and implications for drug therapy. Treatments in Respiratory Medicine, 5, 325-342. doi:10.2165/00151829-200605050-00004 [9] Desmouliere, A., Chaponnier, C. and Gabbiani, G. (2005) Tissue repair, contraction, and the myofibroblast. Wound Repair and Regeneration, 13, 7-12. doi:10.1111/j.1067-1927.2005.130102.x [10] Hinz, B., et al. (2012) Recent developments in myofibro- blast biology: Paradigms for connective tissue remodel- ing. American Journal of Pathology, 180, 1340-1355. doi:10.1016/j.ajpath.2012.02.004 [11] Laurent, G.J., et al. (2008) Escape from the matrix: Mul- tiple mechanisms for fibroblast activation in pulmonary fibrosis. Proceedings of the American Thoracic Society, 5, 311-315. doi:10.1513/pats.200710-159DR [12] Desmouliere, A., et al. (1995) Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. American Journal of Pathol- ogy, 146, 56-66. [13] Tomasek, J.J., et al. (2002) Myofibroblasts and mech- ano-regulation of connective tissue remodelling. Nature Reviews Molecular Cell Biolog, 3, 349-363. doi:10.1038/nrm809 [14] Thannickal, V.J. and Horowitz, J.C. (2006) Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proceedings of the American Thoracic Society, 3, 350- 356. doi:10.1513/pats.200601-001TK [15] Cha, S.I., et al. (2010) Compartmentalized expression of c-FLIP in lung tissues of patients with idiopathic pulmo- nary fibrosis. American Journal of Respiratory Cell and Molecular Biology, 42, 140-148. doi:10.1165/rcmb.2008-0419OC [16] Lepparanta, O., et al. (2010) Transcription factor GATA- 6 is expressed in quiescent myofibroblasts in idiopathic pulmonary fibrosis. American Journal of Respiratory Cell and Molecular Biology, 42, 626-632. doi:10.1165/rcmb.2009-0021OC [17] Korfei, M., et al. (2008) Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fi- brosis. American Journal of Respiratory and Critical Care Medicine, 178, 838-846. doi:10.1164/rccm.200802-313OC [18] Maher, T.M., et al. (2010) Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmo- nary fibrosis. American Journal of Respiratory and Cri- tical Care Medicine, 182, 73-82. doi:10.1164/rccm.200905-0674OC [19] Buhling, F., et al. (2005) Altered expression of mem- brane-bound and soluble CD95/Fas contributes to the re- sistance of fibrotic lung fibroblasts to FasL induced apoptosis. Respiratory Research, 6, 37. doi:10.1186/1465-9921-6-37 Copyright © 2012 SciRes. OPEN ACCESS  T. H. Sisson et al. / Advances in Bioscience and Biotechnology 3 (2012) 657-664 Copyright © 2012 SciRes. 664 OPEN ACCESS [20] Horowitz, J.C., et al. (2012) Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. International Journal of Biochemistry & Cell Biology, 44, 158-169. doi:10.1016/j.biocel.2011.10.011 [21] Horowitz, J.C., et al. (2006) Constitutive activation of prosurvival signaling in alveolar mesenchymal cells iso- lated from patients with nonresolving acute respiratory distress syndrome. American Journal of Physiology— Lung Cellular and Molecular Physiology, 290, L415- L425. doi:10.1152/ajplung.00276.2005 [22] Horowitz, J.C., et al. (2004) Activation of the pro-sur- vival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. Journal of Biological Chemistry, 279, 1359-1367. doi:10.1074/jbc.M306248200 [23] Horowitz, J.C., et al. (2007) Combinatorial activation of FAK and AKT by transforming growth factor-beta1 con- fers an anoikis-resistant phenotype to myofibroblasts. Cell Signal, 19, 761-771. doi:10.1016/j.cellsig.2006.10.001 [24] Huang, S.K., et al. (2009) Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB Journal, 23, 4317-4326. doi:10.1096/fj.08-128801 [25] Xia, H., et al. (2004) Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibro- blast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. Journal of Biological Chemistry, 279, 33024-33034. doi:10.1074/jbc.M313265200 [26] Rumble, J.M. and Duckett, C.S. (2008) Diverse functions within the IAP family. Journal of Cell Science, 121, 3505-3507. doi:10.1242/jcs.040303 [27] Sah, N.K., et al. (2006) Structural, functional and thera- peutic biology of survivin. Cancer Letters, 244, 164-171. doi:10.1016/j.canlet.2006.03.007 [28] Altieri, D.C. (2010) Survivin and IAP proteins in cell- death mechanisms. Biochemical Journal, 430, 199-205. doi:10.1042/BJ20100814 [29] Guha, M. and Altieri, D.C. (2009) Survivin as a global target of intrinsic tumor suppression networks. Cell Cycle, 8, 2708-2710. doi:10.4161/cc.8.17.9457 [30] Altieri, D.C. (2008) New wirings in the survivin networks. Oncogene, 27, 6276-6284. doi:10.1038/onc.2008.303 [31] White, E.S., et al. (2003) Integrin alpha4beta1 regulates migration across basement membranes by lung fibro- blasts: A role for phosphatase and tensin homologue de- leted on chromosome 10. American Journal of Respira- tory and Critical Care Medicine, 168, 436-442. doi:10.1164/rccm.200301-041OC [32] Oikawa, T., et al. (2010) Identification of a small-mole- cule inhibitor of the interaction between Survivin and Smac/DIABLO. Biochemical and Biophysical Research Communications, 393, 253-258. doi:10.1016/j.bbrc.2010.01.113 [33] Ryan, B.M., O’Donovan, N. and Duffy, M.J. (2009) Sur- vivin: A new target for anti-cancer therapy. Cancer Treatment Reviews, 35, 553-562. doi:10.1016/j.ctrv.2009.05.003 [34] Kulasekaran, P., et al. (2009) Endothelin-1 and trans- forming growth factor-beta1 independently induce fibro- blast resistance to apoptosis via AKT activation. Ameri- can Journal of Respiratory Cell and Molecular Biology, 41, 484-493. doi:10.1165/rcmb.2008-0447OC [35] Frankel, S.K., et al. (2006) TNF-alpha sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apop- tosis. American Journal of Respiratory Cell and Molecu- lar Biology, 34, 293-304. doi:10.1165/rcmb.2005-0155OC [36] Huang, S.K., et al. (2008) Variable prostaglandin E2 resistance in fibroblasts from patients with usual intersti- tial pneumonia. American Journal of Respiratory and Critical Care Medicine, 177, 66-74. doi:10.1164/rccm.200706-963OC [37] Andersen, M.H., et al. (2007) The universal character of the tumor-associated antigen survivin. Clinical Cancer Research, 13, 5991-5994. doi:10.1158/1078-0432.CCR-07-0686 [38] Kelly, R.J., et al. (2011) Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mo- lecular Cancer, 10, 35. doi:10.1186/1476-4598-10-35 [39] Pavlyukov, M.S., et al. (2011) Survivin monomer plays an essential role in apoptosis regulation. Journal of Bio- logical Chemistry, 286, 23296-23307. doi:10.1074/jbc.M111.237586 [40] Nakahara, T., et al. (2011) YM155, a novel survivin sup- pressant, enhances taxane-induced apoptosis and tumor regression in a human Calu 6 lung cancer xenograft model. Anticancer Drugs, 22, 454-462. doi:10.1097/CAD.0b013e328344ac68 [41] Anandharaj, A., Cinghu, S. and Park, W.Y. (2011) Ra- pamycin-mediated mTOR inhibition attenuates survivin and sensitizes glioblastoma cells to radiation therapy. Acta Biochim Biophys Sin (Shanghai), 43, 292-300. [42] Hideshima, T., et al. (2007) Inhibition of Akt induces significant downregulation of survivin and cytotoxicity in human multiple myeloma cells. British Journal of Hae- matology, 138, 783-791. doi:10.1111/j.1365-2141.2007.06714.x [43] Kang, H.R., et al. (2007) Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. Journal of Experimental Medicine, 204, 1083-1093. doi:10.1084/jem.20061273 [44] Le Cras, T.D., et al. (2010) Inhibition of PI3K by PX-866 prevents transforming growth factor-alpha-induced pul- monary fibrosis. American Journal of Pathology, 176, 679-686. doi:10.2353/ajpath.2010.090123 [45] Marsillach, J., et al. (2008) Changes in the expression of genes related to apoptosis and fibrosis pathways in CCl4-treated rats. Molecular and Cellular Biochemistry, 308, 101-109. doi:10.1007/s11010-007-9617-0
|