A. Meberg / Open Journal of Pediatrics 2 (2012) 219-227
226
strictive definition of correction. Even repaired CHDs
may have an increased death risk over time, some caused
by letal arrythmias [22]. Probably all patients undergoing
therapeutic interventions should be included in a life-
long follow-up programme.
4.6. Deaths
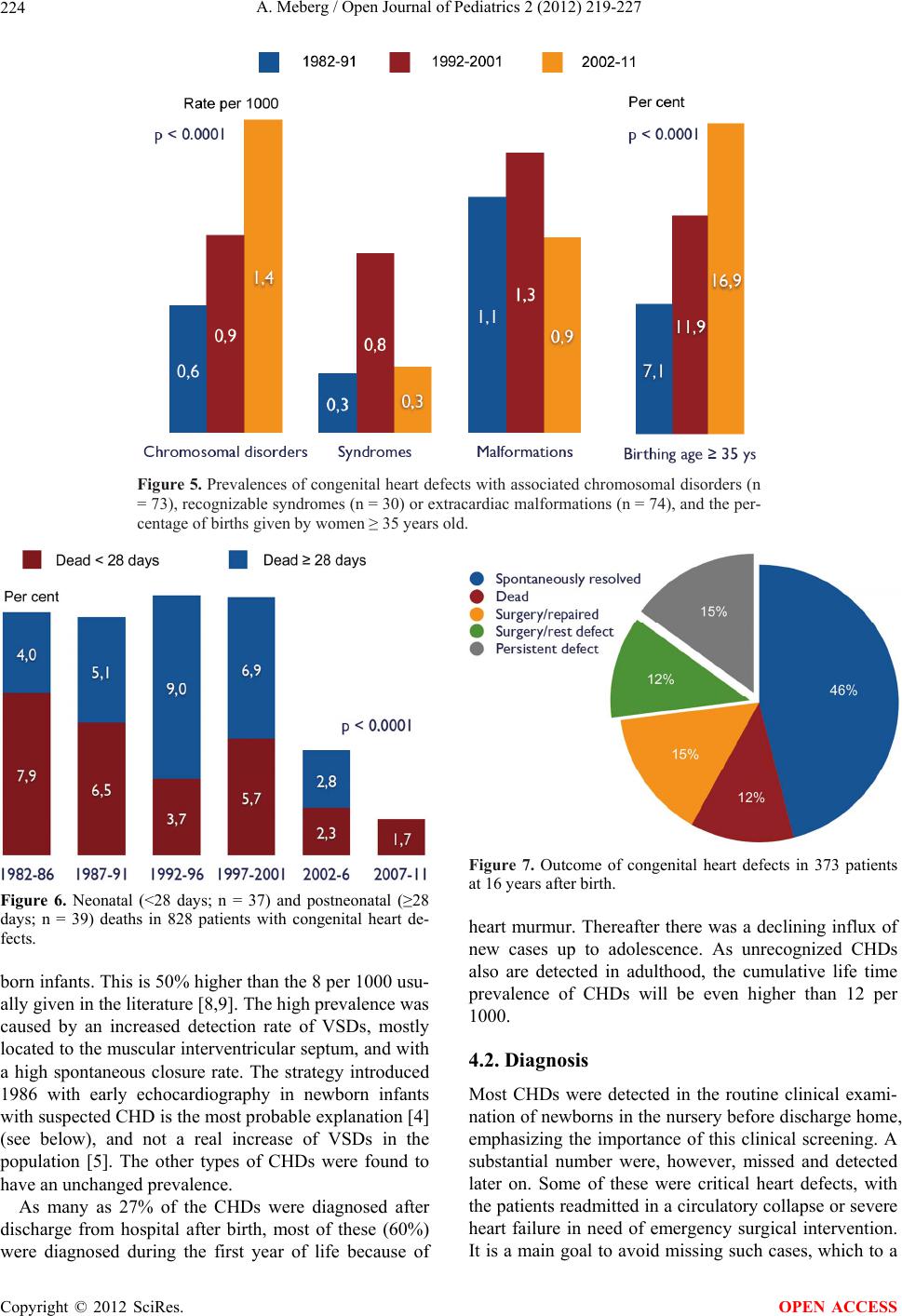
Mortality declined significantly over time, reflecting a
steady improvement in cardiologic diagnostics and thera-
peutic interventions. Nearly half of the deaths occurred
during the first four weeks of life. A substantial number
of these cases died without therapeutic procedures being
undertaken because of the complexity of the CHDs or
severe associated syndromes or extracardiac malforma-
tions. Further deaths occurred during infancy after the
neonatal period, causing a total of 80% of the deaths to
occur during the first year of life.
Some cases died with an unrecognized CHD detected
by autopsy. However, most of these cases died very early
(minutes or hours after birth) from associated extracar-
diac malformations, most of them born prematurely.
None of these deaths were judged to be avoidable. A
positive trend for minimizing deaths of patients with
unrecognized CHDs is found in other studies [23].
4.7. GUCH Programmes
The improved prognosis for CHDs has increased the
number of patients in need for long-term follow-up. Spe-
cial GUCH-programmes have been established to take
care of these patients. Such programmes focus on their
heart condition as well as growth and development, edu-
cation and work, family planning and pregnancy [24]. It
may be disputed what heart conditions need such spe-
cialized follow-up. If all patients with a CHD are defined
as risk cases, even those with spontaneous resolvement,
1% of the general population of children will need a
GUCH-programme. If only risk groups are referred (such
as untreated patients with a persistent CHD and selected
groups passing surgery) 0.3% - 0.5% of the general popu-
lation of children will need a life-long specialized care.
5. CONCLUSION
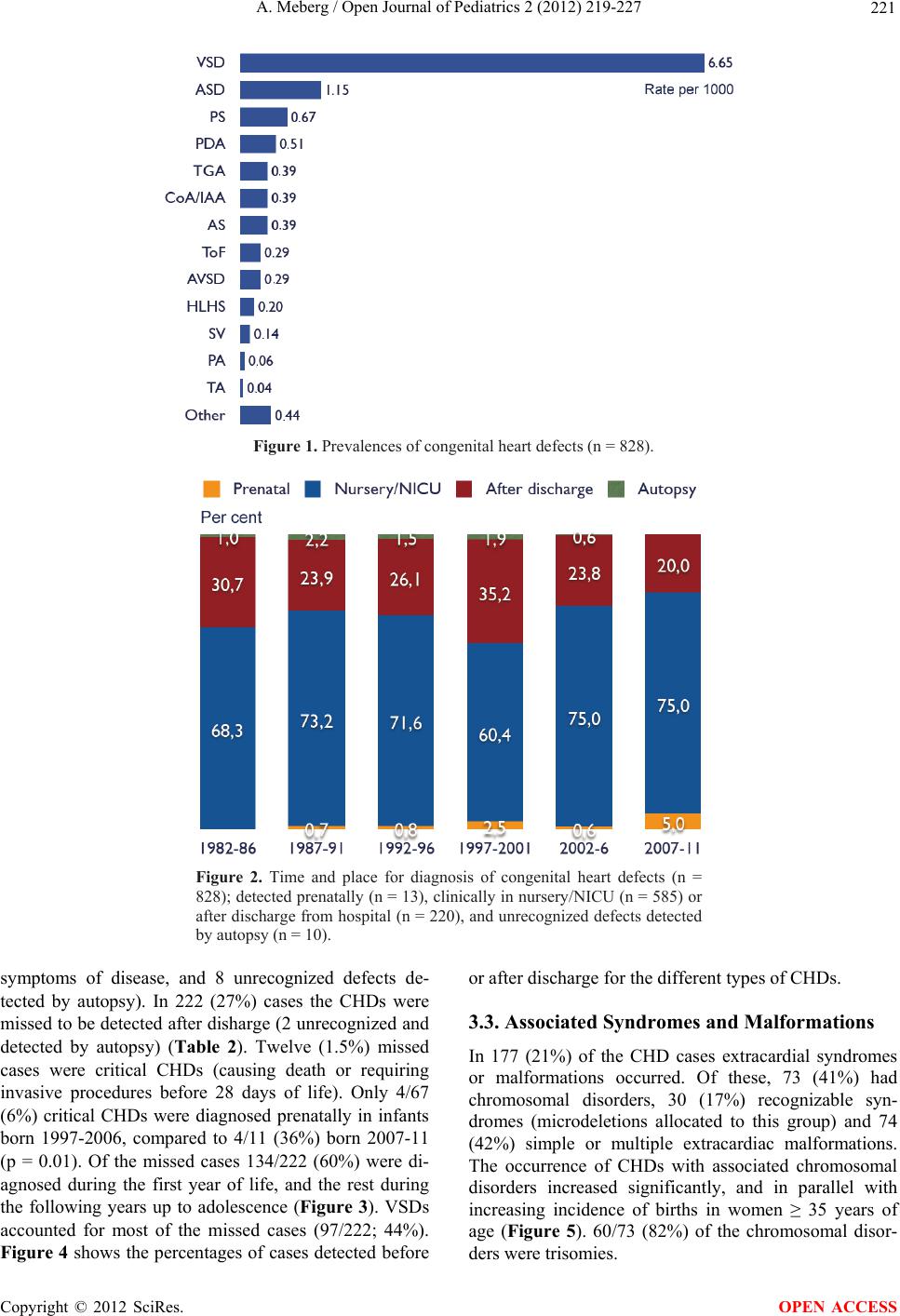
The prevalence of CHDs increased significantly after
introduction of echocardiography in newborns with sus-
pected CHD, all caused by an increased detection rate of
small muscular VSDs. The prevalence of CHDs with
associated chromosomal disorders increased in parallel
with increasing high birthing age. A substantial number
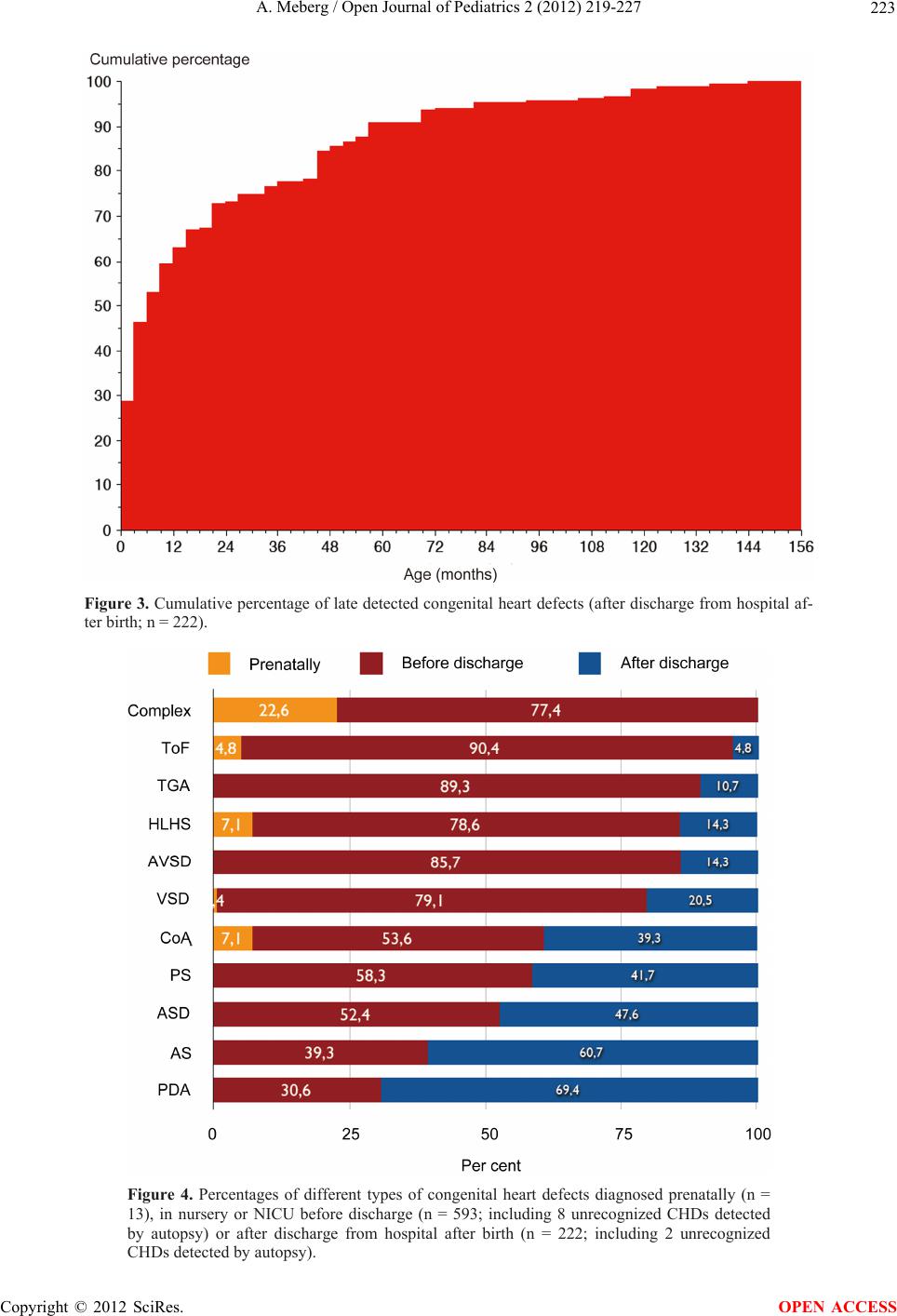
are overlooked in the routine heart screening after birth,
even some critical CHDs. The prenatal detection rate of
critical CHDs increased. Most deaths occurred during
infancy, especially in the neonatal period. Increasing
survival increases the need for referral for long-term fol-
low-up.
6. ACKNOWLEDGEMENTS
The study was supported by a grant from Vestfold Hospital Trust.
REFERENCES
[1] Rice, M.J., McDonald, R.W., Reller, M.D. and Sahn, D.J.
(1996) Pediatric echocardiography: Current role and a
review of technical advances. The Journal of Pediatrics,
128, 1-14. doi:10.1016/S0022-3476(96)70421-3
[2] Morris, C.D. and Menashe, V.D. (1991) 25-year mortality
after surgical repair of congenital heart defect in child-
hood: A population-based cohort study. JAMA: The Jour-
nal of the American Mediacal Association, 266, 3447-
3452. doi:10.1001/jama.1991.03470240069035
[3] Somerville, J. (1990) “Grown-up” survivors of congenital
heart disease: Who knows? Who cares? British Journal of
Hospital Medicine, 43, 132-136.
[4] Meberg, A., Otterstad, J.E., Frøland, G., Sørland, S. and
Nitter-Hauge, S. (1994) Increasing incidence of ventricular
septal defects caused by improved detection rate. Acta
Paediatrica, 83, 653-657.
doi:10.1111/j.1651-2227.1994.tb13102.x
[5] Dickinson, D.F. (1998) Ventricular septal defect: (Not)
another epidemic? Cardiology in the Young, 8, 423-424.
doi:10.1017/S1047951100007046
[6] International Society of Cardiology (1970) Classification
of heart disease in childhood. VBR Offsetdrukkerij,
Groningen.
[7] Fyler, D.C., Buckley, L.P., Hellenbrand, W.E., Cohn, H.E.,
Kirklin, J.W., Nadas, A.S., et al. (1980) Report of the
New England Regional Infant Cardiac Program. Pediatrics,
65, 377-461.
[8] Archer, N. (2005) Cardiovascular disease. In: Rennie, J.M.
Ed., Roberton’s Textbook of Neonatology, 4th Edition,
Elsevier Churchill Livingstone, Philadelphia, 619-660.
[9] Bernstein, D. (2011) Epidemiology and genetic basis of
congenital heart disease. In: Kliegman, R.M., Stanton,
B.F., Schor, N.F., St. Geme, J.W. III and Behrman, R.E.,
Eds., Nelson Tex tbook of Pediatrics, 19th Edition, Elsevier
Saunders, Philadelphia, 1549.
doi:10.1016/B978-1-4377-0755-7.00418-8
[10] Mellander, M. and Sunnegårdh, J. (2006) Failure to diag-
nose critical heart malformations in newborns before dis-
charge—An increasing problem? Acta Pædiatrica, 95,
407-413. doi:10.1080/08035250500541910
[11] Tegnander, E., Eik-Nes, S.H., Johansen, O.J. and Linker,
D.T. (1995) Prenatal detection of heart defects at the
routine fetal examination at 18 weeks in a non-selected
population. Ultrasound in Obstetrics & Gynecology, 5,
372-380. doi:10.1046/j.1469-0705.1995.05060372.x
[12] Meberg, A., Brügmann-Pieper, S., Due, R. Jr., Eskedal,
L., Fagerli, I., Farstad, T., et al. (2008) First day of life
pulse oximetry screening to detect congenital heart defects.
The Journal of Pediatrics, 152, 761-765.
doi:10.1016/j.jpeds.2007.12.043
Copyright © 2012 SciRes. OPEN ACCESS