American Journal of Plant Sciences
Vol. 4 No. 3 (2013) , Article ID: 29155 , 6 pages DOI:10.4236/ajps.2013.43082
Small RNAs and Transgenerational Epigenetic Variation
![]()
Commonwealth Scientific and Industrial Research Organization Plant Industry, Canberra, Australia.
Email: Joanne.lee@csiro.au, Zane.Duxbury@sainsbury-laboratory.ac.uk, *Ming-Bo.Wang@Csiro.au
Received January 5th, 2013; revised February 18th, 2013; accepted February 28th, 2013
Keywords: siRNA; Epigenetics; Arabidopsis Thaliana; Transgenerational Epigenetic Variation; RNA Directed DNA Methylation (RdDM)
ABSTRACT
Small RNAs are found in eukaryotes and are responsible for regulation of chromatin structure, RNA processing and stability, translation and transcription. 24-nt small interfering RNA (siRNA) are known to mediate gene inactivation via the RNA-directed DNA methylation pathway (RdDM) and are important for natural heritable changes in plant species. DNA cytosine methylation can be maintained between generations and this may be important for accelerated adaption to stress conditions. Research is currently focused toward the epigenetic response to disease, the stability of DNA methylation over generations, the elucidation of newly discovered pathways for de novo DNA methylation, and the application of epigenetic variation to breeding programs. This review aims to give a brief but comprehensive examination on small RNAs and transgenerational epigenetic variation.
1. Introduction
RNA directed gene silencing has been studied since the early 1990s and is quickly becoming the hot topic of the decade. With the multitude of papers being published and the numerous interests from epigenetic research groups around the world, it is no wonder that small non-coding RNA has been thrust into the spotlight.
Epigenetics can be described as “control of gene expression based on chromatin organization rather than on primary (genetic) DNA sequence information” [1]. Short and long noncoding RNAs are now known to be key regulators of chromatin structures. Small non-coding RNAs are important regulatory molecules in eukaryotes and, as a general rule, exert inhibitory regulation of gene expression. Small RNA regulation occurs in chromosome segregation, chromatin structure, RNA processing and stability, translation and transcription.
Understanding RNA-mediated regulatory pathways is therefore pivotal to understanding epigenetics. Environmental changes, such as those associated with global warming or disease, impart associated stress to flora and fauna. Evidence is building which suggests that epigenetic alterations, or epimutations, are occurring against protein-coding genes at high frequencies to enable rapid adaption of complex traits ultimately leading to genetic assimilation [2]. This rapid adaption to the environment via epigenetic alleles (epialleles) is not possible with classical genetics.
Plants are the best system to study epigenetic mechanisms as, unlike mammals, they are capable of transferring DNA methylation between generations and their cytosine methylation occurs at all sequence contexts [3]. This is important on the functional level because plant development, silencing of alien genes, reproductive transition, preservation of chromatin structures and evasion of homologous recombination all rely on DNA methylation.
2. SiRNAs and De Novo DNA Methylation
There are three major classes of small RNAs (sRNAs) in eukaryotes: piwi-interacting RNAs (piRNAs), small interfering RNAs (siRNAs) and microRNAs (miRNAs). Minor classes of small RNAs have been found in various organisms. Specific to animals, piRNAs are derived from single-stranded RNA and inactive transposons in the germ line. Functionally analogous to the piRNAs are the siRNAs, which are the most abundant small RNAs in plants. SiRNAs are generally derived from repetitive sequences, viruses and transposon rich regions and are processed into 21 - 24 nucleotide (nt) from long doublestranded RNA (dsRNA) or long hairpin RNA (hpRNA). MiRNAs are processed from single-stranded transcripts derived from MIR genes which form double-stranded secondary structures that are cleaved to generate the mature miRNA usually 21 or 22 nt in size. MiRNAs and siRNAs act in both somatic and germ lines to regulate endogenous genes involved in growth and development in addition to defending the genome from invasive nucleic acids. The main difference between miRNA and siRNA is that siRNA, with the exception of trans-acting siRNA (tasiRNA), silences the same locus from which they were derived whereas the miRNA silences in trans [4]. TasiRNA is a class of plant endogenous siRNA that can silences genes in trans.
The processing of siRNA from dsRNA is performed by a ribonuclease (RNase) III-like endonuclease termed Dicer [5]. Arabidopsis encodes four DICER-LIKE (DCL) proteins responsible for processing 21-nt (DCL4), 22-nt (DCL2) or 24-nt (DCL3) siRNAs and ~21-nt microRNA (DCL1). The 24-nt siRNAs, also known as heterochromatic-siRNAs, are particularly important for heritable epialleles. This class of siRNA is processed from dsRNA generated by the plant-specific DNA-DEPENDENT RNA POLYMERASE IV (Pol IV) and RNA-DEPENDENT RNA POLYMERASE 2 (RDR2), and exclusively mediate gene inactivation via the RNA-directed DNA methylation (RdDM) pathway (Figure 1). Here, a single 24-nt siRNA strand in the ARGONAUTE (AGO) 4-POLYMERASE V (AGO4-Pol V) complex directs de novo methylation of DNA homologous to the loaded siRNA via the action of DOMAINS REARRANGED METHYLASE 2 (DRM2). The nomenclature for both polymerases involved in RdDM has changed over time, see Table 1 for nomenclature comparisons [6]. The main role of RdDM is to methylate DNA to silence transposable elements and repetitive sequences, RdDM has been shown to affect leaf senescence and response to abiotic stress such as drought, cold, salt and hypoxia [7-10]. The RdDM response to stress is poorly understood, but this could be elucidated with the potential discovery of novel stressinduced proteins involved in RdDM. Likewise the involvement of RdDM in other traumatic situations may be determined in plants.
Recently, it has been proposed that INVOLVED IN DE NOVO 2 (IDN2), an RNA-binding protein, is required for DNA methylation establishment [11,12]. Known to bind to dsRNA, the IDN2 protein is thought to act downstream of initial siRNA biogenesis in the RdDM pathway. It was found that either IDN2-LIKE1 (IDNL 1)
Table 1. Nomenclature for plants to describe the Polymerase promoter.
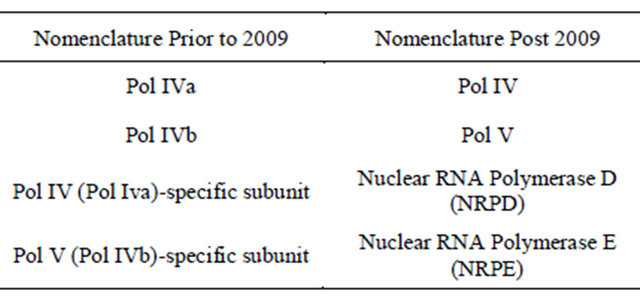
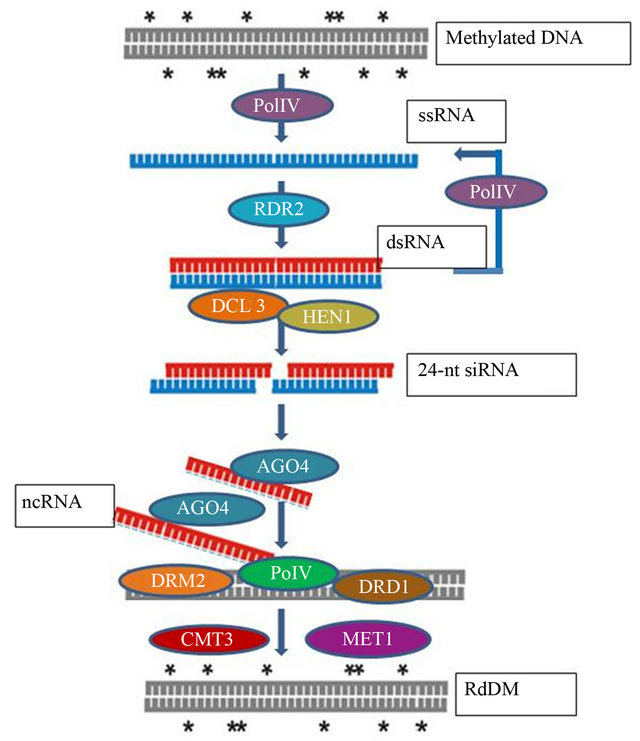
Figure 1. RdDM pathway—adapted from Eamens et al., 2008; Methylated DNA acts as a template for single stranded RNA (ssRNA) to be transcribed by RNA polymerase IV (Pol IV). RNA is then converted to double stranded (dsRNA) by RNA-directed RNA polymerase 2 (RDR2). Additional RNA molecules can be formed by Pol IV in a self-perpetuating loop. The dsRNA is diced by DICER LIKE 3 (DCL3) into 24-nucleotide siRNA duplexes which are then methylated at the 3’ termini by HUA EN-HANCER 1 (HEN1) to protect them from degradation. ARGONAUTE 4 (AGO4) then bind to one strand of the siRNA duplexes and interacts with nascent RNA transcript synthesized by RNA polymerase V (PolV) to direct cytosine methylation in the DNA by DOMAINS RE-ARRANGED METHYLASE2 (DRM2) and DEFECTIVE IN RNA-DI-RECTED DNA METHYLATION1 (DRD1). The de novo methylation can be maintained by METHYLTRANS-FERASE (MET1) and CHROMOMETHYLASE3 (CMT3).
or IDN2-LIKE 2 (IDNL 2) is required in cooperation with IDN2 to complete DRM2-mediated genome methylation. Only minimal activity of DRM2 can occur with IDN2 complex mutants. Due to the complexity of the RdDM pathway it is likely that more proteins will be uncovered that help process DNA methylation.
3. DNA Methylation in Plants
DNA methylation is widespread in the Arabidopsis genome; 24% of every CG dinucleotide, 6.7% of every CHG combination, and 1.7% of every cytosine in a CHH context is methylated [13]. DNA methylation is mainly in transposable elements and repetitive sequences [14]. Methylation is common in endogenous gene promoters or within their transcribed regions in Arabidopsis thaliana [8,13].
Repeat pericentromeric regions rich in siRNAs show high levels of CG, CHG and CHH methylation [13]. However, between 20% - 35% of genes contain significant levels of CG methylation within their transcribed regions, which is known as gene body methylation [13- 15]. The exact cytosine methylation status can be determined by single-base resolution bisulfite sequencing [16]. Bisulfite treatment converts unmethylated cytosine residues to uracil but does not affect methylated cytosines. Bisulfite treated DNA sequences can be compared with published genome sequences to determine which cytosines are methylated. This procedure is simple and can be easily scaled to whole genome coverage, but relies on the availability of high-quality genome sequences.
4. Methylation Maintenance
In Arabidopsis thaliana at least three pathways exist to control maintenance of DNA cytosine methylation, but DRM2 is solely responsible for de novo cytosine methylation under the guidance of siRNA via the RdDM pathway [11]. De novo methylation at CG and CHG sites is maintained when DNA replicates, which is catalysed by the methyltransferases METHYLTRANSFERASE (MET1) and CHROMOMETHYLASE3 (CMT3), respectively [17, 18]. MET1 methylates the nascent strand using hemimethylated DNA as a template [17]. CHG methylation via CMT3 is directed by histone modifications catalysed by the H3K9 methyltransferase KRYPTONITE/SUVH4 (KYP) [19,20]. Methylated CHH residues are not maintained between generations or during DNA replication and must instead be re-established de novo by RdDM after every replication event.
Arabidopsis methytransferase mutants were used in a study [13] to look at the genome wide effects of DNA methylation in plants. Using a variety of single, double and triple mutants they found that any line containing a MET1 mutation had completely lost CG methylation and greatly reduced CHH methylation throughout the genome. This suggests that CHH methylation requires, to some extent, the presence of CG methylation to enable redundant behaviour from the various DNA methyltransferases. Disrupting chromatin remodelling enzymes can significantly reduce all contexts of DNA methylation. For example, the ddm1 mutant lacks the function of an ATPase chromatin remodeller and as a result has global DNA methylation reduced by ~70% [2]. Gene body methylation, which usually occurs at the CG context, persists in RdDM mutants but is disrupted in met1 mutants [21]. Gene body CG methylation was rescued by transforming met1 mutant with a MET1 cDNA transgene. In these transformants, MET1 was able to remethylate DNA without a homologous hemimethylated DNA template, suggesting a role for MET1 in de novo CG methylation. This putative function provides an explanation for the reduced de novo CG methylation in met1 mutants previously observed [22].
5. Alternative RdDM Pathways
While most angiosperms rely on DCL3 to cleave double stranded RNA to produce 24-nt siRNA other plant families have developed their own RNA-mediated silencing pathway. The gymnosperm conifers, such as the Norway Spruce or the Western Red Cedar, are related to angiosperms such as Arabidopsis thaliana but have no detectable ability to produce 24-nt small RNA capable of directing chromatin modification [23,24]. This is believed to be due to the absence of DCL3. No DCL3-like expressed sequence tags were found in conifers but a new DCL family was detected that is not known to exist in angiosperms. These conifers do however have abundant levels of diverse, rapidly evolving 21-nt miRNAs which are documented in angiosperms. This may mean a new RNA-mediated silencing pathway has evolved in conifers to allow the novel DCL family and diverse 21-nt RNA to regulate heterochromatin [23,24].
Another recent study has identified a potential de novo DNA methylation pathway which has a heritable effect known to accumulate in ddm1 methylation deficient mutants [25]. The locus for the BONSAI (BNS) gene was investigated, which, contrary to global methylation patterns, is locally hypermethylated in inbred homozygous ddm1 backgrounds. This hypermethylation is associated with siRNAs and is in CG and non-CG contexts. Double homozygote mutants for ddm1 and any one of the RdDM genes retain BNS hypermethylation. However, double mutants for ddm1 and cmt3 or kyp are hypomethylated at the BNS locus, suggesting that CMT3, directed by H3K9 methylation, is necessary for de novo CG and non-CG DNA methylation at this locus.
6. Remethylation
Epialleles allow variation in traits beyond the genetic sequence alone and therefore are attractive targets for breeding new agriculturally favorable cultivars. Disrupting methylation patterns by knocking-out essential genes, such as met1 or ddm1, and reintroducing WT methylation function through outcrossing is a simple method to introduce methylation diversity in plants with DNA sequences near isogenic to WT. Two groups have recently used this method to generate epigenetic recombinant inbred lines (epiRILS) [2,26]. Restoring WT MET1 function to the progeny of met1 mutants does not completely recover WT methylation patterns [26]. Instead, remethylation is directed predominantly at centromeric regions and is a result of RdDM [27]. In ddm1-derived epiRILs it was found that ~30% of variability in flowering time and plant height was due to heritable factors and not environmental conditions, which is comparable to values considered in breeding programs [2]. Interestingly, continual selfing of ddm1 leads to a gradual reduction in DNA methylation over generations instead of the immediate loss of methylation observed in met1 plants [25]. This could be utilised to produce a severely hypomethylated progenitor that could potentially generate epiRILs with increasingly diverse patterns of methylation. These new hypomethylated cultivars could for example have a higher level of gene expression in disease responsive genes ultimately leading to resistant cultivars to pathogens.
7. Spontaneous Methylation (New Epialleles)
The frequency and extent of spontaneous methylation variation across generations has been recently analyzed. DNA methylation is stable over numerous generations but the number of generations over which stability is maintained depends on the trait. For example, flowering time and plant height epialleles were stable when monitored for eight generations [2]. In a more comprehensive, genome-wide study of both ancestral and descendant Arabidopsis lines using MethylC-Seq (genome-wide bisulphite sequencing), epialleles were stable for up to thirty generations [28]. The study of identical sequences with differentially methylated regions between the ancestral state and descendant lines also found many new spontaneous epialleles. This work indicates that while DNA methylation can be stably inherited across generations, new methylation epialleles are still able to form to enable continued rapid adaption beyond that allowed by genetic mutations. This is a great advantage for a plant under stress conditions. However, not all epialleles are stable; RdDM is a dynamic process where demethylation and remethylation continuously occur. For example, some of the ddm1 mutant induced hypomethylation variants regained wild type DNA methylation patterns conferred by RdDM after two to five generations [2]. It can be speculated that the varying stability of epialleles could help explain the disparity of disease-causing allelic transcription responsible for heritable diseases that develop in response to environmental cues.
The classical definition of a complex heritable trait is a phenotype that is influenced by alleles of multiple genes and the environment [29]. The complex trait would be passed from parents to offspring in a stable and causative manner. It is becoming evident that a phenotype may change across generations without alteration of the DNA sequence in a manner that defies traditional Mendelian inheritance. Chromatin modification, such as through the loss or gain of DNA methylation, is one such epigenetic mechanism capable of exerting an influence on gene expression transmitted between generations [2].
8. Effect of Stress on Methylation
The ability to adapt to unfavorable conditions is a plants greatest asset. Epigenetic adjustments to metabolism, energy allocation and next generation growth grants an adaptive advantage to progeny growing in the same environment as the parent [9]. A recent study has examined the methylation patterns of Arabidopsis thaliana progeny exposed to 25 and 75 mM sodium chloride [8]. Most gene promoters with changes in methylation were hypermethylated and were enriched with regulators of chromatin structure. The progeny were hypermethylated upstream and downstream of the gene and within exons. These findings supported the reduced gene expression, increased levels of H3K9me2 (dimethylated histone 3 lysine 9) and diminished H3K9ac (histone 3 lysine 9 acetylation) found in the methylated gene bodies of saltstressed progeny. Unfortunately no information on the successive generations was given.
The effect of temperature and UV-B stress on Arabidopsis also shows a temporary transgenerational influence on epigenetic regulatory mechanisms [9]. Progeny to the third generation showed this result. No later generations were examined. Transmission of stress effects to progeny in non-stressed environments occurred in a small number of cells but was reset during seed maturation. The plants did not show DNA methylation changes but instead showed strong increases in histone-acetylation, causing an expansion of transcriptionally active chromatin. This histone modification is said to overcome the hypermethylated DNA loci and reduce silencing of stress-mediated genes. The increase in H3K9 acetylation was also observed in a similar study under drought [30] and UV-B [31]. The transient transmission of epigenetic control mechanisms is likely to protect genome integrity while allowing the plant to focus energy on other factors more important to the current generation.
Evidence exists that plants can prime their immune system to provide faster and stronger defence mechanisms after a localized pathogen attack through the mechanism of systemic acquired resistance (SAR) [32]. The role of epigenetics in SAR is an emerging area for study. It may help to answer questions such as; “What happens when a plant has a short generation with a limited ability to outlive disease outbreaks” and “can priming be inherited epigenetically?” It has recently been shown that epigenetic variation can influence plant defence via hormones such as salicylic acid (SA) which acts against fungal, bacterial and viral pathogen attack, and jasmonic acid (JA) which is responsible for defence against herbivorous insects [33].
Chromatin immunoprecipitation demonstrated that the progeny of Arabidopsis plants infected with Pseudomonas syringae pv tomato DC3000 (PstDC3000) have increased H3K9 acetylation in the promoters of SA-inducible genes, a histone mark that is associated with active transcription of the genes resulting in resistance to the hemibiotrophic pathogen [34]. Conversely, these progeny plants show increased tri-methylation of H3K27 in the promoters of JA-responsive genes, which denotes a repressed state of transcription. SAR has been shown in Arabidopsis to be maintained epigenetically over one stressfree generation from plants originally exposed to the PstDC3000 bacterium [34]. The DNA hypomethylated loci induced by PstDC3000 are thought to direct priming of SA-dependent defenses in the SAR of subsequent generations via histone modification. This could possibly occur through siRNAs and the RdDM pathway response to pathogen infection.
9. Future Directions in Breeding
The importance of stable epialleles through generations becomes clear when traits, determined by methylation status, are selected during breeding. A study in Brassica napus (rapeseed) found an increase in yield potential from populations which were artificially selected for particular epigenomic states [35]. They found not only could energy use efficiency be artificially selected epigeneticcally but that DNA methylation patterns and the important agronomical and physiological characteristics were heritable. Epigenetic marker technology is a promising area of research which could greatly enhance breeding for complex traits such as disease resistance and flowering time. The mechanisms responsible for stable epigenetics are still unclear, and a better understanding of the function of DNA methylation, histone modification and siRNAs in transcription and translation processes would undoubtedly enhance our ability to use epigenetics in breeding programs [35].
10. Conclusion
There are many pieces in the puzzle of small RNA and epigenetics. With the era of next-generation sequencing, bioinformatics and more technologies continually becoming available and affordable, the rate at which we can accumulate knowledge is astounding. It is with this knowledge that the mechanics behind sRNA-mediated epigenetics in plants will be fully unravelled. An understanding of transgenerational instability and the mechanisms associated with epiallelic states will lead the way for future studies in plant biotechnology.
11. Acknowledgements
The authors would like to thank Tony Millar for critical review of this manuscript. This work was supported by an Australian National University PhD scholarship and CSIRO top-up scholarship (to J.R.M.L.). MBW was supported by an Australian Research Council Future Fellowship (FT0991956).
REFERENCES
- J. Bender, “DNA Methylation and Epigenetics,” Annual Review of Plant Biology, Vol. 55, 2004, pp. 41-68. doi:10.1146/annurev.arplant.55.031903.141641
- F. Johannes, et al., “Assessing the Impact of Transgenerational Epigenetic Variation on Complex Traits,” Plos Genetics, Vol. 5, No. 6, 2009, Article ID: e1000530. doi:10.1371/journal.pgen.1000530
- V. Pavet, et al., “Arabidopsis Displays Centromeric DNA Hypomethylation and Cytological Alterations of Heterochromatin upon Attack by Pseudomonas syringae,” Molecular Plant-Microbe Interactions, Vol. 19, No. 6, 2006, pp. 577-587. doi:10.1094/MPMI-19-0577
- R. W. Carthew and E. J. Sontheimer, “Origins and Mechanisms of miRNAs and siRNAs,” Cell, Vol. 136, No. 4, 2009, pp. 642-655. doi:10.1016/j.cell.2009.01.035
- A. Eamens, et al., “RNA Silencing in Plants: Yesterday, Today, and Tomorrow,” Plant Physiology, Vol. 147, No. 2, 2008, pp. 456-468. doi:10.1104/pp.108.117275
- S. Lahmy, et al., “PolV(PolIVb) Function in RNA-Directed DNA Methylation Requires the Conserved Active Site and an Additional Plant-Specific Subunit,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 106, No. 3, 2009, pp. 941-946. doi:10.1073/pnas.0810310106
- J. A. Brusslan, et al., “Genome-Wide Evaluation of Histone Methylation Changes Associated with Leaf Senescence in Arabidopsis,” Plos One, Vol. 7, No. 3, 2012, Article ID: e33151. doi:10.1371/journal.pone.0033151
- A. Bilichak, et al., “The Progeny of Arabidopsis thaliana Plants Exposed to Salt Exhibit Changes in DNA Methylation, Histone Modifications and Gene Expression,” Plos One, Vol. 7, No. 1, 2012, Article ID: e30515. doi:10.1371/journal.pone.0030515
- C. Lang-Mladek, et al., “Transgenerational Inheritance and Resetting of Stress-Induced Loss of Epigenetic Gene Silencing in Arabidopsis,” Molecular Plant, Vol. 3, No. 3, 2010, pp. 594-602. doi:10.1093/mp/ssq014
- J.-M. Kim, et al., “Chromatin Regulation Functions in Plant Abiotic Stress Responses,” Plant Cell and Environment, Vol. 33, No. 4, 2010, pp. 604-611. doi:10.1111/j.1365-3040.2009.02076.x
- I. Ausin, et al., “INVOLVED IN DE NOVO 2-Containing Complex Involved in RNA-Directed DNA Methylation in Arabidopsis,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 109, No. 22, 2012, pp. 8374-8381. doi:10.1073/pnas.1206638109
- M. Xie, et al., “A Subgroup of SGS3-Like Proteins Act Redundantly in RNA-Directed DNA Methylation,” Nucleic Acids Research, Vol. 40, No. 10, 2012, pp. 4422- 4431. doi:10.1093/nar/gks034
- S. J. Cokus, et al., “Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning,” Nature, Vol. 452, No. 7184, 2008, pp. 215-219. doi:10.1038/nature06745
- D. Zilberman, et al., “Genome-Wide Analysis of Arabidopsis Thaliana DNA Methylation Uncovers an Interdependence between Methylation and Transcription,” Nature Genetics, Vol. 39, No. 1, 2007, pp. 61-69. doi:10.1038/ng1929
- X. Zhang, et al., “Genome-Wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis,” Cell, Vol. 126, No. 6, 2006, pp. 1189-1201. doi:10.1016/j.cell.2006.08.003
- J. Reinders, et al., “Genome-Wide, High-Resolution DNA Methylation Profiling Using Bisulfate-Mediated Cytosine Conversion,” Genome Research, Vol. 18, No. 3, 2008, pp. 469-476. doi:10.1101/gr.7073008
- H. Saze, O. M. Scheid and J. Paszkowski, “Maintenance of CpG Methylation Is Essential for Epigenetic Inheritance during Plant Gametogenesis,” Nature Genetics, Vol. 34, No. 1, 2003, pp. 65-69. doi:10.1038/ng1138
- L. Bartee, F. Malagnac and J. Bender, “Arabidopsis cmt3 Chromomethylase Mutations Block Non-CG Methylation and Silencing of an Endogenous Gene,” Genes & Development, Vol. 15, No. 14, 2001, pp. 1753-1758. doi:10.1101/gad.905701
- J. P. Jackson, et al., “Control of CpNpG DNA Methylation by the KRYPTONITE Histone H3 Methyltransferase,” Nature, Vol. 416, No. 6880, 2002, pp. 556-560. doi:10.1038/nature731
- F. Malagnac, L. Bartee and J. Bender, “An Arabidopsis SET Domain Protein Required for Maintenance but Not Establishment of DNA Methylation,” Embo Journal, Vol. 21, No. 24, 2002, pp. 6842-6852. doi:10.1093/emboj/cdf687
- A. Miura, et al., “An Arabidopsis jmjC Domain Protein Protects Transcribed Genes from DNA Methylation at CHG Sites,” Embo Journal, Vol. 28, No. 8, 2009, pp. 1078-1086. doi:10.1038/emboj.2009.59
- W. Aufsatz, et al., “The Role of MET1 in RNA-Directed De Novo and Maintenance Methylation of CG Dinucleotides,” Plant Molecular Biology, Vol. 54, No. 6, 2004, pp. 793-804. doi:10.1007/s11103-004-0179-1
- E. V. Dolgosheina, et al., “Conifers Have a Unique Small RNA Silencing Signature,” RNA—A Publication of the RNA Society, Vol. 14, No. 8, 2008, pp. 1508-1515. doi:10.1261/rna.1052008
- R. D. Morin, et al., “Comparative Analysis of the Small RNA Transcriptomes of Pinus contorta and Oryza sativa,” Genome Research, Vol. 18, No. 4, 2008, pp. 571- 584. doi:10.1101/gr.6897308
- T. Sasaki, et al., “RNAi-Independent De Novo DNA Methylation Revealed in Arabidopsis Mutants of Chromatin Remodeling Gene DDM1,” Plant Journal, Vol. 70, No. 5, 2012, pp. 750-758. doi:10.1111/j.1365-313X.2012.04911.x
- J. Reinders, et al., “Compromised Stability of DNA Methylation and Transposon Immobilization in Mosaic Arabidopsis epigenomes,” Genes & Development, Vol. 23, No. 8, 2009, pp. 939-950. doi:10.1101/gad.524609
- F. K. Teixeira, et al., “A Role for RNAi in the Selective Correction of DNA Methylation Defects,” Science, Vol. 323, No. 5921, 2009, pp. 1600-1604. doi:10.1126/science.1165313
- R. J. Schmitz, et al., “Transgenerational Epigenetic Instability Is a Source of Novel Methylation Variants,” Science, Vol. 334, No. 6054, 2011, pp. 369-373. doi:10.1126/science.1212959
- D. L. Hartl and E. W. Jones, “GENETICS Analysis of Genes and Genomes,” 6th Edition, Jones and Bartlett Publishers, Sudbury, 2005, p. 854.
- J.-M. Kim, et al., “Alterations of Lysine Modifications on the Histone H3 N-Tail under Drought Stress Conditions in Arabidopsis thaliana,” Plant and Cell Physiology, Vol. 49, No. 10, 2008, pp. 1580-1588. doi:10.1093/pcp/pcn133
- P. Casati, et al., “Histone Acetylation and Chromatin Remodeling Are Required for UV-B-Dependent Transcriptional Activation of Regulated Genes in Maize,” Plant Cell, Vol. 20, No. 4, 2008, pp. 827-842. doi:10.1105/tpc.107.056457
- M. Jaskiewicz, U. Conrath and C. Peterhaensel, “Chromatin Modification Acts as a Memory for Systemic Acquired Resistance in the Plant Stress Response,” Embo Reports, Vol. 12, No. 1, 2011, pp. 50-55. doi:10.1038/embor.2010.186
- V. Latzel, et al., “Epigenetic Variation in Plant Responses to Defence Hormones,” Annals of Botany, Vol. 110, No. 7, 2012, pp. 1423-1428. doi:10.1093/aob/mcs088
- E. Luna, et al., “Next-Generation Systemic Acquired Resistance,” Plant Physiology, Vol. 158, No. 2, 2012, pp. 844-853. doi:10.1104/pp.111.187468
- M. Hauben, et al., “Energy Use Efficiency Is Characterized by an Epigenetic Component That Can Be Directed through Artificial Selection to Increase Yield,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 106, No. 47, 2009, pp. 20109- 20114.
Abbreviations
DCL Dicer-like DNA Deoxyribonucleic acid epiRIL Epigenetic recombinant inbred line miRNA Micro RNA piRNA Piwi-interacting RNAs RNA Ribonucleic acid RdDM RNA directed DNA methylation sRNA Small RNA siRNA Small-interfering RNA
NOTES
*Corresponding author.