 American Journal of Plant Sciences, 2012, 3, 1322-1327 http://dx.doi.org/10.4236/ajps.2012.39159 Published Online September 2012 (http://www.SciRP.org/journal/ajps) Effective Procedure for Development of EST-SSR Markers Using cDNA Library Kyung A Kim1, Hee-Cheon Park1, Jae-Keun Sohn2, Kyung-Min Kim2* 1Department of Biology, College of Natural Sciences, Kyungpook National University, Daegu, Korea (South); 2School of Applied Biosciences, College of Agriculture and Life Science, Kyungpook National University, Daegu, Korea (South). Email: Kyung-AKim, hikyunga@knu.ac.kr, Hee-Cheon Park, heecheon@knu.ac.kr, Jae-Keun Sohn, jhsohn@knu.ac.kr, *Kyung-Min Kim, kkm@knu.ac.kr Received August 8th, 2012; revised September 5th, 2012; accepted September 13th, 2012 ABSTRACT The present study was conducted to develop EST-SSR markers using the cDNA library from rice plant. Total RNA ex- tracted from the leaves of brown plant hopper resistance gene originated from a rice cultivar “Cheongcheong” and sen- sitive rice cultivar “Nakdong” were used to synthesize a cDNA library. As a result of analyzing the cDNA library, the 17 EST-SSR primer sets were developed. This study enables to provide effective marker assisted selection (MAS) methods on the selection of white-backed planthopper resistance gene originated from a rice plant more simply, quickly and precisely. Furthermore, using this marker’s advantage of deriving from cDNA, it is possible to identify the white-backed planthopper resistance gene. In addition, this study introduces a technique for construction of a cDNA library safely without using radioactivity. Keywords: cDNA; EST-SSR Markers; Radioactivity; Rice 1. Introduction Several protocols for cDNA library construction have been investigated to date; however, these are each com- plicated and difficult to control [1-6]. Moreover, the procedure for size fraction selection requires the use of radioactivity. Therefore, this study was conducted to de- velop expressed sequence tag-simple sequence repeat (EST-SSR) markers of the brown plant hopper resistance gene for the “Cheongcheong” and the sensitive rice culti- var, “Nakdong”. Furthermore, we introduce a safe and easy method for construction of a cDNA library without the use of radioactivity. The white-backed planthopper is one of the most harmful insects to crop growth [7]. Most farmers are currently cultivating a japonica sensitive rice cultivar in Korea; however, it is very difficult to examine brown plant hopper resistance in rice [8,9]. Marker as- sisted selection (MAS) is able to select brown plant hop- per resistant rice cultivars based on DNA markers [10- 13]. Using MAS, it is possible to overcome the complex- ity and imprecision associated with traditional methods of selecting brown plant hopper resistant rice cultivars [14]. So, we carried out the development of EST-SSR markers for molecular breeding in rice. 2. Materials and Methods 2.1. Extraction of Total RNA and Genomic DNA Total RNA was extracted using the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen GmbH, Hilden, Germany).Briefly, RLT Buffer contain- ing β-mercaptoethanol (β-ME) was added to fresh leaves of “Cheongcheong” and “Nakdong” and thoroughly mixed. The mixture was then transferred to a supplied spin column and centrifuged for 5 minutes. Next, the supernatant from the spin column was carefully trans- ferred to a new 1.5 ml tube, to which an equal volume of 70% ethanol was added and mixed by pipetting. The sample was then immediately transferred to an RNeasy spin column and centrifuged for 1 minute at 13,000 rpm, after which the flow-through was discarded. Next, 700 μl of buffer RW1 was added to the spin column and the sample was centrifuged for 1 minute at 13,000 rpm to wash the column membrane. The flow-through was then discarded, after which 500 μl of buffer RPE was added to the RNeasy spin column, which was then centrifuged for 1 minute at 13,000 rpm to wash the membrane. The flow-through was again discarded and this process was repeated. Next, the RNeasy spin column was placed in a new 1.5 ml collection tube and 30 µl of DEPC water was added directly to membrane. After incubating for 10 *Corresponding author. Copyright © 2012 SciRes. AJPS  Effective Procedure for Development of EST-SSR Markers Using cDNA Library 1323 minutes, the column was centrifuged for 1 minute at 13,000 rpm to elute the RNA and the extracted RNA was then stored at –80˚C. The genomic DNA was extracted using a DNeasy Mini Kit according to the manufacturer’s instructions (Qiagen GmbH, Hilden, Germany). 2.2. Construction of cDNA Library A cDNA library was produced using a cDNA Synthesis Kit (TaKaRa, Shiga, Japan) and a cDNA PCR Library Kit (TaKaRa, Shiga, Japan). Briefly, template RNA was added to a mixture composed of 2 μl 5× first strand syn- thesis buffer, 0.5 μl dNTPs mixture (10 mM), 0.5 μl RNase inhibitor (20 unit·μl–1), 1 μl oligodT RA primer (1 μg·μl–1) and 0.5 μl of M-MLV reverse transcriptase. Upon adding the template RNA, the volume was adjusted to 10 μl. The mixture was then reacted for 10 minutes at room temperature, after which it was incubated at 42˚C for 1 hour, then heated to 80˚C for 5 minutes to produce the first strand of cDNA. Next, 10 μl of the first-strand cDNA synthesis mixture was mixed with 15 μl of 5× second strand synthesis buffer and 1.5 μl of dNTP mix- ture (10 mM), after which the volume was adjusted to 71 μl with DEPC water. In the next step, 1 μl of E. coli DNA polymerase I (20 unit·μl–1) and 1 μl of E. coli- RNase H/E. coli DNA ligase mixture was added, mixed well and reacted at 16˚C for 2 hours. The mixture was then heated to 72˚C, after which 2 μl of T4 DNA poly- merase (1 unit·μl–1) was added and thoroughly mixed. The mixture was then allowed to react at 37˚C for 10 minutes, at which time 6 μl of stop solution (0.2M EDTA, 2 mg·ml–1 glycogen, pH 8.0) was added. An equal vo- lume of PCI (phenol: chloroform: isoamyl alcohol = 25:24:1) was then added, after which the mixture was stirred for 1 minute and centrifuged at 15,000 rpm for 5 minutes to generate pure second-strand cDNA. The su- pernatant was subsequently transferred to a new 1.5 ml tube, after which an equal volume of CI (chloroform: isoamylalcohol 24:1) was added and the mixture was stirred for 1 minute and then centrifuged at 15,000 rpm for 5 minutes. The supernatant was subsequently trans- ferred to a new 1.5 ml tube and equal volumes of 4 M ammonium acetate and isopropanol were added. Follow- ing incubation at –80˚C for 1 hour, the mixture was cen- trifuged at 15,000 rpm for 5 minutes. 2.3. Construction of Second-Strand cDNA without Radioactivity The supernatant was then removed and 1 ml of 70% ethanol was added to wash the precipitate. After the pre- cipitate dried, it was dissolved in 5 μl of RNase-free wa- ter and ExTaq polymerase (TaKaRa, Shiga, Japan) was used to synthesize an adaptor sequence with a restriction enzyme site at each terminal of the second-strand cDNA. To accomplish this, 30 μl of second strand cDNA, 5 μl of 10× ExTaq buffer, 4 μl of dNTP mixture (2.5 mM), 1 μl of CA primer (20 p mole·μl–1, 5’-CGTGGTACCATG- GTCTAGAGT-3’), 1 μl of RA primer (20 pmole·μl–1, 5’-CTGATCTAGACCTGCAGGCTC-3’), 0.5 μl TaKa- RaExTaq polymerase (5 unit·μl–1) (TaKaRa, Shiga, Japan) and 8.5 μl nuclear free water were mixed to give a final volume of 50 μl, after which PCR was performed by sub- jecting the samples tothe following conditions: 2 minutes at 94˚C followed, by 35 cycles of 30 seconds at 94˚C, 30 seconds at 60˚C and 3 minutes at 4˚C and then final ex- tension for 10 minutes at 72˚C. To select 500 bp to 1 kb for the cDNA library, the cDNA PCR amplicons were electrophoresed in 1.0% agarose gel, after which selected bands were cut out and extracted from the gel (Qiagen- Co., Germany). After the gel extraction procedure, PCR was performed (TaKaRa, Shiga, Japan) by subjecting the samples to the same conditions as described above. 2.4. cDNA Library Transformation to Vector The cDNA librarywas purified using a PCR purification kit (QiagenCo., Germany) and then ligated into the pGEM-T Easy Vector (Promega Co., USA). After adding 5 μl of 2× rapid ligation buffer and 1 μl of T4 DNA li- gase (unit·μl–1), the pGEM-T Easy Vector (50 ng·μl–1) and cDNA PCR products were adjusted to the ratio. The volume was then adjusted to 10 μl with nuclease-free water, after which the sample was allowed to react at 4˚C for 12 hours. Next, 5 μl of ligation product and 50 μl of DH 5α competent cells were mixed and reacted on ice for 30 minutes and then incubated at 42˚C for 1 minute. The mixture was again incubated on ice for 2 minutes, after which 250 μl of LB broth (10 g Bactotryptone, 5 g Bacto yeast extract and 5 g NaCl) was added and the samples were cultured at 37˚C for 1 hour while shaking at 180 rpm. The culture was then incubated at 37˚C for 12 hours after platingon LB agar amended with 100 ppm·l–1 ampi- cillin, 0.5 mM IPTG and 80 µg·ml–1 X-Gal. Blue-colored and white-colored colonies appeared after culture, with white indicating that transformation had occurred. Ac- cordingly, white colonieswere selected, added to 5 ml of LB broth, and then cultured at 37˚C for 14 hours. Fol- lowing incubation, the culture medium was centrifuged at 3000 rpm for 10 minutes and then extracted using a Qiaprep Spin Miniprep Kit (QiagenCo., Germany). After removing the supernatant, 250 μl of P1 buffer was added to dissolve the colonies, and the sample was then trans- ferred to a 1.5 ml tube and mixed carefully with 250 μl of P2 buffer. Next, 350 μl of N3 buffer was added and the sample was centrifuged at 13,000 rpm for 10 minutes. The supernatant was then transferred to a new 1.5 ml tube, centrifuged again at 13,000 rpm for 5 minutes, and then transferred to a new 1.5 ml tube. Next, 700 μl of supernatant was transferred to a spin column and centri- Copyright © 2012 SciRes. AJPS  Effective Procedure for Development of EST-SSR Markers Using cDNA Library 1324 fuged at 13,000 rpm for 1 minute, after which the flow through was discarded. PB buffer (5 μl) was then added to the column, after which it was centrifuged at 13,000 rpm for 1 minute, and 750 μl of PE buffer was added to wash the plasmid DNA. The flow through from the col- lection tube was then discarded, after which the column was centrifuged to remove any residual buffer. The spin column was then added to a 1.5 ml tube containing 30 μl of nuclease-free water to extract the plasmid DNA. 2.5. DNA Sequencing and Primer Design The plasmid DNA sequence was analyzed by SolGent (SolGent Co., Korea), which verified that the cDNA was inserted into the pGEM-T Easy Vector (Promega Co., USA). The DNA sequencing analysis results were then used for SSR-site analysis in the EST sequence using a microsatellite analysis program (http://www.wsmartins.net/websat/) [15], and the SSR sites analyzed were used to prepare the forward and re- verse primers. 2.6. Polymorphism Analysis Using an EST-SSR Marker Polymorphism analysis was conducted using the markers of EST-SSR primer sets developed from this study. Briefly, PCR was used to investigate genomic crane DNA using TaKaRaExTaq polymerase (TaKaRa, Shiga, Japan) and the EST-SSR primer set (forward and reverse primers) developed in this study. The PCR conditions were as follows: initial denaturation at 94˚C for 5 min- utes, followed by 35 cycles of 94˚C for 30 seconds, 56˚C - 58˚C for 30 seconds and 72˚C for 45 seconds and then final extension at 72˚C for 5 minutes. QIAxcel (Qiagen- Co., Germany) was used to verify the amplified band in the PCR product, and the size was verified using the QX DNA size marker (FX174/HaeⅢ). Only bands that were verified with a 0% noise cut-off (0% - 99%) and 100% contrast (1% - 100%) were selected; the red portion of the band peak size (bp) and concentration (ng·μl–1) were referenced in a data table. 3. Results It is laborious and time-consuming to examine brown plant hopper resistance in rice cultivars. To obtain good quality results and reduce labor, we developed a simple and safe method of cDNA construction for development of EST-SSR markers. This procedure is able to select a cDNA library of hopping size, about 500 bp to 1 kbp. Fragments of this size can be ligated into the pGEM- T-Easy vector efficiently, thereby enabling a high yield of good quality ligated products. This procedure can also be carried out safely without the need to use radioactivity to determine size fraction. The EST-SSR markers of the brown plant hopper resistance gene originating from the “Cheongcheong” and the sensitive rice cultivar “Nak- dong” were developed. Seventeen EST-SSR primer sets were designed the expected size was shown (Table 1). The definite PCR bands of the amplified DNA from rice cultivarsusing the EST-SSR primer sets was achieved (Figure 1(a)). For the mapping of resistant gene to white-backed planthopper, the doubled-haploid (DB) po- pulations was developed by anther culture of F1 plants from a cross “Cheongcheong/Nagdong”. PCR analysis of the DNA from the 22 “Cheongcheong/Nakdong” Dou- bled Haploid (CNDH) populations using the CC24-2F2R primer set showed clear bands between each populations (Figure 1(b)). All patterns of the PCR bands using EST- SSR primers were shown very clearly and precisely, re- spectively. 4. Discussions The several protocols for cDNA library constructionwere previously reported that exploit the mRNA cap structure to enrich for full-length sequence [3], the “oligo-capping” method [4,5], CAPture [2], CAP-trapper [1] and SMARTTM technology of constructing full-length cDNA library [6]. We also obtained good quality cDNA library using safe size fraction method. Overall, the methods presented here should reduce the labor and effort re- quired for identification or resistant strains of rice while increasing safety by removing the need to use radioactive materials. Sequencing was performed on a total of 147 cDNA insertions confirmed through EcoRІ digestion. Using the DNA STAR SeqManⅡ expert sequence analysis software (version 5.01), the forward and reverse primers were selected as EST-SSR sites were found. As a result of analyzing the EST-SSR sites of the sequence using the Microsatellite analysis program (http://www.wsmartins.net/websat/) [15] on the 20 se- lected primer sets and used to develop the EST-SSR primers (Table 1). To verify their availability as markers, PCR was performed on each of the designed EST-SSR primer sets and seventeen primer sets with high repro- ducibility as EST-SSR markers were selected. Many of ESTs are available in public databases, which offer an opportunity to identify SSRs in ESTs by data mining. These sequences may provide an estimate of diversity in the expressed portion of the genome and may be useful for comparative mapping, for tagging important traits of interest, and for additional map-based cloning of impor- tant genes [16]. For effective conservation and the use of genetic resources, evaluation of the genetic variation is crucial and could be dramatically enhanced by using mo- lecular genotyping tools. Evaluation of germplasm with SSRs derived from ESTs (EST-SSRs) may enhance the role of genetic markers by assaying variation in trans- cribed and known-function genes. Furthermore, since the Copyright © 2012 SciRes. AJPS 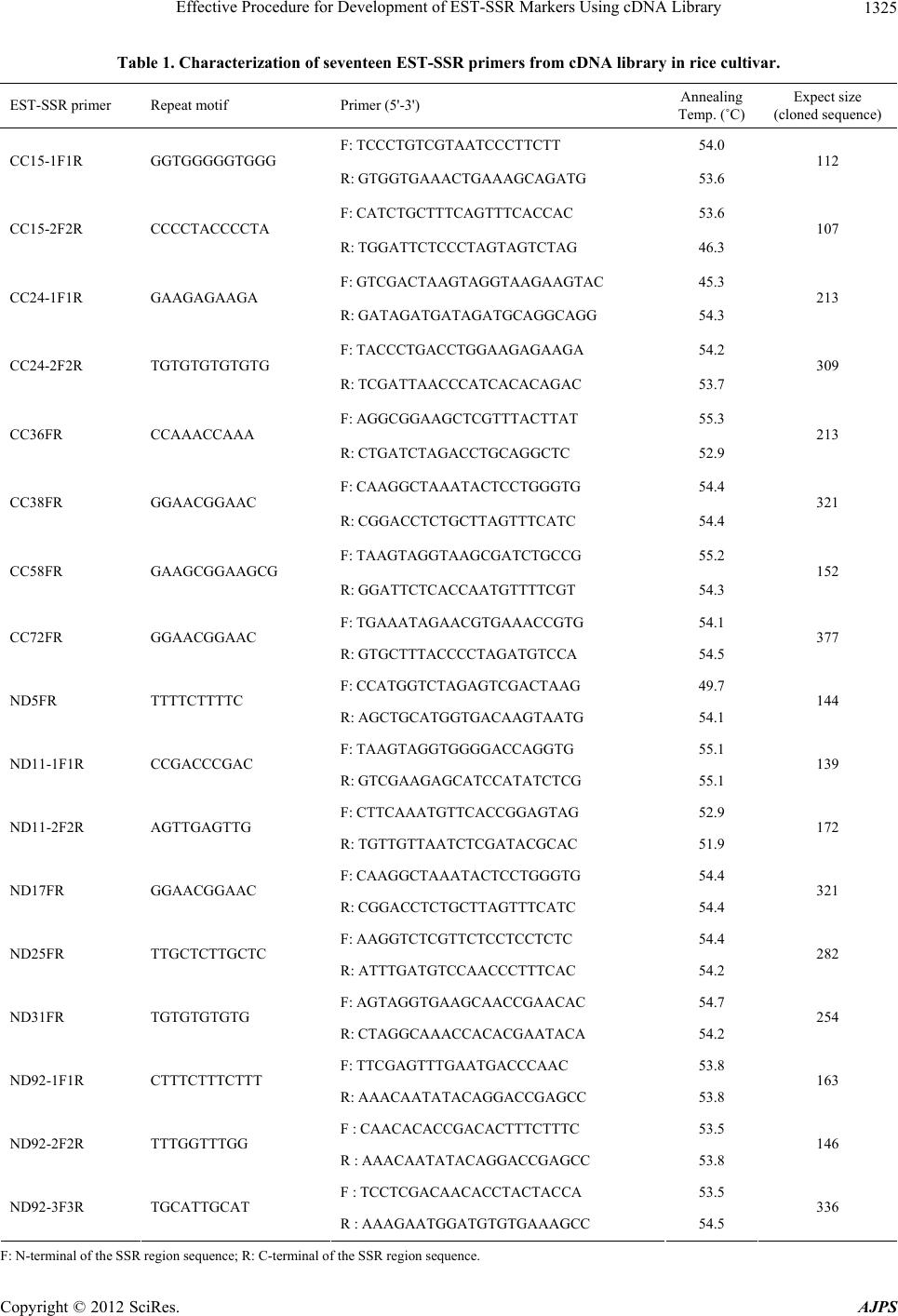 Effective Procedure for Development of EST-SSR Markers Using cDNA Library Copyright © 2012 SciRes. AJPS 1325 Table 1. Characterization of seventeen EST-SSR primers from cDNA library in rice cultivar. EST-SSR primer Repeat motif Primer (5'-3') Annealing Temp. (˚C) Expect size (cloned sequence) F: TCCCTGTCGTAATCCCTTCTT 54.0 CC15-1F1R GGTGGGGGTGGG R: GTGGTGAAACTGAAAGCAGATG 53.6 112 F: CATCTGCTTTCAGTTTCACCAC 53.6 CC15-2F2R CCCCTACCCCTA R: TGGATTCTCCCTAGTAGTCTAG 46.3 107 F: GTCGACTAAGTAGGTAAGAAGTAC 45.3 CC24-1F1R GAAGAGAAGA R: GATAGATGATAGATGCAGGCAGG 54.3 213 F: TACCCTGACCTGGAAGAGAAGA 54.2 CC24-2F2R TGTGTGTGTGTG R: TCGATTAACCCATCACACAGAC 53.7 309 F: AGGCGGAAGCTCGTTTACTTAT 55.3 CC36FR CCAAACCAAA R: CTGATCTAGACCTGCAGGCTC 52.9 213 F: CAAGGCTAAATACTCCTGGGTG 54.4 CC38FR GGAACGGAAC R: CGGACCTCTGCTTAGTTTCATC 54.4 321 F: TAAGTAGGTAAGCGATCTGCCG 55.2 CC58FR GAAGCGGAAGCG R: GGATTCTCACCAATGTTTTCGT 54.3 152 F: TGAAATAGAACGTGAAACCGTG 54.1 CC72FR GGAACGGAAC R: GTGCTTTACCCCTAGATGTCCA 54.5 377 F: CCATGGTCTAGAGTCGACTAAG 49.7 ND5FR TTTTCTTTTC R: AGCTGCATGGTGACAAGTAATG 54.1 144 F: TAAGTAGGTGGGGACCAGGTG 55.1 ND11-1F1R CCGACCCGAC R: GTCGAAGAGCATCCATATCTCG 55.1 139 F: CTTCAAATGTTCACCGGAGTAG 52.9 ND11-2F2R AGTTGAGTTG R: TGTTGTTAATCTCGATACGCAC 51.9 172 F: CAAGGCTAAATACTCCTGGGTG 54.4 ND17FR GGAACGGAAC R: CGGACCTCTGCTTAGTTTCATC 54.4 321 F: AAGGTCTCGTTCTCCTCCTCTC 54.4 ND25FR TTGCTCTTGCTC R: ATTTGATGTCCAACCCTTTCAC 54.2 282 F: AGTAGGTGAAGCAACCGAACAC 54.7 ND31FR TGTGTGTGTG R: CTAGGCAAACCACACGAATACA 54.2 254 F: TTCGAGTTTGAATGACCCAAC 53.8 ND92-1F1R CTTTCTTTCTTT R: AAACAATATACAGGACCGAGCC 53.8 163 F : CAACACACCGACACTTTCTTTC 53.5 ND92-2F2R TTTGGTTTGG R : AAACAATATACAGGACCGAGCC 53.8 146 F : TCCTCGACAACACCTACTACCA 53.5 ND92-3F3R TGCATTGCAT R : AAAGAATGGATGTGTGAAAGCC 54.5 336 F: N-terminal of the SSR region sequence; R: C-terminal of the SSR region sequence. 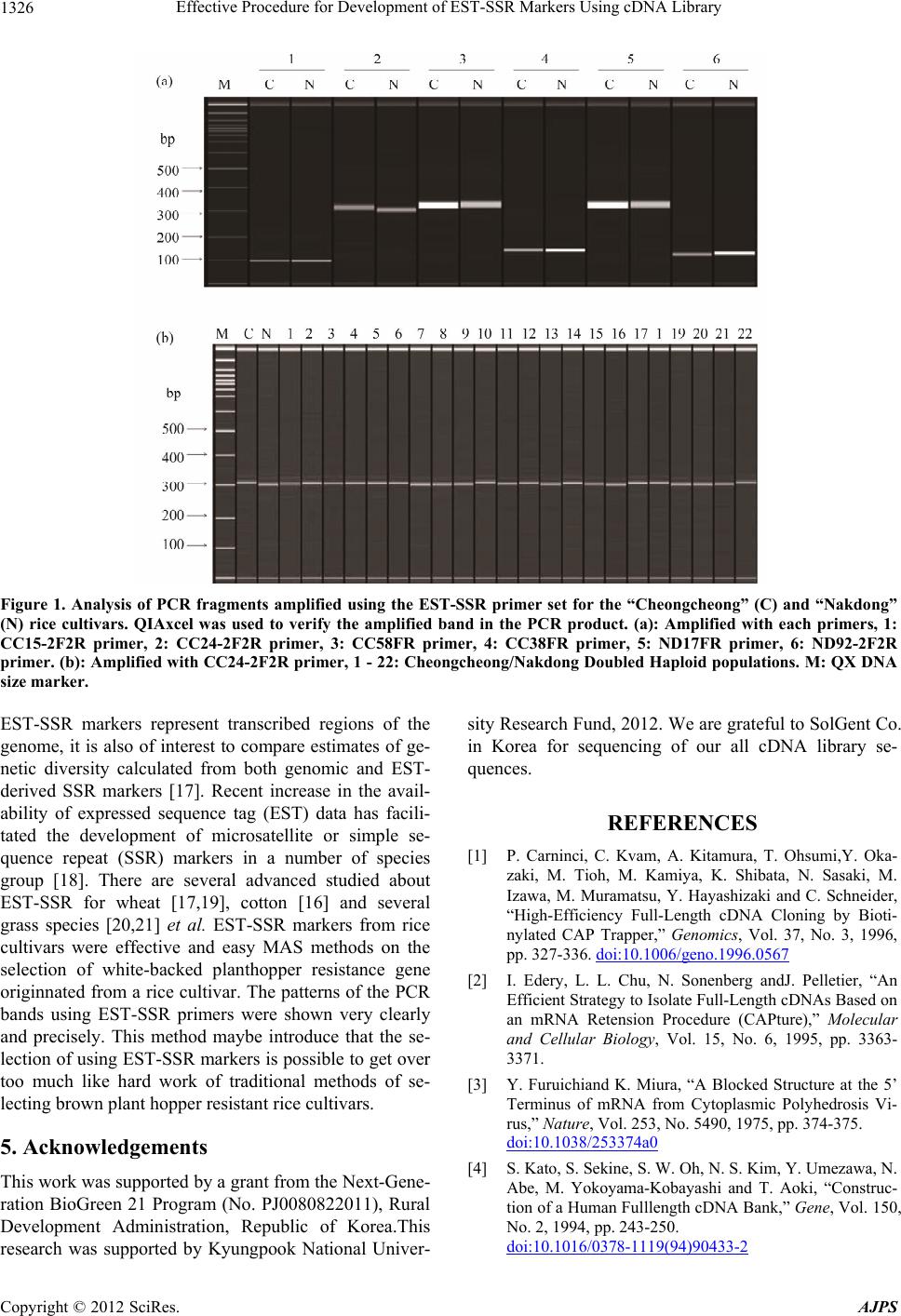 Effective Procedure for Development of EST-SSR Markers Using cDNA Library 1326 Figure 1. Analysis of PCR fragments amplified using the EST-SSR primer set for the “Cheongcheong” (C) and “Nakdong” (N) rice cultivars. QIAxcel was used to verify the amplified band in the PCR product. (a): Amplified with each primers, 1: CC15-2F2R primer, 2: CC24-2F2R primer, 3: CC58FR primer, 4: CC38FR primer, 5: ND17FR primer, 6: ND92-2F2R primer. (b): Amplified with CC24-2F2R primer, 1 - 22: Cheongcheong/Nakdong Doubled Haploid populations. M: QX DNA size marker. EST-SSR markers represent transcribed regions of the genome, it is also of interest to compare estimates of ge- netic diversity calculated from both genomic and EST- derived SSR markers [17]. Recent increase in the avail- ability of expressed sequence tag (EST) data has facili- tated the development of microsatellite or simple se- quence repeat (SSR) markers in a number of species group [18]. There are several advanced studied about EST-SSR for wheat [17,19], cotton [16] and several grass species [20,21] et al. EST-SSR markers from rice cultivars were effective and easy MAS methods on the selection of white-backed planthopper resistance gene originnated from a rice cultivar. The patterns of the PCR bands using EST-SSR primers were shown very clearly and precisely. This method maybe introduce that the se- lection of using EST-SSR markers is possible to get over too much like hard work of traditional methods of se- lecting brown plant hopper resistant rice cultivars. 5. Acknowledgements This work was supported by a grant from the Next-Gene- ration BioGreen 21 Program (No. PJ0080822011), Rural Development Administration, Republic of Korea.This research was supported by Kyungpook National Univer- sity Research Fund, 2012. We are grateful to SolGent Co. in Korea for sequencing of our all cDNA library se- quences. REFERENCES [1] P. Carninci, C. Kvam, A. Kitamura, T. Ohsumi,Y. Oka- zaki, M. Tioh, M. Kamiya, K. Shibata, N. Sasaki, M. Izawa, M. Muramatsu, Y. Hayashizaki and C. Schneider, “High-Efficiency Full-Length cDNA Cloning by Bioti- nylated CAP Trapper,” Genomics, Vol. 37, No. 3, 1996, pp. 327-336. doi:10.1006/geno.1996.0567 [2] I. Edery, L. L. Chu, N. Sonenberg andJ. Pelletier, “An Efficient Strategy to Isolate Full-Length cDNAs Based on an mRNA Retension Procedure (CAPture),” Molecular and Cellular Biology, Vol. 15, No. 6, 1995, pp. 3363- 3371. [3] Y. Furuichiand K. Miura, “A Blocked Structure at the 5’ Terminus of mRNA from Cytoplasmic Polyhedrosis Vi- rus,” Nature, Vol. 253, No. 5490, 1975, pp. 374-375. doi:10.1038/253374a0 [4] S. Kato, S. Sekine, S. W. Oh, N. S. Kim, Y. Umezawa, N. Abe, M. Yokoyama-Kobayashi and T. Aoki, “Construc- tion of a Human Fulllength cDNA Bank,” Gene, Vol. 150, No. 2, 1994, pp. 243-250. doi:10.1016/0378-1119(94)90433-2 Copyright © 2012 SciRes. AJPS  Effective Procedure for Development of EST-SSR Markers Using cDNA Library 1327 [5] K. Maruyama and S. Sugano, “Oligo-Capping: A Simple Method to Replace the Cap Structure of Eucaryotic mRNAs with Oligoribonucleotides,” Gene, Vol. 138, No. 1-2, 1994, pp. 171-174. doi:10.1016/0378-1119(94)90802-8 [6] Y. Y. Zhu, E. M. Machleder, A. Chenchik, R. Li and P. D. Siebert, “Reverse Transcriptase Template Switching: A SMARTTM Approach for Full-Length cDNA Library Construction,” BioTechniques, Vol. 30, No. 4, 2001, pp. 892-897. [7] J. H. Seo, K. M. Kim, S. M. Kim and J. K. Sohn, “De- velopment of RAPD Marker Related to Brown Planthop- per Resistance Gene Derived from Rice Cultivar, Cheongcheongbyeo,” Korean Journal of Crop Science, Vol. 50, No. 6, 2005, pp. 453-456. [8] S. M. Kim, S. J. Yang and J. K. Sohn, “Development of a DNA Marker for Selection of Resistant Plants to Brown Planthopper (BPH) in Rice,” Korean Journal of Breeding, Vol. 35, No. 5, 2003, pp. 277-282. [9] U. S. Yeo, D. Y. Kwak, S. J. Lim, W. G. Ha, H. J. Cho and J. K. Sohn, “Relationship between Agronomic Traits and Resistance to Brown Planthopper in Japonica RIL Population,” Korean Journal of Breeding, Vol. 34, No. 3, 2002, pp. 148-152. [10] H. Hirabayashi and O. Tsugufumi, “RFLP Mapping of Bph 1 (Brown Planthopper Resistance Gene) in Rice,” Breeding Science, Vol. 45, No. 3, 1995, pp. 369-371. [11] Z. Huang, G. He, L. Shu, X. Li and Q. Zhang, “Identifi- cation and Mapping of Two Brown Plantopper Resistance Genes in Rice,” TAG Theoretical and Applied Genetetics, Vol. 102, No. 6-7, 2001, pp. 929-934. doi:10.1007/s001220000455 [12] K. K. Jena, I. C. Pasalu, Y. K. Rao, Y. Varalaxmi, K. Krishnaiah, G. S. Khush and G. Kochert, “Molecular Tagging of a Gene for Resistance to Brown Planthopper in Rice (Oryza sativa L.),” Euphytica, Vol. 129, No. 1, 2002, pp. 81-88. doi:10.1023/A:1021590025240 [13] K. Murata, M. Fujiware, C. Kaneda, S. Takumi, N. Mori and C. Nakamura, “RFLP Mapping of a Brown Planthop- per (Nilaparvatalugens Stal) Resistance Gene bph 2 of Indica Rice Introgressed into a Japonica Breeding Line 'Norin-PL4',” Genes &Genetics Systems, Vol. 73, No. 6, 1998, pp. 359-364. doi:10.1266/ggs.73.359 [14] P. N. Sharma, A. Torii, S. Takumi, N. Mori andC. Naka- mura, “Marker-Assisted Pyramiding of Brown Planthop- per (Nilaparvatalugens Stal) Resistance Genes bph 1 and bph 2 on Rice Chromosome 12,” Hereditas, Vol. 140, No. 1, 2004, pp. 61-69. doi:10.1111/j.1601-5223.2004.01726.x [15] Microsatellite Analysis Program. http://www.wsmartins.net/websat/ [16] S. N. Qureshi, S. Saha, R. V. Kantety and J. N. Jenkins, “Molecular Biology and Physiology. EST-SSR: A New Class of Genetic Markers in Cotton,” The Journal of Cotton Science, Vol. 8, No. 2, 2004, pp. 112-123. [17] I. Eujayl, M. E. Sorrells, M. Baum, P. Wolters and W. Powell, “Isolation of EST-Derived Microsatellite Markers for Genotyping the A and B Genomes of Wheat,” Tag Theoretical and Applied Genetics, Vol. 104, No. 2-3, 2002, pp. 399-407. doi:10.1007/s001220100738 [18] R. K. Varshney, R. Sigmund, A. Börner, V. Korzun, N. Stein, M. E. Sorrells, P. Langridge and A. Graner, “Inter- specific Transferability and Comparative Mapping of Barley EST-SSR Markers in Wheat, Rye and Rice,” Plant Science, Vol. 168, No. 1, 2005, pp. 195-202. doi:10.1016/j.plantsci.2004.08.001 [19] I. Eujayl, M. Sorrells, M. Baum, P. Wolters and W. Pow- ell, “Assessment of Genotypic Variation among Culti- vated Durum Wheat Based on EST-SSRS and Genomic SSRS,” Euphytica, Vol. 119, No. 1-2, 2001, pp. 39-43. doi:10.1023/A:1017537720475 [20] M. A. R. Mian, M. C. Saha, A. A. Hopkins and Z. Y. Wang, “Use of Tall Fescue EST-SSR Markers in Phy- logenetic Analysis of Cool-Season Forage Grasses,” Ge- nome, Vol. 48, No. 4, 2005, pp. 637-647. doi:10.1139/g05-029 [21] M. C. Saha, M. A. R. Mian, I. Eujayl, J. C. Zwonitzer, L. Wang and G. D. May, “Tall Fescue EST-SSR Markers with Transferability across Several Grass Species,” TAG Theoretical and Applied Genetics, Vo. 109, No. 4, 2004, pp. 783-791. doi:10.1007/s00122-004-1681-1 Copyright © 2012 SciRes. AJPS
|