T. Fujita, H. Fujii / Advances in Bioscience and Biotechnology 3 (2012) 626-629 627
into p3XFLAG-CMV-7.1 (Sigma-Aldrich) that was
cleaved with Bgl II and blunted to generate 3×FNLD/
pCMV-7.1. Subsequently, the DNA sequence encoding
DB and the dimerization domain of LexA was amplified
with the LexA-N (26081) (5'-ccctttCCTGAGGGAATG
AAAGCGTTAACG-3') and LexA-C w/D (26628) (5'-aa
atgtcgaCTACAGCCAGTCGCCGTTGCG-3') primers us-
ing pBTM116 [4] as template. The PCR product was
cleaved with Mlu I and Sal I and inserted into Mlu I- and
Sal I-cleaved 3×FNLD/pCMV-7.1 to generate 3×FNLDD/
pCMV-7.1.
To construct pMXs-I2, the coding sequence of en-
hanced green fluorescent protein (EGFP) of pMXs-IG [5]
was replaced with that of human CD2 antigen (hCD2) [6].
To construct 3×FNLDD/pMXs-I2, the DNA sequence
encoding 3×FNLDD was cleaved with Sac I and Sal I,
blunted, and inserted into the pMXs-I2 vector that was
cleaved with EcoR I and Not I and blunted.
All PCR-derived DNA sequences were verified by
DNA sequencing.
2.2. Cell Lines
293T was maintained in DMEM supplemented with 10%
fetal calf serum (FCS). Ba/F3 [7]-derived cells were
maintained in RPMI-1640 supplemented with 10% FCS,
10 mM Hepes (pH 7.2), 1 × non-essential amino acid, 1
mM sodium pyruvate, 5 nM 2-mercaptoethanol, 1 ng/ml
interleukin-3.
1 × 107 of Ba/F3 were transfected with Mlu I-digested
pGL3C-Neo-cHS4c × 24-LexA × 2 [3] (100 µg) together
with the hygromycin resistance gene (3 µg) by electro-
poration using Gene Pulser II (Bio-Rad) at 250 V, 975
μF. The transfected cells were selected in the presence of
hygromycin (1 mg/ml) to establish the cHS4-core-1.2k
cell line. Subsequently, 1 × 107 of cHS4-core-1.2k were
transfected with Sca I-digested 3×FNLDD/pCMV-7.1
(100 µg) or Hind III-digested FCNLD/pEF (100 µg)
together with the puromycin resistance gene (3 µg) by
electroporation. The transfected cells were selected in the
presence of hygromycin (1 mg/ml) and puromycin (0.6
µg/ml) to establish the 3×FNLDD/cHS4-core-1.2k or
FCNLD/cHS4-core-1.2k cell line.
2.3. Immunoblot Analysis
Nuclear extracts were prepared with NE-PER Nuclear and
Cytoplasmic Extraction Reagents (Pierce). Immunoblot
analysis was performed as described before [6]. Anti-
FLAG M2 Ab was purchased from Sigma-Aldrich.
2.4. Flowcytometry
2 µg of pMXs-I2 or 3×FNLDD/pMXs-I2 was transfected
into 1 × 106 of 293T cells with Lipofectamine 2000 (In-
vitrogen). Two days after transfection, cells were har-
vested and stained with phycoerythrin (PE)-conjugated
anti-hCD2 Ab (BD Biosciences). Subsequently, cells
were intracellularly stained with fluorescein isothiocy-
anate (FITC)-conjugated anti-FLAG M2 (Sigma-Aldrich)
using the Fixation/Permeabilization and Permeabilization
buffer set (eBioscicence). Flowcytometric analysis was
performed on FACS Calibur (BD Biosciences), and data
was analyzed with FlowJo software (TreeStar).
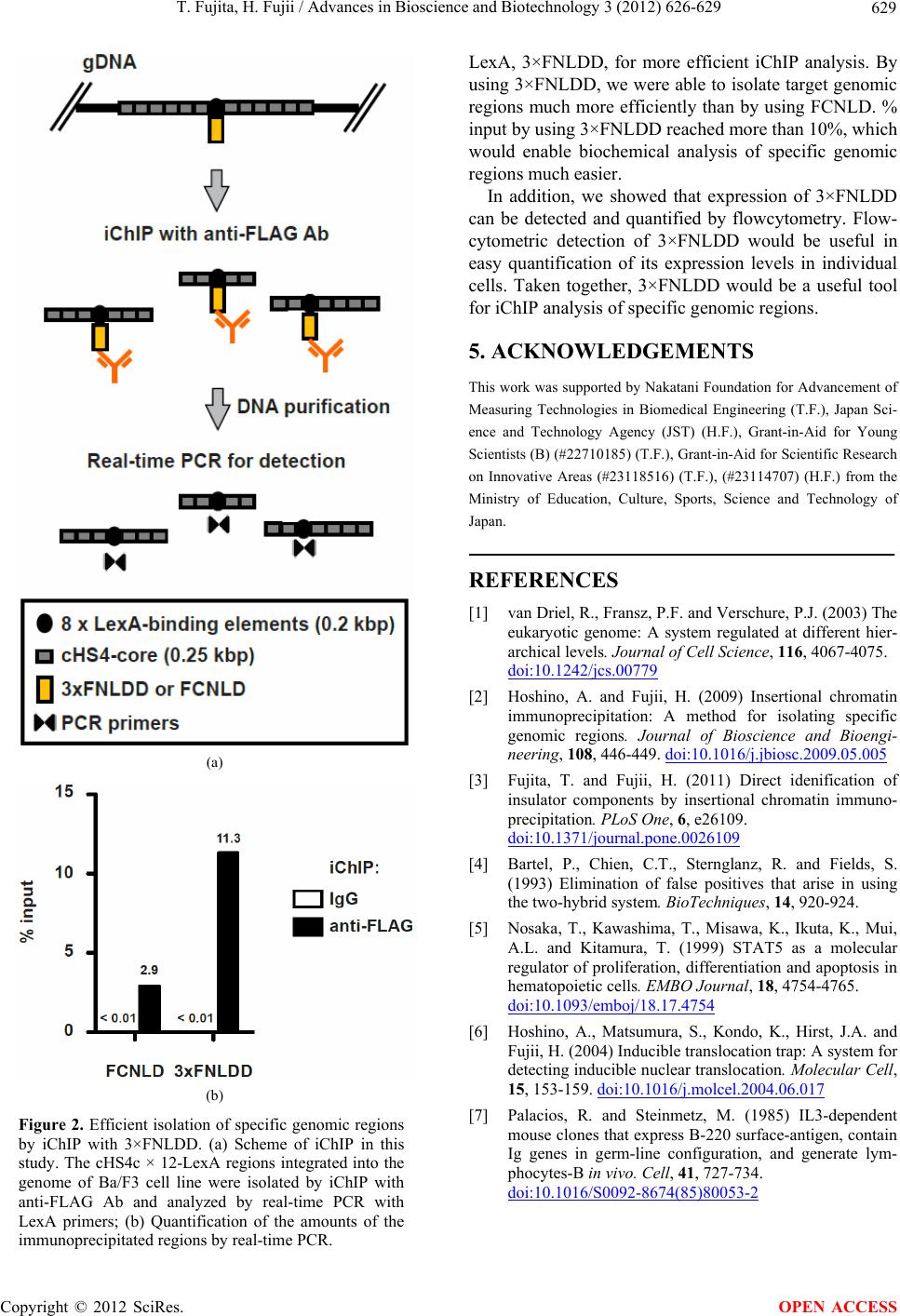
2.5. Chromatin Preparation and iChIP
Cells (4 × 106) were fixed with 1% formaldehyde at 37˚C
for 5 min. The chromatin fraction was extracted and
fragmented (2 kbp-long on average) by sonication and
subjected to iChIP as described previously [3]. The DNA
purified by phenol-chloroform extraction and ethanol
precipitation was used as a template for real-time PCR
with Power SYBR Green PCR Master Mix (Applied
Biosystems) using the Applied Biosystems 7900HT Fast
Real-Time PCR System. PCR cycles were as follows:
heating at 50˚C for 2 min followed by 95˚C for 10 min;
40 cycles of 95˚C for 15 sec and 60˚C for 1 min. The
primers used in this experiment are LexA-N2 (26572)
(5'-ttctctatcgataggtacctcg-3') and LexA-C (26573) (5'-tct
attcagcggatctcgagcg-3').
3. RESULTS AND DISCUSSION
3.1. Generation of the Expression System of
3×FNLDD
For iChIP analysis, we have used the FCNLD protein [2]
consisting of 2 × FLAG tags, a tobacco etch virus (TEV)
protease cleavage site, calmodulin-binidng peptide, the
NLS of SV40-T-antigen, and LexA DB. To generate
more efficient tagged LexA proteins for iChIP analysis,
we constructed a plasmid expressing the 3×FNLDD pro-
tein consisting of 3 × FLAG tags, the NLS of SV40-
T-antigen, and DB as well as the dimerization domain of
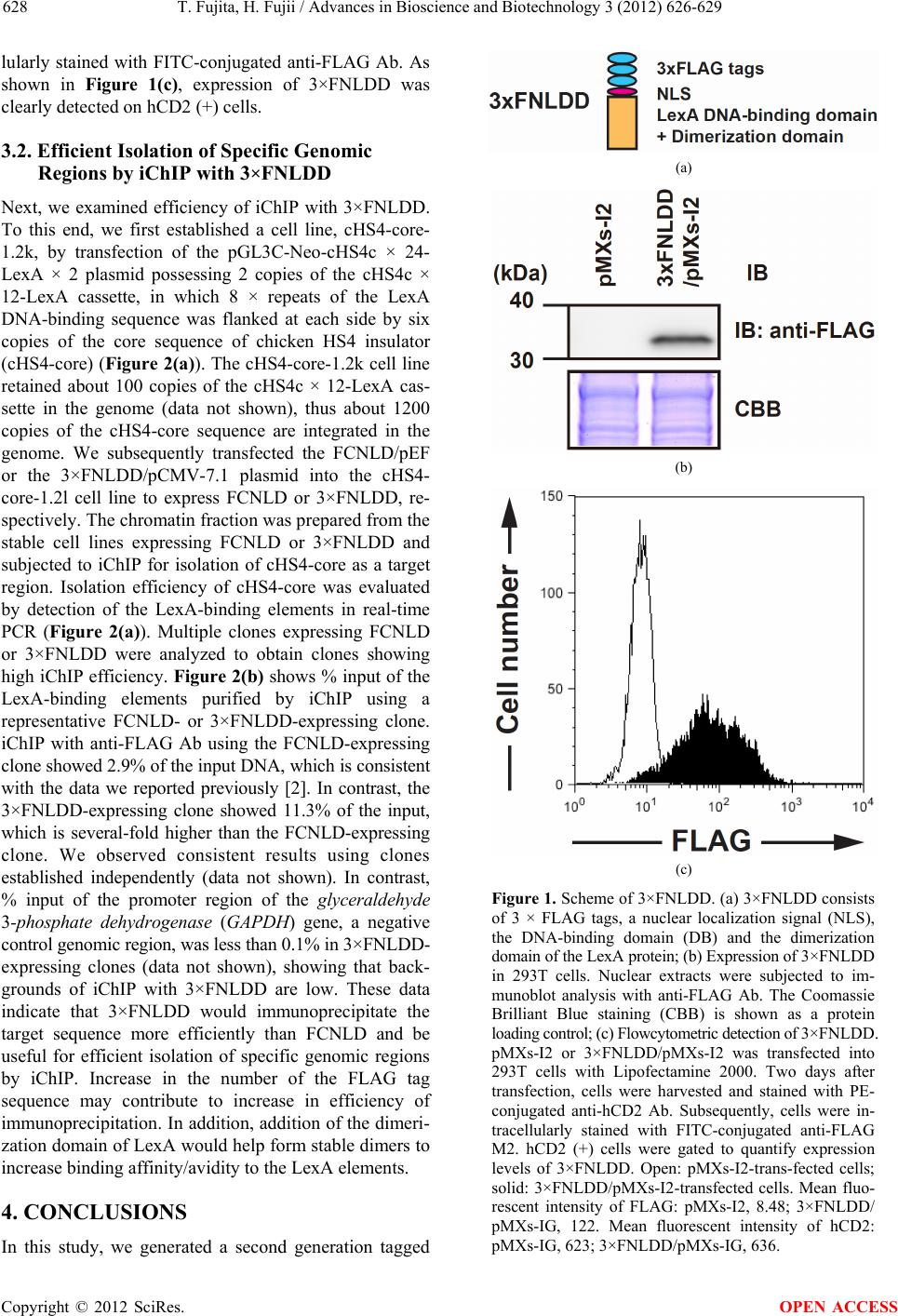
LexA (Figure 1(a)). To increase efficiency of immuno-
precipitation, 3 × FLAG tags were used instead of 2 ×
FLAG tags. In addition, we removed the TEV protease
cleavage site and calmodulin-binding peptide because
efficiency of cleavage by TEV was low in crosslinked
chromatin (data not shown).
The pMXs-I2 vector expressing hCD2 or 3×FNLDD/
pMXs-I2 bicistronically expressing 3×FNLDD and hCD2
was transfected into 293T cells. Nuclear extracts were
prepared and subjected to immunoblot analysis using
anti-FLAG Ab. As shown in Figure 1(b), expression of
3×FNLDD was detected.
Next, we examined expression of 3×FNLDD in indi-
vidual cells by flowcytometry. 293T cells transfected
with pMXs-I2 or 3×FNLDD/pMXs-I2 were stained with
PE-conjugated anti-hCD2 Ab and subsequently intracel-
Copyright © 2012 SciRes. OPEN ACCESS