Journal of Cancer Therapy
Vol.2 No.5(2011), Article ID:16679,12 pages DOI:10.4236/jct.2011.25102
Phase I Dose Escalation Study with the Lewis Y Carbohydrate Specific Humanized Antibody IGN311
![]()
1Zentralklinikum, Augsburg, Germany; 2Igeneon, Vienna, Austria; 3AGES Pharmed, Vienna, Austria; 4S-TARget, Vienna, Austria; 5Apeiron, Vienna, Austria; 6CDMS, Vienna, Austria; 7Immatics, Tübingen, Germany; 8f-Star, Vienna, Austria; 9ViroLogik, Erlangen, Germany; 10Meridian Biopharmaceuticals, Vienna, Austria.
E-mail: *a.nechansky@meridian-biopharm.at
Received October 3rd, 2011; revised November 6th, 2011; accepted November 16th, 2011.
Keywords: Passive immunotherapy, Therapeutic monoclonal antibody, Disseminated tumor cells, Phase I study, Lewis Y carbohydrate, HAHA (Human Anti-Human Antibodies)
ABSTRACT
Purpose: Investigation of safety, tolerability, pharmacokinetics, and anti-tumor activity of the Lewis Y-specific, fully humanized monoclonal antibody (mAb) IGN311 in patients with Lewis Y positive tumors in a Phase I clinical trial. Experimental Design: Twelve patients (pts) were enrolled in an open-label, uncontrolled, dose escalating Phase I study. Three pts received 50 mg, three pts 100 mg and six pts 200 mg IGN311 by i.v. infusion on days 1 and 15. Blood samples were taken immediately before infusion, and 0.5, 4, 8, 24 hours post infusion, as well as on days 3, 5 and 8 after the first and second infusion, respectively, and day 29. A final visit was scheduled for day 43. Results: No drug related adverse events were observed in the 50 mg and 100 mg dose groups. Three out of six patients in the 200 mg dose group showed drug related adverse reactions with nausea, vomiting and hypotension in one patient (NCI CTC grade 3) being the dose limiting toxicities. t1/2 of IGN311 was ~20 days after second infusion of IGN311. Sera of patients receiving IGN311 were capable of lysing Lewis Y positive tumor cells in vitro by both, complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC). Circulating tumor cells found in the peripheral blood in two out of twelve pts prior to treatment were reduced after treatment to below the quantification limit of the detection method. None of the patients showed an increase in the number of disseminated tumor cells during treatment period. Conclusions: The good safety and PK profile, the biological activity regarding CDC and ADCC mediated tumor cell lysis, and the elimination of circulating tumor cells warrant further clinical investigation of IGN311.
1. Introduction
Passive immunotherapy of cancer is based on the administration of antibodies or immune effector cells that have the ability to directly mediate anti-tumor responses. The humanized mAb IGN311 which is specific for the tumor-associated carbohydrate antigen Lewis Y. (LeY) has been developed for passive immunotherapy of patients with LeY expressing tumors. The LeY antigen is expressed on the majority of human cancers of epithelial origin whereas expression on normal tissue is limited to epithelial cells of the esophagus, stomach, the proximal small intestine, some acinar cells of the pancreas and resting granulocytes [1-5]. Regarding the expression profile on tumor cells, predominantly adenocarcinomas of the lung, breast, colorectal, gastric, pancreatic, prostate and ovarian cancers have been tested positive for LeY [2,6-9]. Pronounced expression of LeY in different tumors is associated with decreased survival and higher metastatic potential [10-14].
Because LeY represents an attractive target for immune therapy, a variety of murine mAbs has been generated against this carbohydrate antigen and their anti-tumor activities have been explored and have demonstrated anti-tumor activity in tumor cell models in vitro, animal models and clinical studies [1,2,6,15-22].
Based on the promising effects found with murine mAbs, several LeY-specific humanized mAb have been designed with improved pharmacokinetics in humans [23-27]. Recently, a Phase I biodistribution and pharmacokinetic trial with the humanized mAb Hu3s193 in patients with LeY positive, advanced epithelial cancer has demonstrated selective targeting of tumors with no evidence of any consistent normal tissue uptake [26]. Another promising humanized LeY specific mAb is the hABL364—designated IGN311—that is the humanized version of the murine mAb BR55-2 [1,6,15-17]. During humanization, the affinity to the target cells and the effector functions were optimized by molecular modeling [23]. After pharmacological and toxicological studies in rhesus monkeys which have indicated favorable pharmacokinetic features of IGN311, a Phase I dose escalation study with IGN311 administered by intravenous infusion to patients with LeY expressing tumors was conducted. The primary objective of this study was to assess safety, tolerability and pharmacokinetics. As secondary objective, anti-tumor activity and immunologic parameters were investigated.
2. Patients and Methods
2.1. Eligibility Criteria
Male or female pts (aged between 18 and 80 years) with biopsy proven LeY positive carcinoma refractory to standard therapies were eligible for this phase I study. LeY expression was determined using formalin-fixed primary tissue. Pts with an anticipated life expectancy ≥4 months and a Karnofsky Performance Score of ≥70 were included. The presence of at least one objective disease marker within the last 6 weeks prior to inclusion was obligatory. No other therapies for the treatment of carcinoma (e.g. chemotherapy, radiation, immune therapy or any investigational agent other than IGN311) during the study period and within 6 weeks prior to therapy with the exceptions of bisphosphonates and hormon therapy was to be administered. Informed consent from all pts was obtained before enrollment into the study.
2.2. Study Design and Treatment
The study was designed as an open-label, uncontrolled, dose escalating Phase I study, enrolling 12 evaluable pts. Pts received IGN311 intravenously during a two hour infusion on day 1 and 15 without pre-medication. Three pts were included for each dose level and had to be extended by two additional patients in case of observed grade 3 toxicities. The first three evaluable pts received 50 mg IGN311 per infusion (cohort I), the next three evaluable pts received 100 mg IGN311 per infusion (cohort II) and five pts received 200 mg IGN311 per infusion (cohort III, because of grade 3 toxicities at this dose level). The only significant amendment to the original protocol was applied to pt 12 with regard to a prolongation of the infusion time from 2 to 4 hours (cohort IV). Pts were under close monitoring of blood pressure, heart rate, respiratory rate, body temperature every 30 minutes during the infusion of IGN311 and 0.5, 1, 1.5, 3, 4 and 8h post infusion. Blood samples were taken immediately before infusion, and 0.5, 4, 8, 24 hours post infusion, as well as on days 3, 5 and 8 after the first and second infusion, respectively, and days 29 and 43. The study was conducted in accordance with the latest revision of the Declaration of Helsinki, the requirements of Good Clinical Practice of the European Community (CPMP/ICH/ 135195) and European Clinical Trials Directive 2001/ 20/EC. Written, voluntary informed consent to participate in this study was obtained prior to enrollment into the study and for performing of any study specific evaluations. The primary objectives of the study were to assess safety, tolerability and pharmacokinetics and to determine dose limiting toxicity (DLT) and maximum-tolerated dose (MTD) of intravenous infusions of two doses of IGN311 administered on day 1 and day 15 to pts with LeY expressing carcinomas. All drug related adverse events were classified using the NCI toxicity criteria. Safety evaluations included clinical and laboratory assessments (physical examination, vital signs, hematology, chemistry profile, urinalysis, ECG, and adverse events). The secondary objectives of the study were to monitor anti-tumor activity and immunological parameters. RECIST criteria were used to evaluate the tumor response using CT scans. Different tumor markers were measured. As surrogate parameter for tumor response the amount of disseminated tumor cells in peripheral blood was used [28-31]. Tumor assessments were performed at baseline and on day 43.
2.3. Manufacturing IGN311
IGN311 drug substance was produced according to GMP guidelines by BioInvent International AB (Lund, Sweden). The final product was filled in glass syringes according to GMP guidelines at the Statens Serum Institute (Copenhagen, Denmark).
2.4. Pharmacokinetics
For pharmacokinetic analysis of IGN311, blood samples were taken before and 0.5, 4, 8, 24 hours after infusion, and on days 3, 5 and 8 after the first and second infusion. Additional blood samples were taken on days 29 and 43. Concentrations of IGN311 in the serum were measured using a specific sandwich ELISA with the anti-idiotypic mAb MMA383 specifically recognizing the idiotype of IGN311 [32]. Pharmacokinetic parameters were calculated [33] separately for each of the two infusions: The area under the concentration-time curve (AUC 0-t) was calculated from the original data using the trapezoidal rule. Kel (slope) was fitted using the log-transformed last three measurements of infusion 1 or infusion 2, respecttively.
2.5. Human Anti-Human Antibody (HAHA) Response
HAHA reactivity was assessed using a BIACORE assay as described previously [34]. Briefly, the highly sensitive assay monitors binding to immobilized IGN311 and to an isotype matched control antibody. A positive HAHA response was defined as at least two-fold increase in RUs compared to the matched control antibody and an absolute value > 50 RUs (Relative Response Units).
2.6. Complement Dependent Cytotoxicity (CDC)
Briefly, LeY positive SKBR3 tumor cells were labeled with 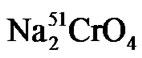 (Amersham, Germany), washed and incubated for 60 minutes at 37˚C with serial dilutions of patient sera, thereby using the patient’s complement. Release of 51Cr from lysed target cells into the supernatant of the samples was measured using a gamma-counter (Cobra 5005, Canberra-Packard, Australia). Cytotoxicity was calculated as published [23].
(Amersham, Germany), washed and incubated for 60 minutes at 37˚C with serial dilutions of patient sera, thereby using the patient’s complement. Release of 51Cr from lysed target cells into the supernatant of the samples was measured using a gamma-counter (Cobra 5005, Canberra-Packard, Australia). Cytotoxicity was calculated as published [23].
2.7. Antibody Dependent Cellular Cytotoxicity (ADCC)
Briefly, LeY positive SKBR3 tumor cells were labeled with 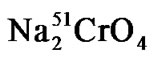 (Amersham, Germany), washed and plated together with 5% heat inactivated serum samples into 96-well microtiter plates. Effector cells derived from peripheral blood mononuclear cells from a healthy volunteer donor were freshly prepared and added to the target cells at an effector to target (E:T) ratio of 40:1. Release of 51Cr from lysed target cells into the supernatant of the samples was measured using a gamma-counter (Cobra 5005, Canberra-Packard, Australia). Cytotoxicity was calculated as published [23].
(Amersham, Germany), washed and plated together with 5% heat inactivated serum samples into 96-well microtiter plates. Effector cells derived from peripheral blood mononuclear cells from a healthy volunteer donor were freshly prepared and added to the target cells at an effector to target (E:T) ratio of 40:1. Release of 51Cr from lysed target cells into the supernatant of the samples was measured using a gamma-counter (Cobra 5005, Canberra-Packard, Australia). Cytotoxicity was calculated as published [23].
2.8. Determination of Disseminated Tumor Cells in Blood
Approximately 28 ml blood was collected on day 1 and day 15 before start of infusion and on day 43, respecttively. Detection of circulating tumor cells in blood was performed by Immunicon Inc. (Enschede, the Netherlands) using a FDA approved method [28-30] based on the magnetic enrichment of EpCAM-positive cells which are subsequently stained with anti-cytokeratin antibodies.
3. Results
Twelve pts (11 male, 1 female) with biopsy proven LeY positive tumors were included in the clinical study at the II. Medical Klinik, Klinikum Augsburg, Germany. The characteristics of the enrolled pts are listed in Table 1.
3.1. Safety and Tolerability
Treatment cohorts I and II with three pts each received 50 mg or 100 mg IGN311, respectively, and did not show any drug related adverse events. Within cohort III (200 mg dose) the third patient (pt 9) experienced drug related adverse events with grade 3 (according to NCI CTC) nausea/vomiting after both administrations of IGN311. Therefore, according to the protocol two additional patients were recruited at this dose group. After the reporting of the first SAE (nausea) classified as “probable drug related” with moderate severity in pt 11 (receiving only one 200 mg infusion) the study protocol was amended regarding an increase of the infusion time from 2 to 4 hours (based on the assumption that toxicities were induced by peak levels of IGN311 in the blood). Because of the occurrence of adverse events such as vomiting, dyspepsia and nausea in pt 12 rated as possibly drug related it was indicated that the MTD had been exceeded. From the data of the 200 mg dose group nausea and vomiting (NCI CTC grade 3) were regarded as dose limiting toxicities and the 100 mg dose level was defined as the maximal tolerated dose in the absence of pre-medication (see Table 2).
Four pts (with 5 reported SAEs) experienced serious adverse events (urethral obstruction and rectal bleeding in pt 2; death due to tumor progression in pts 8 and 10, hospitalization for observation because of vomiting in pt 11, see Table 3). The SAEs of pts 2, 8 and 10 had apparently no relationship with the drug but were disease related. Pts 8 and 10 died during therapy—the death was disease related. The SAE of pt 11 (Cohort III) was classified as “probable related” to the study drug with moderate severity. Twenty minutes after start of infusion vomiting and the symptom hypotension (90 mm Hg) set on and the infusion was interrupted for 2 hours after which the hypotension resolved.
Only one laboratory deviation was classified as a possibly drug-related adverse event in pt 11: the lipase value increased asymptomatically up to 929 U/L two days after application of IGN311. All other laboratory deviations were classified as related to the underlying cancer disease.
3.2. Pharmacokinetics
Nine pts were assessable for pharmacokinetic analysis after the 1st and 2nd infusion of IGN311. Whereas all patients of the 50 mg and 100 mg dose groups were available for analysis, in the 200 mg dose group serum samples from only a limited number of pts (n = 3) were available. Pt 11 showed adverse drug reactions during the first infusion of IGN311 causing interruption of the infusion for 2 hours and finally withdraw the consent for the second infusion. Consequently the results of pt 11 were excluded from the final calculation. The results are listed in Table 4. Peak serum concentrations were measured 30 minutes after each infusion and were found to decrease over time (Figure 1(a)). The t1/2 after the 1st infusion was in accordance with concentrations observed in toxicological studies with IGN311 in rhesus monkeys with up to 10 days for the 50 mg dose and up to 12 days for the 100 and 200 mg dose. Mean peak serum concentrations of IGN311 were in the range of 8 µg/ml for 50 mg, 17 µg/ml for the 100 mg and 52 µg/ml for 200 mg IGN311 dose. AUC0-t values ranged from 892 for 50 mg, 2711 for 100 mg to 5879 for 200 mg IGN311. T1/2 after the 2nd infusion was 13, 24 and 21 days for the 50, 100 and 200 mg dose groups, respectively. Mean peak serum concentrations ranged from 9 µg/ml (50 mg), 23 µg/ml (100 mg) to 71 µg/ml for the 200 mg IGN311 dose group. AUC0-t values ranged from 1678, 6549 to 12053 µg/ml·h for the 50, 100 and 200 mg dose group, respectively (Table 4). The increase of Cmax and the AUC0-t value was proportional to the applied dose indicating a linear pharmacokinetics (Figure 1(a), Table 4).
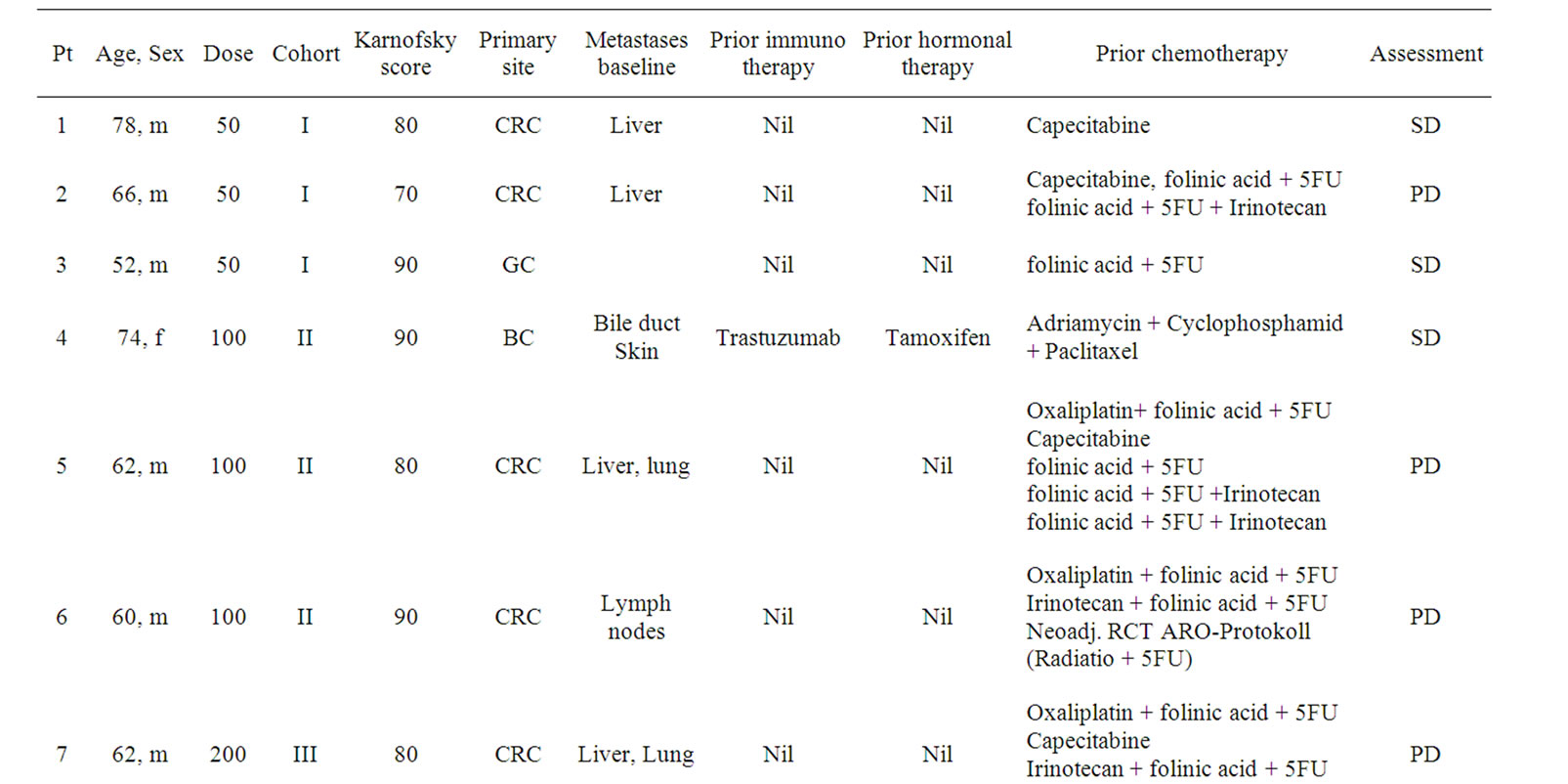
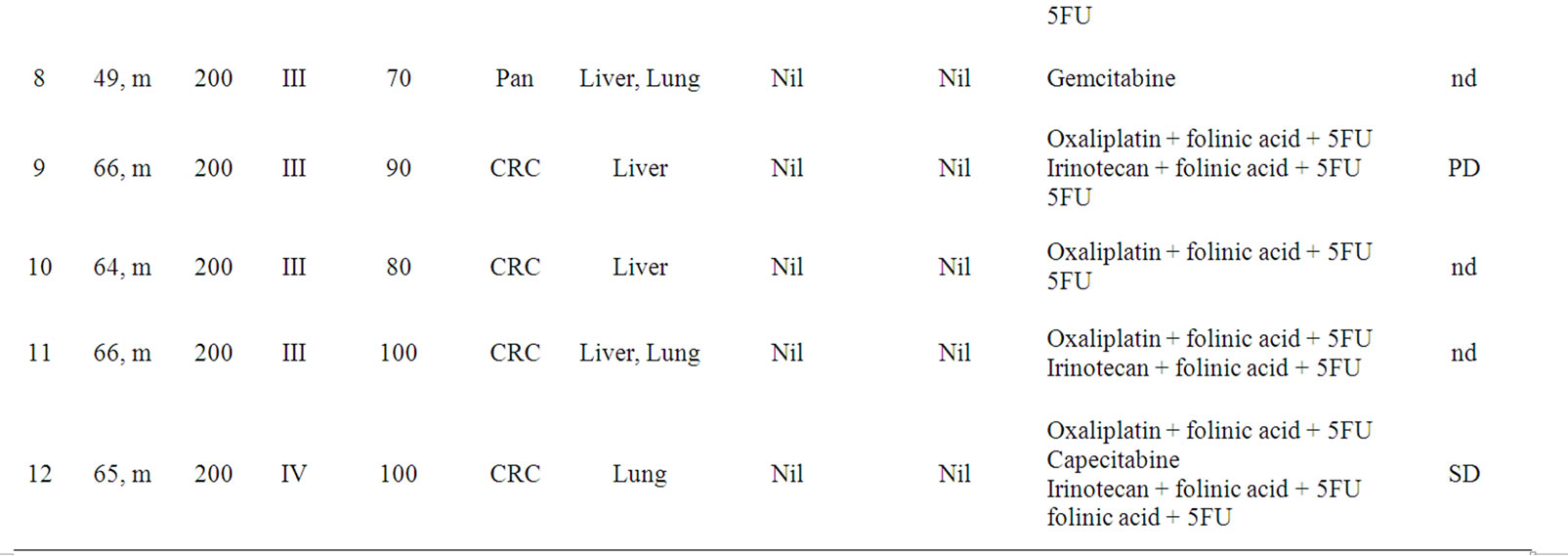
Table 1. Age, Sex, Dose, Cohort, Karnofsky performance status, Primary site and site of Metastasis at baseline, prior immunoor hormonal therapy and tumor assessment of all patients of the study. Nine out of 12 pats were assessable for tumor assessment. nd = not determined.
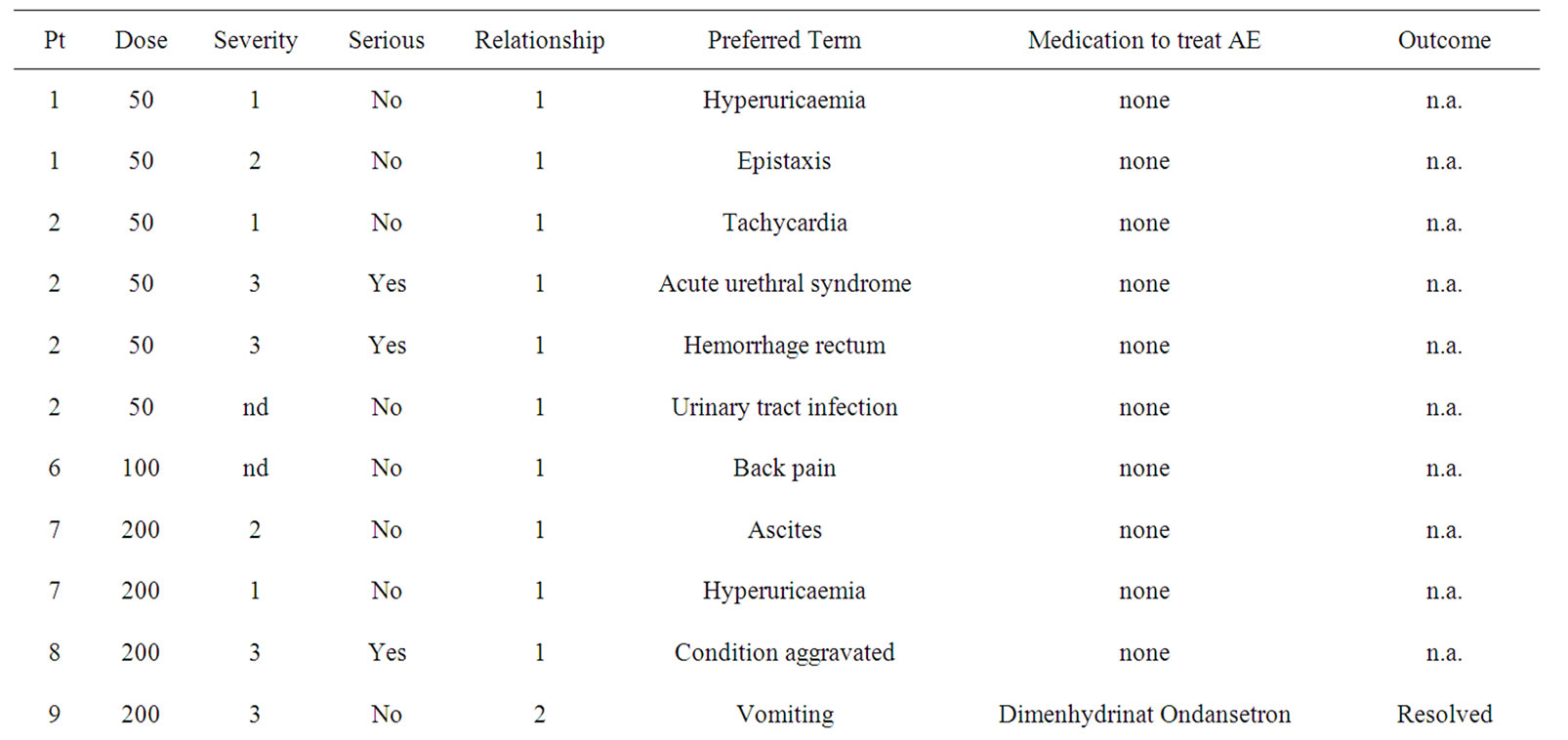
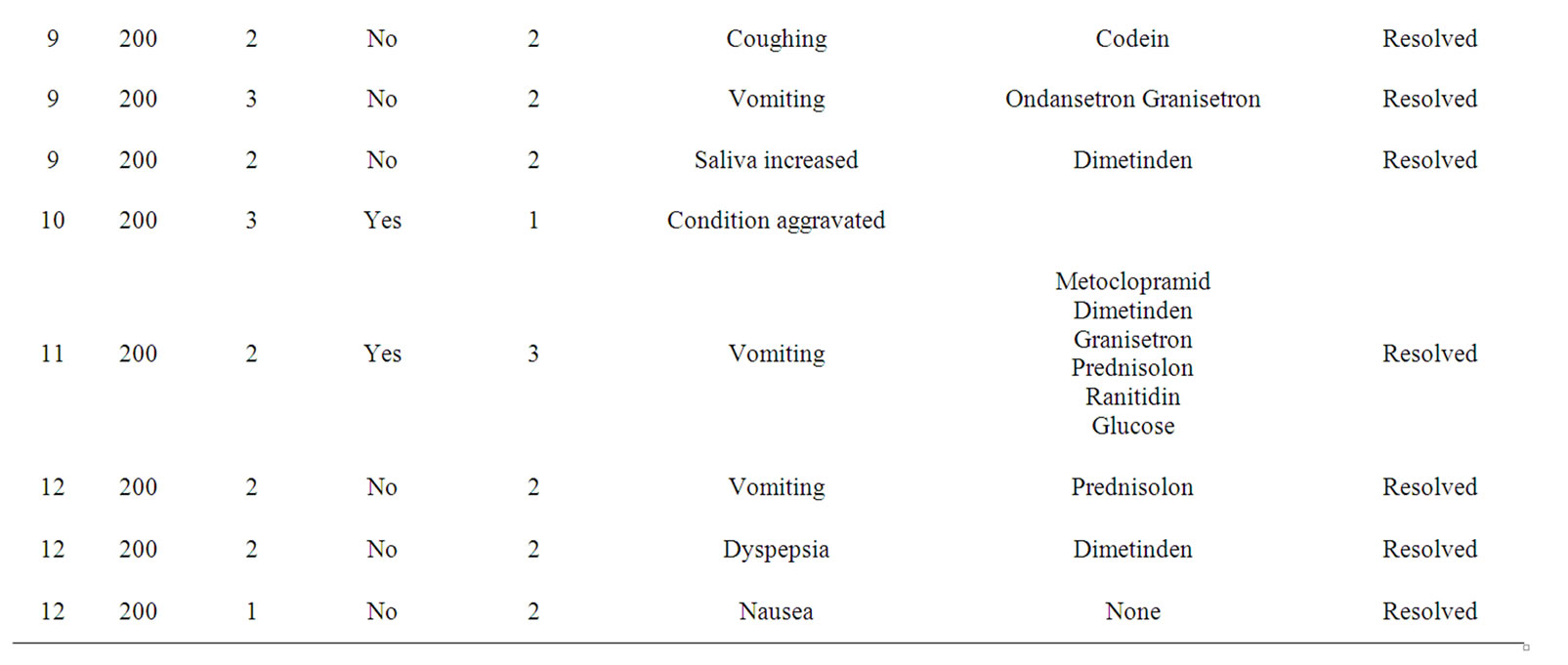
Table 2. Adverse Events (AE) grouped by severity, preferred term, medication applied and the outcome are shown. Severity was graded according to common toxicity criteria (CTC). Relationship to study drug was assigned: 1 = unlikely; 2 = possible; 3 = probable.
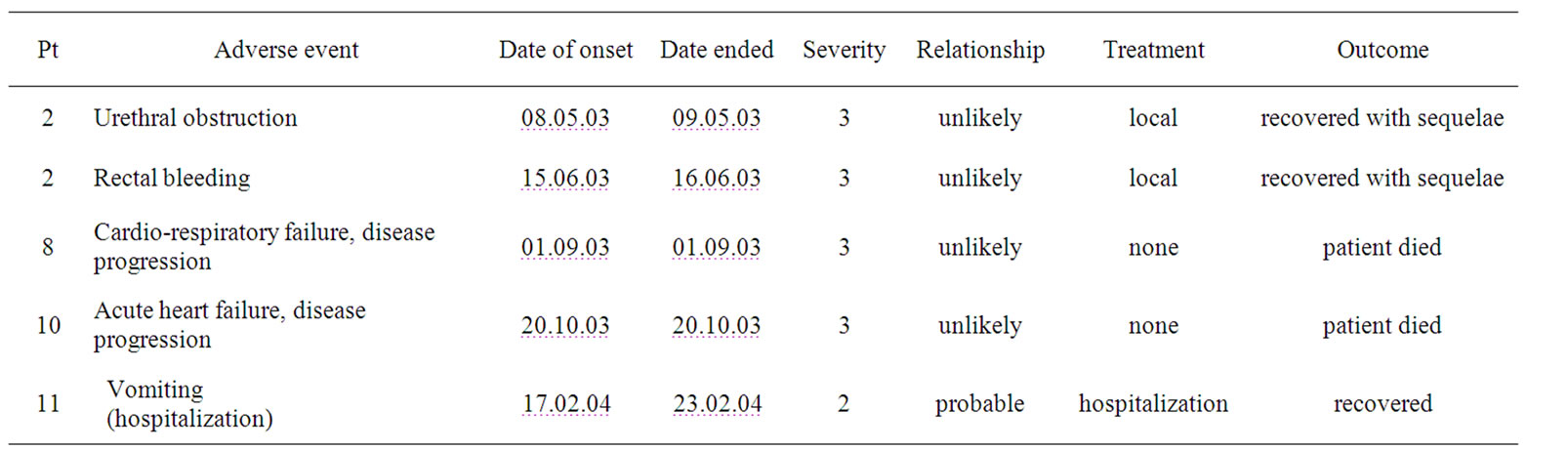
Table 3. Serious adverse events (SAE).
3.3. Complement Dependent Cytotoxicity (CDC)
CDC data are shown as percentage of specific lysis of LeY positive SKBR3 target cells. Significant CDC activity was measured in the pts serum in all three dose groups (Figure 1(b)). The kinetics of CDC activity were found to follow the pharmacokinetics measured for IGN311 in the pts sera (Figure 1(a)), with peak activities shortly after the first and second application. For the 50 mg dose group the CDC activity returned to baseline at the end of the study. In contrast, at the two higher dose groups of 100 mg and 200 mg, respectively, CDC activity remained above background at all time-points of the study (up to 43 days which was defined as the end of the study) and was in accordance with IGN311 titers.
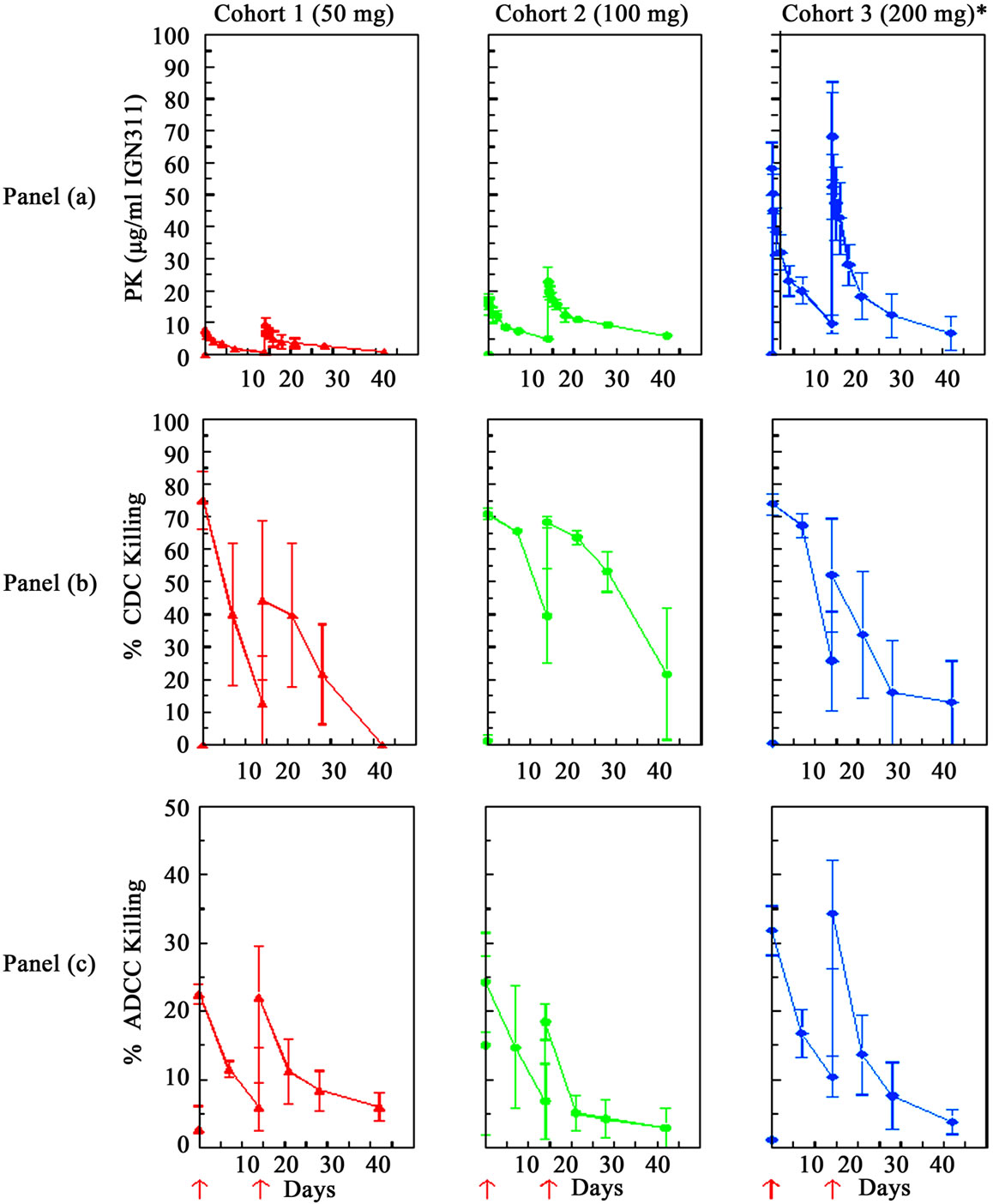
Figure 1. Pharmacokinetics and Pharmacodynamics. Three pts per dose level (*also in cohort III)) were assessable for PK and PD analysis of IGN311. Blood samples were taken before and 0.5, 4, 8, 24 hours after start of infusion, and on days 3, 5 and 8 after the first and second infusion. Additional blood samples were taken on days 29 and 43. Infusion time points at day 1 and day 15 are indicated as red arrows. Panel (a): Concentrations of IGN311 in the serum were measured using a specific sandwich ELISA with mAb MMA383 which specifically recognizes the idiotype of IGN311. Pharmacokinetic parameters were calculated separately for each of the two infusions. Panel (b): CDC was measured using the breast cancer cell line SKBR3 as target cell line and the patient sera as complement source. Blood samples were taken before and 0.5 hours after start of infusion, on days 8 after the first and second infusion, and on days 29 and 43. Panel (c): ADCC was measured using the breast cancer cell line SKBR3 as target cell line and PBMCs of healthy donors as effector cells (effector to target ratio of 40:1). Blood samples were taken before and 0.5 hours after start of infusion, on days 8 after the first and second infusion, and on days 29 and 43.
3.4. Antibody Dependent Cellular Cytotoxicity (ADCC)
ADCC data are shown as percentage of specific lysis of LeY positive SKBR3 target cells after incubation with (heat inactivated patient derived) serum and effector cells derived from PBMCs from a healthy donor at an E:T ratio of 40:1 (Figure 1(c)). The kinetics of ADCC activity were found to follow the pharmacokinetics data measured for IGN311 in the pts sera (Figure 1(a)), and was in accordance with the kinetics of CDC activity (Figure 1(b)) with peak activities shortly after the first and second application.
3.5. Human Anti-Human Antibody (HAHA) Response
First infusion of IGN311 induced a HAHA response (defined as at least two-fold increase in RUs and absolute value > 50 RUs) in six out of 12 pts. Onset of the HAHA response was detectable at day 7 and peak levels were reached before the second infusion at day 15 (Figure 2). Importantly, the second application did not further increase HAHA titers but rather the HAHA response decreased.
3.6. Disseminated Tumor Cells (DTCs) in Blood
Because disseminated tumor cells are the anticipated target of an IGN311 therapy, the blood of patients was analyzed for DTCs using antibodies specific for EpCAM —a marker frequently expressed on cells of epithelial origin, but absent on blood cells. EpCAM positive cells (i.e. indicative for DTCs) were found in the peripheral blood in two out of twelve pts at day 1 before treatment. Notably, in both patients (pt 5 from the 100 mg dose group, and pt 7 from the 200 mg dose group) the numbers of DTCs were reduced after two applications of IGN311 to below the quantification limit of the method (Figure 3).
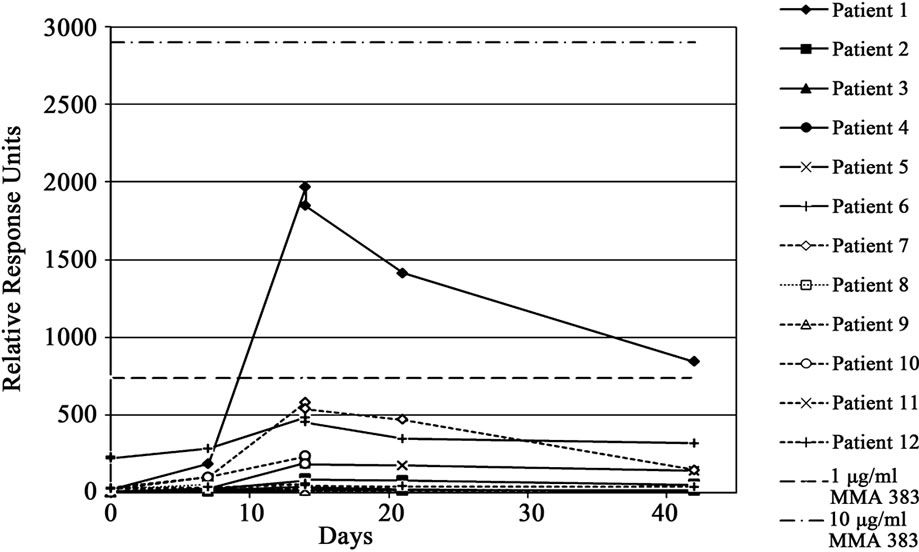
Figure 2. HAHA response was assayed using SPR (BIAcore). Relative response units (RUs) obtained with patient sera are shown. The straight dotted line indicates the response obtained with 1 µg/ml of the IGN311 anti-id antibody MMA383.
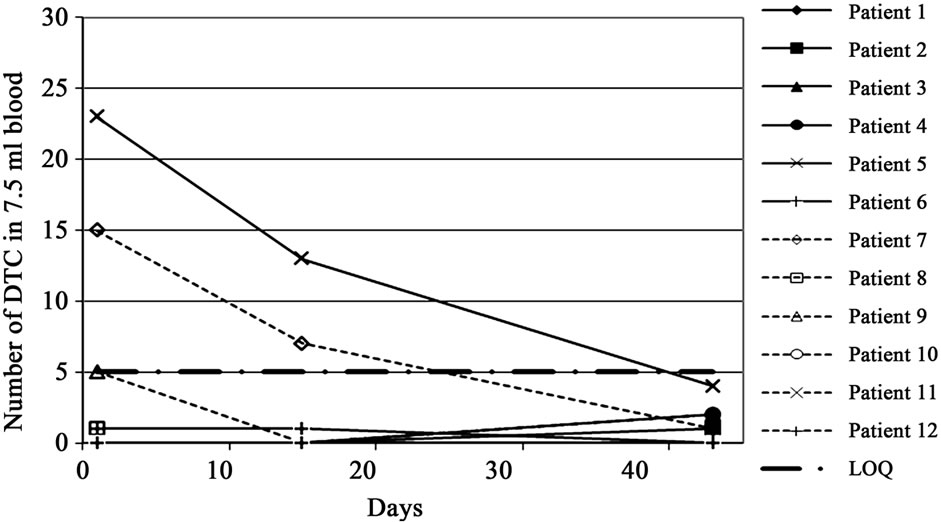
Figure 3. Number of disseminated tumor cells (DTC) detected in 7.5 ml blood of each patient before the 1st and 2nd application of IGN311 and at the end of study. EpCAM positive cells were magnetically enriched and subsequently stained with anticytokeratin antibodies and counter-stained with anti-CD45 antibodies confirming the epithelial origin of selected cells.
3.7. Tumor Assessment
Only nine out of 12 pts were evaluable for tumor assessment at the end of the study. Of these nine pts, four pts (1, 3, 4 and 12) showed a stabilization of disease according to RECIST and 5 pts (2, 5-7 and 9) showed progressive disease (Table 1).
4. Discussion
This Phase I dose escalation study demonstrated that IGN311 administered twice at biweekly intervals was well tolerated at doses up to 100 mg. The MTD of IGN311 was found to be exceeded at 200 mg with NCI-CTC grade 3 nausea and vomiting being dose limiting. Furthermore, drug related adverse reaction like hypotension, coughing, hypersalivation and heartburn were found in the 200 mg cohort but were successfully treated with appropriate medication. It should be noted that no premedication was given. In earlier clinical trials, the parental murine mAb ABL 364 displayed similar toxicities already at lower dose levels [17-19]. The increase of the infusion time from 2 to 4 hours for pt 12 (second infusion) did not change DLT. Glucocorticoids, antihistamines and serotonin (5HT3) antagonists were successfully applied in case of grade 2 or grade 3 DLT (nausea and vomiting). Grade 1 toxicities of vomiting and nausea resolved without any treatment. Notably, treatment with IGN311 did not result in any hematological toxicity. With the exception of pt 11 (in the 200 mg dose group) who showed an asymptomatic increase in the lipase values (rated as a symptom of a drug-related adverse event) all other deviations of laboratory parameters were classified as related to the underlying cancer disease.
The fully humanized mAb IGN311 has shown a favorable serum half-life in this study with a t1/2 of 10 - 12 days after the first infusion and of ~20 days following the second infusion of 100 mg IGN311/patient, being close to the reported t1/2 of 21 days of natural IgG and other therapeutic anti-tumor mAbs like Rituximab or Trastuzumab [35,36] (Figure 1(a), Table 4). Regarding the patient’s safety, monitoring of the induction of HAHA responses is important and also recommended by the FDA. A BIACORE-based assay was applied that is able to also detect weak HAHA responses [34]. After IGN311 application, HAHA reactivity was detected in six out of 12 pts defined by an at least two-fold increase in RUs over pre-serum values and the absolute values being >50 RUs. Because no increase of binding to the isotype matched control antibody was observed it can be concluded that the observed HAHA response has been directed against the specific binding regions of IGN311.
With the exception of one study [37] clinical trials with humanized mAbs have so far not reported such high incidence of HAHA responses—e.g. application of CAMPATH (anti-CD52 humanized Ab, Alemtuzumab) [38] or Herceptin (humanized anti-HER2 mAb) [39] was reported to induce almost no HAHA responses after i.v. application (0 out of 30) or (1 out of 211), respectively. It has to be stressed here that these studies applied ELISA based techniques therefore using a different detection system than applied in the current study. The BIACORE —surface plasmon resonance—based method monitors HAHA binding in real time without the need of a secondary, enzyme-labeled detection antibody and is therefore capable to detect low affinity antibodies which results in improved sensitivity. Within the present study, 50% HAHA positive patients were identified which is in accordance with results published by Ritter et al. using a similar detection system [37]. Regarding the detected anti-drug antibodies it should be noted that the SPRbased Biacore method is generally more sensitive than ELISA because with ELISA low-affinity antibodies can hardly be detected [40].
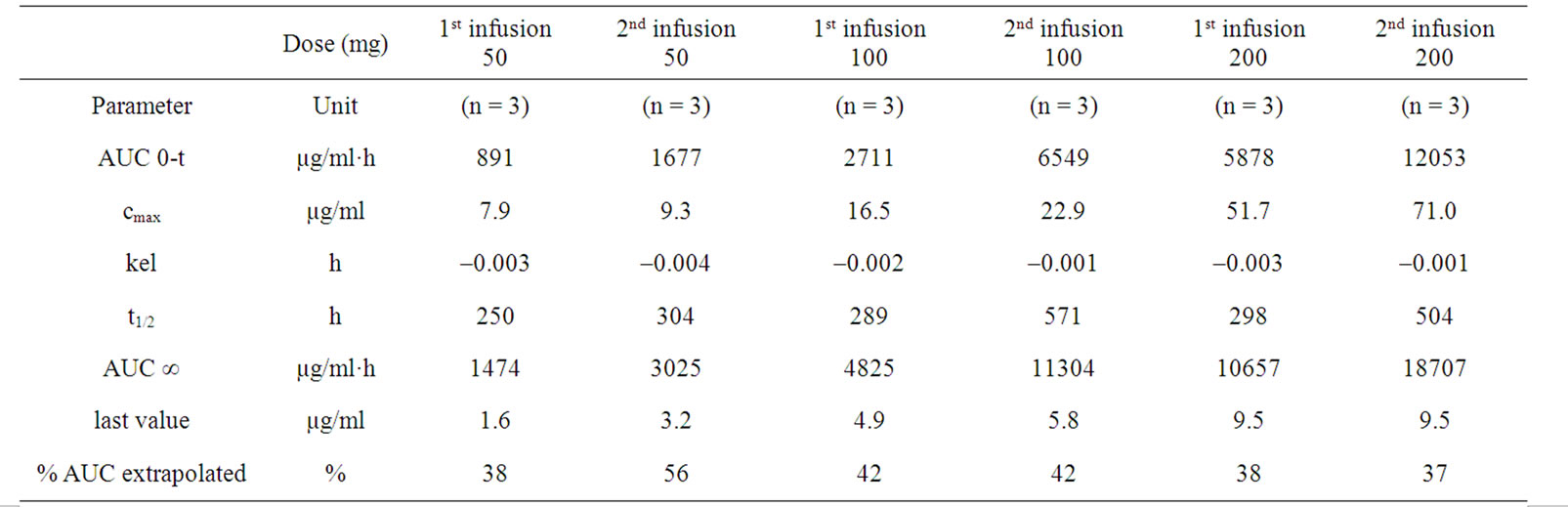
Table 4. Pharmacokinetic parameters were calculated for each dose level, separated for 1st and 2nd infusion. Only three out of six pats of the 200 mg dose level group were evaluable for pharmacokinetic analysis.
Importantly, with regard to the therapeutic potential, sera of patients receiving IGN311 were capable of lysing LeY positive tumor cells ex vivo by both, CDC and ADCC. Whereas IGN311 has been also shown to block signal transduction through LeY glycosylated growth factor receptors such as EGFR and Her2-neu on tumor cells [24,41], the effector functions mediated by this fully humanized antibody, i.e. CDC and ADCC may be considered to be pivotal for the anticipated anti-tumor effects of IGN311 in vivo.
Regarding anti-tumor efficacy, EpCAM positive cells in the peripheral blood—indicative for DTC’s—detectable in two out of twelve patients (pts 5 and 7) before treatment—were eliminated after treatment to below the quantification limit of the detection method. Cristofanilli et al. [28-31] have shown that DTC’s in peripheral blood of pts with breast cancer are of prognostic significance and can be used as a surrogate parameter for therapeutic efficacy of anti-cancer therapy. In recent publications, the concept of CTC’s as prognostic factor for metastatic disease has been confirmed. Krebs et al. [42] demonstrated that in patients with Non-Small-Cell Lung Cancer (NSCLC) progression-free survival inversely correlates with the amount of detectable CTC’s. This is in accordance with Tanaka et al. [43] who showed that in lung cancer patients, CTC counts significantly increased along with tumor progression, especially with development of distant metastasis. For breast cancer, Pachmann et al. [44] showed that an increase of circulating epithelial tumor cells was a strong indicator for a relapse. In summary, the isolation and characterization of individual tumor cells from the blood of patients known to have metastatic cancer hold tremendous potential for new biological insight with very real clinical applications [45]. Traditionally, CTC’s are isolated using the well-characterized surface molecule EpCAM that is expressed on various epithelial tumors [46]. Therefore, the recovery/detection might be improved if a combination with additional tumor cell markers is applied [47,48].
5. Conclusions
The assessment of tumor markers and objective tumor responses by RECIST criteria did not reveal clear results. First hints for an in vivo anti-tumor activity of IGN311 are the induced ex vivo effector functions and the disappearance of detectable DTCs in peripheral blood (evident after only two applications). It is the opinion of the authors that the therapeutic potential of IGN311 may primarily reside in eliminating DTC’s cells and micrometastases thereby preventing progression of disease, rather than in direct effects on bulky tumors which are preferably removed by surgery. Possibly, the application of IGN311 in an adjuvant, neoadjuvant or perioperative setting, or in combination with additional treatment modalities, e.g. radiotherapy and chemotherapy, may finally translate into a significant improvement in patients disease status and long term survival of the patients. Based on the presented data further clinical (Phase II) testing of IGN311 in a metastatic disease set-up is planned.
6. Acknowledgements
A.N. thanks Klaus Hajszan and Katherina Olsacher for help with formatting the manuscript.
REFERENCES
- M. Blaszczyk-Thurin, J. Thurin, O. Hindsgaul, K. A. Karlsson, Z. Steplewski and H. Koprowski, “Y and Blood Group B Type 2 Glycolipid Antigens Accumulate in a Human Gastric Carcinoma Cell Line as Detected by Monoclonal Antibody. Isolation and Characterization by Mass Spectrometry and NMR Spectroscopy,” Journal of Biological Chemistry, Vol. 262, No. 1, 1987, pp. 372- 379.
- I. Hellström, H. J. Garrigues, U. Garrigues and K. E Hellström, “Highly Tumor-Reactive, Internalizing, Mouse Monoclonal Antibodies to Le(y)-Related Cell Surface Antigens,” Cancer Research, Vol. 50, No. 7, 1990, pp. 2183- 2190.
- S. Zhang, H. S. Zhang, C. Cordon-Cardo, G. Ragupathi and P. O. Livingston, “Selection of Tumor Antigens as Targets for Immune Attack Using Immunohistochemistry: II. Blood Group-Related Antigens,” International Journal of Cancer, Vol. 73, No. 1, 1997, pp. 50-56. doi:10.1002/(SICI)1097-0215(19970926)73:1<50::AID-IJC9>3.0.CO;2-0
- Y. S. Kim, M. Yuan, S. H. Itzkowitz, Q. B. Sun, T. Kaizu, A. Palekar, B. F. Trump and S. Hakomori, “Expression of LeY and Extended LeY Blood Group-Related Antigens in Human Malignant, Premalignant, and nonmalignant Colonic Tissues,” Cancer Research, Vol. 46, No. 11, 1986, 5985-5992.
- M. Dettke, G. Palfi and H. Loibner, “Activation-Dependent Expression of the Blood Group-Related Lewis Y Antigen on Peripheral Blood Granulocytes,” Journal of Leukocyte Biology, Vol. 68, No. 4, 2000, pp. 511-514.
- Z. Steplewski, M. Blaszczyk-Thurin, M. Lubeck, H. Loibner, D. Scholz and H. Koprowski, “Oligosaccharide Y Specific Monoclonal Antibody and Its Isotype Switch Variants,” Hybridoma, Vol. 9, No. 2, 1990, pp. 201-210. doi:10.1089/hyb.1990.9.201
- H. Inagaki, J. Sakamoto, H. Nakazato, H, A. E. Bishop and J. Yura, “Expression of Lewisa, Lewisb, and Sialated Lewisa Antigens in Early and Advanced Human Gastric Cancers,” Journal of Surgery Oncology, Vol. 44, No. 4, 1990, pp. 208-213. doi:10.1002/jso.2930440404
- J. Sakamoto, K. Furukawa, C. Cordon-Cardo, B. W. Yin, W. J. Rettig, H. F. Öttgen, L. J. Old and K. O. Lloyd, “Expression of Lewisa, Lewisb, X, and Y Blood Group Antigens in Human Colonic Tumors and Normal Tissue and in Human Tumor-Derived Cell Lines,” Cancer Research, Vol. 46, No. 3, 1986, pp. 1553-1561.
- K. Murata, H. Egami, Y. Shibata, K. Sakamoto, A. Misumi and M. Ogawa, “Expression of Blood GroupRelated Antigens, ABH, Lewis(a), Lewis(b), Lewis(x), Lewis(y), CA19-9, and CSLEX1 in Early Cancer, Intestinal Metaplasia, and Uninvolved Mucosa of the Stomach,” American Journal of Clinical Pathology, Vol. 98, No. 1, 1992, pp. 67-75.
- M. Miyake, T. Taki, S. Hitomi and S. Hakomori, “Correlation of Expression of H/Le(y)/Le(b) Antigens with SurVival in Patients with Carcinoma of the Lung,” New England Journal of Medicine, Vol. 327, No. 1, 1992, pp. 14-18. doi:10.1056/NEJM199207023270103
- A. Larena, M. Vierbuchen, S. Schroder, A. Larena-Avellaneda, I. Hadshiew and R. Fischer, “Blood Group Antigen Expression in Papillary Carcinoma of the Thyroid Gland. An Immunohistochemical and Clinical Study of Expression of Lewis, ABO and Related Antigens,” Langenbecks Archiv für Chirurgie, Vol. 381, No. 2, 1996, pp. 102-113.
- K. Steplewska-Mazur, A. Gabriel, W. Zajecki, M. Wylezol and M. Glück, “Breast Cancer Progression and Expression of Blood Group-Related Tumor-Associated Antigens,” Hybridoma, Vol. 19, No. 2, 2000, pp. 129-133. doi:10.1089/02724570050031167
- H. Inufusa, Y. Nakatani, T. Adachi, T. Wakano, A. Nakajima, M. Nakamura, M. Suzuki, O. Ando, M. Kurimoto, M. Miyake, K. Shindo and M. Yasutomi, “Correlation of Prognosis of Breast Cancer Patients and Expression of Ley Which Acts as a Cofactor of Tumor Procoagulant,” International Journal of Oncology, Vol. 13, No. 3, 1998, pp. 481-487.
- Z. Madjd, T. Parsons, N. F. Watson, I. Spendlove, I. Ellis and L. G. Durrant, “High Expression of Lewis y/b AntiGens Is Associated with decreased survival in Lymph Node Negative Breast Carcinomas,” Breast Cancer Research, Vol. 7, No. 5, 2005, pp. R780-R787. doi:10.1186/bcr1305
- K. Kitamura, E. Stockert, P. Garin-Chesa, S. Welt, K. O. Lloyd, K. L. Armour, T. P. Wallace, W. J. Harris, F. J. Karr and L. J. Old, “Specificity Analysis of Blood Group Lewis-y (Le(y)) Antibodies Generated against Synthetic and Natural Le(y) Determinants,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 91, No. 26, 1994, pp. 12957-12961. doi:10.1073/pnas.91.26.12957
- D. Scholz, M. Lubeck, H. Loibner, J. McDonald-Smith, Y. Kimoto, H. Koprowski and Z. Steplewski, “Biological Activity in the Human System of Isotype Variants of Oligosaccharide-Y-Specific Murine Monoclonal Antibodies,” Cancer Immunology, Immunotherapy, Vol. 33, No. 3, 1991, pp. 153-157. doi:10.1007/BF01756135
- Z. Steplewski, M. D. Lubeck, D. Scholz, H. Loibner, McDonald-Smith and H. Koprowski, “Tumor Cell Lysis and Tumor Growth Inhibition by the Isotype Variants of MAb BR55-2 Directed against Y Oligosaccharide,” In Vivo, Vol. 5, No. 2, 1991, pp. 79-83.
- G. Schlimok, K. Pantel, H. Loibner, I. Fackler-Schwalbe and G. Riethmüller, “ Reduction of Metastatic Carcinoma Cells in Bone Marrow by Intravenously Administered Monoclonal Antibody: Towards a Novel Surrogate Test to Monitor Adjuvant Therapies of Solid Tumours,” European Journal of Cancer, Vol. 31A, No. 11, 1995, pp. 1799-1803. doi:10.1016/0959-8049(95)00317-C
- R. A. Stahel, H. Lacroix, J. P. Sculier, R. Morant, J. Richner, E. Janzek, H. Loibner and H. Blythman, “Phase I/II Study of Monoclonal Antibody against Lewis Y Hapten in Relapsed Small-Cell Lung Cancer,” Annals of Oncology, Vol. 3, No. 4, 1992, pp. 319-320.
- G. Schlimok, G. Riethmüller, K. Pantel, et al., “Tumor Cell Cytotoxicity Induced in Patients by a Murine Monoclonal Antibody against the Lewis Y Antigen,” Proceeding of Annual Meeting of the American Society of Clinical Oncology, Vol. 10, 1991, pp. 212-214.
- I. Pastan, E. T. Lovelace, M. G. Gallo, A. V. Rutherford, J. L. Magnani and M. C. Willingham, “Characterization of Monoclonal Antibodies B1 and B3 That React with Mucinous Adenocarcinomas,” Cancer Research, Vol. 51, No. 14, 1991, pp. 3781-3787.
- G. J. Schreiber, K. E. Hellström and I. Hellström, “An Unmodified Anticarcinoma Antibody, BR96, Localizes to and Inhibits the Outgrowth of Human Tumors in Nude Mice,” Cancer Research, Vol. 52, No. 12, 1992, pp. 3262-3266.
- M. S. Co, J. Baker, K. Bednarik, E. Janzek, W. Neruda, P. Mayer, R. Plot, B. Stumper, M. Vasquez, C. Queen and H. Loibner, “Humanized Anti-Lewis Y Antibodies: In Vitro Properties and Pharmacokinetics in Rhesus Monkeys,” Cancer Research, Vol. 56, No. 5, 1996, pp. 1118-1125.
- H. Farhan, C. Schuster, M. Klinger, E. Weisz, G. Waxenecker, M. Schuster, V. Sexl, G. C. Mudde, M. Freissmuth and R. Kircheis, “Inhibition of Xenograft Tumor Growth and Down-Regulation of ErbB Receptors by an Antibody Directed against Lewis Y Antigen,” Journal of Pharmacology and Experimental Therapy, Vol. 319, No. 3, 2006, pp. 1459-1466. doi:10.1124/jpet.106.107318
- A. M. Scott, D. Geleick, M. Rubira, K. Clarke, E. C. Nice, F. E. Smyth, E. Stockert, E. C. Richards, F. J. Carr, W. J. Harris, K. L. Armour, J. Rood, A. Kypridis, V. Kronina, R. Murphy, F. T. Lee, Z. Liu, K. Kitamura, G. Ritter, K. Laughton, E. Hoffmann, A. W. Burgess and L. J. Old, “Construction, Production, and Characterization of Humanized Anti-Lewis Y Monoclonal Antibody 3S193 for Targeted Immunotherapy of Solid Tumors,” Cancer Research, Vol. 60, No. 12, 2000, pp. 3254-3261.
- A. M. Scott, N. Tebbutt, F. T. Lee, T. Cavicchiolo, Z. Liu, S. Gill, A. M. Poon, W. Hopkins, F. E. Smyth, C. Murone, D. MacGregor, A. T. Papenfuss, B. Chappell, T. H. Saunder, M. W. Brechbiel, I. D. Davies, R. Murphy, G. Chong, E. W. Hoffmann and L. J. Old, “A Phase I Biodistribution and Pharmacokinetic Trial of Humanized Monoclonal Antibody Hu3s193 in Patients with Advanced Epithelial Cancers That Express the Lewis-Y Antigen,” Clinical Cancer Research, Vol. 13, No. 11, 2007, pp. 3286-3292. doi:10.1158/1078-0432.CCR-07-0284
- L. M. Krug, D. T. Milton, A. A. Jungbluth, L. C. Chen, E. Quaia, N. Pandit-Taskar, A. Nagel, J. Jones, M. G. Kris, R. Finn, P. Smith-Jones, A. M. Scott, L. Old and C. Divgi, “Targeting Lewis Y (Le(y)) in Small Cell Lung Cancer with a Humanized Monoclonal Antibody, hu3S193: A Pilot Trial Testing Two Dose Levels,” Journal Thorac Oncology, Vol. 2, No. 10, 2007, pp. 947-952. doi:10.1097/JTO.0b013e3181560dcc
- M. Cristofanilli, G. T. Budd, M. J. Elliset, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. Terstappen and D. F. Hayes, “Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer,” New England Journal of Medicine, Vol. 351, No. 8, 2004, pp. 781-791. doi:10.1056/NEJMoa040766
- M. Cristofanilli, D. F. Hayes, G. T. Budd, M. J. Ellis, A. Stopeck, J. M. Reuben, G. V. Doyle, J. Matera, W. J. Allard, M. C. Miller, H. A. Fritsche, G. N. Hortybagyi and L. W. Terstappen, “Circulating Tumor Cells: A Novel Prognostic Factor for Newly Diagnosed Metastatic Breast Cancer,” Journal of Clinical Oncology, Vol. 23, No. 7, 2005, pp. 1420-1430. doi:10.1200/JCO.2005.08.140
- S. Riethdorf, H. Fritsche, V. Müller, T. Rau, C. Schindlbeck, B. Rack, W. Janni, C. Coith, K. Beck, F. Jänicke, S. Jackson, T. Gornet, M. Christofanilli and K. Pantel, “Detection of Circulating Tumor Cells in Peripheral Blood of Patients with Metastatic Breast Cancer: A Validation Study of the Cell Search System,” Clinical Cancer Research, Vol. 13, No. 3, 2007, pp. 920-928. doi:10.1158/1078-0432.CCR-06-1695
- V. Müller, N. Stahmann, S. Riethdorf, T. Rau, T. Zabel, A. Goetz, F. Jänicke and K. Pantel, “Circulating Tumor Cells in Breast Cancer: Correlation to Bone Marrow Micrometastases, Heterogeneous Response to Systemic Therapy and Low Proliferative Activity,” Clinical Cancer Research, Vol. 11, No. 10, 2005, pp. 3678-3685. doi:10.1158/1078-0432.CCR-04-2469
- M. Perkins, R. Theiler, S. Lunte and M. Jeschke, “Determination of the Origin of Charge Heterogeneity in a Murine Monoclonal Antibody,” Pharmaceutical Research, Vol. 17, No. 9, 2000, pp. 1110-1117. doi:10.1023/A:1026461830617
- B. W. Cheung, L. L. Cartier, H. Q. Russlie and R. J. Sawchuk, “The Application of Sample Pooling Methods for Determining AUC, AUMC and Mean Residence Times in Pharmacokinetic Studies,” Fundamental & Clinical Pharmacology, Vol. 19, No. 3, 2005, pp. 347-354. doi:10.1111/j.1472-8206.2005.00329.x
- O. Szolar, S. Stranner, I. Zinoecker, G. C. Mudde, G. Himmler, G. Waxenecker and A. Nechansky, “Qualification and Application of a Surface Plasmon ResonanceBased Assay for Monitoring Potential HAHA Responses Induced after Passive Administration of a Humanized Anti Lewis-Y Antibody,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 41, No. 4, 2006, pp. 1347- 1353. doi:10.1016/j.jpba.2006.03.026
- K. Tobinai, T. Igarashi, K. Itoh, Y. Kobayashi, M. Taniwaki, M. Ogura, T. Kinoshita, T. Hotta, K. Aikawa, K. Tsushita, A. Hiraoka, Y. Matsuno, S. Nakamura, S. Mori and Y. Ohashi, “Japanese Multicenter Phase II and Pharmacokinetic Study of Rituximab in Relapsed or Refractory Patients with Aggressive B-Cell Lymphoma,” Annals of Oncology, Vol. 15, No. 5, 2004, pp. 821-830. doi:10.1093/annonc/mdh176
- R. Bruno, C. B. Washington , J. F. Lu, G. Liebermann, L. Banken and P. Klein, “Population Pharmacokinetics of Trastuzumab in Patients with HER2+ Metastatic Breast Cancer,” Cancer Chemotherapy and Pharmacology, Vol. 56, No. 4, 2005, pp. 361-369. doi:10.1007/s00280-005-1026-z
- G. Ritter, L. S. Cohen, C. Williams Jr., E. C. Richard, L. J. Old and S. Welt, “Serological Analysis of Human AntiHuman Antibody Responses in Colon Cancer Patients Treated with Repeated Doses of Humanized Monoclonal Antibody A33,” Cancer Research, Vol. 61, No. 18, 2001, pp. 6851-6859.
- G. Hale, P. Rebello, L.R. Brettman, C. Fegan, B. Kennedy, E. Kimby, M. Leach, J. Lundin, H. Mellstedt, P. Moreton, A. C. Rawstron, H. Waldmann, A. Osterborg and P. Hillmen, “Blood Concentrations of Alemtuzumab and Antiglobulin Responses in Patients with Chronic Lymphocytic Leukemia Following Intravenous or Subcutaneous Routes of Administration,” Blood, Vol. 104, No. 4, 2004, pp. 948-955. doi:10.1182/blood-2004-02-0593
- M. A. Cobleigh, C. L. Vogel, D. Tripathy, N. J. Robert, S. Scholl, L. Fehrenbacher, J. M. Wolter, V. Paton, S. Shak, G. Lieberman and D. J. Slamon, “Multinational Study of the Efficacy and Safety of Humanized Anti-HER2 Monoclonal Antibody in Women Who Have HER2-Overexpressing Metastatic Breast Cancer That Has Progressed after Chemotherapy for Metastatic Disease,” Journal of Clinical Oncology, Vol. 17, No. 9, 1999, pp. 2639-2648.
- A. Nechansky, “HAHA-Nothing to Laugh about Measuring the Immunogenicity (Human Anti-Human Antibody Response) Induced by Humanized Monoclonal Antibodies Applying ELISA and SPR Technology,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 51, No. 1, 2010, pp. 252-254. doi:10.1016/j.jpba.2009.07.013
- M. Klinger, H. Farhan, H. Just, H. Drobny, G. Himmler, G. C. Mudde, M. Freissmuth and V. Sexl, “Antibodies Directed against Lewis-Y Antigen Inhibit Signaling of Lewis-Y Modified ErbB Receptors,” Cancer Research, Vol. 64, No. 3, pp. 1087-1093. doi:10.1158/0008-5472.CAN-03-2435
- M. G. Krebs, R. Sloane, L. Priest, L. Lancashire, J. M. Hou, A. Greystoke, T. H. Ward, R. Ferraldeschi, A. Hughes, G. Clack, M. Ranson, C. Dive and F. H. Blackhall, “Evaluation and Prognostic Significance of Circulating Tumor Cells in Patients with Non-Small-Cell Lung Cancer,” Journal Clinical Oncology, Vol. 29, No. 12, 2011, pp. 1556-1563. doi:10.1200/JCO.2010.28.7045
- F. Tanaka, K. Yoneda, N. Kondo, M. Hashimoto, T. Takuwa, S. Matsumoto, Y. Okumura, S. Rahman, N. Tsubota, T. Tsujimura, K. Kuribayashi, K. Fukuoka, T. Nakano and S. Hasegawa, “Circulating Tumor Cell as a Diagnostic Marker in Primary Lung Cancer,” Clinical Cancer Research, Vol. 15, No. 22, 2009, pp. 6980-6986. doi:10.1158/1078-0432.CCR-09-1095
- K. Pachmann, O. Camara, A. Kohlhase, C. Rabenstein, T. Kroll, I. B. Runnebaum and K. Hoeffken, “Assessing the Efficacy of Targeted Therapy Using Circulating Epithelial Tumor Cells (CETC): The Example of SERM Therapy Monitoring as a Unique Tool to Individualize Therapy,” Journal Cancer Research Clinical Oncology, Vol. 137, No. 5, 2010, pp. 821-828. doi:10.1007/s00432-010-0942-4
- S. Maheswaran and D. A. Haber, “Circulating Tumor Cells: A Window into Cancer Biology and Metastasis,” Current Opinion Genetics Development, Vol. 20, No. 1, 2010, pp. 96-99. doi:10.1016/j.gde.2009.12.002
- M. Trzpis, P. M. J. McLaughlin, L. M. de Leij and M. C. Harmsen, “Epithelial Cell Adhesion Molecule. More than a Carcinoma Marker and Adhesion Molecule,” American Journal Pathology, Vol. 171, No. 2, 2007, pp. 386-395. doi:10.2353/ajpath.2007.070152
- E. A. Punnoose, S. K. Atwal, J. M. Spoerke, H. Savage, A. Pandita, R. F. Yeh, A. Pirzkall, B. M. Fine, L. C. Amler, D. S. Chen and M. R. Lackner, “Molecular Biomarker Analyses Using Circulating Tumor Cells,” PLoS One, Vol. 5, No. 9, 2010, p. e12517. doi:10.1371/journal.pone.0012517
- E. Obermayr, F. Sanchez-Cabo, M. K. Tea, C. F. Singer, M. Krainer, M. B. Fischer, J. Sehouli, A. Reinthaller, R. Horvat, G. Heinze, D. Tong and R. Zeillinger, “Assessment of a Six Gene Panel for the Molecular Detection of Circulating Tumor Cells in the blood of Female Cancer Patients,” BMC Cancer, Vol. 10, pp. 666-672. doi:10.1186/1471-2407-10-666