Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2010, 3, 684-699 JBiSE doi:10.4236/jbise.2010.37093 Published Online July 2010 (http://www.SciRP.org/journal/jbise/). Published Online July 2010 in SciRes. http://www.scirp.org/journal/jbise Nucleotide host markers in the influenza A viruses Wei Hu Department of Computer Science, Houghton College, Houghton, USA. Email: wei.hu@houghton.edu Received 7 May 2010; revised 23 May 2010; accepted 25 May 2010. ABSTRACT In the efforts to understand the molecular character- istics responsible for the ability of influenza viruses to cross species, various amino acid host markers in influenza viruses were uncovered. Our previous study identified a collection of novel amino acid host markers in ten proteins of 2009 pandemic H1N1. As an extension of our prior work, the objective of the current study was to employ Random Forests, a ro- bust pattern recognition technique, to discover nu- cleotide host makers in the ten corresponding genes of 2009 pandemic H1N1, along with those in the genes of avian and swine viruses. Although different, there was an association between the amino acid markers in proteins and the nucleotide markers in the related genes due to codon translations. Moreover, nucleotide host markers have the capability to indi- cate important positions within a codon for host switches as well as the significance of synonymous mutations on host shifts, all of which amino acid markers could not provide. Our findings highlighted that two or even three nucleotide markers could co- exist within a single codon, and the different impor- tance values of these markers could further discri- minate the multiple markers within a codon. The nucleotide markers found in this study rendered a comprehensive genomic view of the complex and sys- temic nature of host adaptation. They verified and enriched the known amino acid markers and offered a larger set of finer host markers for further experi- mental confirmation. Keywords: 2009 Pandemic H1N1; Host Switch Marker; Influenza; Mutation; Random Forests 1. INTRODUCTION The swine-origin 2009 pandemic H1N1 was a clear re- minder that understanding the biological mechanisms of cross-species transmission of influenza viruses remained an urgent and crucial research topic. Extensive search of host-shift markers in the influenza viruses resulted in a rich set of avian-human or swine-human markers [1-7]. However, sequence analysis of the recently emerged 2009 pandemic H1N1 virus suggested the absence of these well-known host switch markers [8]. Although the symptoms of 2009 pandemic H1N1 were mild, the fear was that the new virus might mutate to a more virulent virus. A recent experiment [9] indicated that the intro- duction of traditional virulence markers (mutations) in PB2 of 2009 pandemic H1N1 did not confer increased virulence or transmission, implying that these markers had minimum impact on this new virus. To tackle the question of where to find the host mark- ers in 2009 pandemic H1N1, it was hypothesized in [8] that they might exist outside of the space of the previ- ously discovered markers. A new procedure using Ran- dom Forests was designed to identify a collection of novel amino acid host markers in ten proteins of 2009 pandemic H1N1, which included, in addition to the SR polymorphism found in [10], a set of markers in PB2 that might play compensatory roles in efficient replica- tion and transmission of this novel virus. The purpose of this study was to uncover nucleotide ho st markers in the ten corresponding genes of 2009 pandemic H1N1 to provide finer and complement information about the host adaptation of this new virus. Furthermore, the nu- cleotide host markers in the ten corresponding genes of avian and swine viruses were also included in this report. Using nucleotide sequences, it was found in [11,12] that mononucleotide composition, rather than the higher- order compositions, was sufficient to distinguish the human and avian viruses with high accuracy. The vi- ruses that replicated in mammals including 2009 pan- demic H1N1 were more likely to change G to A in the mRNA than vice versa. The patterns of nucleotide fre- quency according to host species demonstrated that the 2009 pandemic H1N1 virus had been evolving in swine prior to its emergence. Another separate report [13] con- firmed that the pattern of nucleotide composition of HA and NA genes of 2009 pandemic H1N1 was closest to that of swine H1N1 compared with the viruses of other  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 685 origins and this novel viru s originated from swine H1N1 based on the codon usage bias. To study the selective pressure acting on each gene segment of 2009 pandemic H1N1 [14], the ratio between the rate of non synony- mous substitution s per non synonymous site and the rate of synonymous substitutions per synonymous sites was computed, exhibiting an active pu rifying selection on all segments. Specially, purifying selection was extreme on NP, MP, PA and PB1, moderate on NS and HA. PB1-F2 protein is a virulence factor in influenza viruses. How- ever, genomic annotations of 2009 pandemic H1N1 [15] discovered a nucleotide mutation (C → A) to render a stop codon at position 12, which resulted in a truncated PB1-F2 protein for this new virus. Many host markers are amino acid markers including the ones in [8]. However, amino acids and nucleotides are related because of codon translations. Some codon substitutions are more likely than others due to the ge- netic code structure and selective pressures favor some codons for enhanced translation speed and fidelity. Therefore, it is not realistic to assume that each amino acid is equally likely to be encoded by any of its codons. In general codon-based host shift information is more accurate than the amino acid-based. Based on this ob- servation, the current study aimed to identify nucleotide host markers through a large-scale comparative analysis of ten genes of influenza viruses. These markers could demonstrate which positions within a codon were im- portant and uncover the synonymous mutations that might be crucial for host switches. To facilitate the dis- covery of these markers, this report proposed to employ Random Forests, a robust pattern recognition technique that was previously applied successfully as a cost effec- tive approach to the study of ten proteins of influenza viruses in [8 ]. 2. MATERIALS AND METHODS 2.1. Sequence Data All influenza virus nucleotide sequences corresponding to the protein sequences used in [8] were retrieved from the Influenza Virus Resource (http://www.ncbi/nlm.nih.giv / genomes/FLU/FLU.html) of the National Center for Biotechnology Information (NCBI). All the sequences used in the study were aligned with MAFFT [16 ]. 2.2. Random Forests Random Forest, proposed by Leo Breiman in 1999 [17], is an ensemble classifier based on many decision trees. Each tree is built on a bootstrap sample from the origin al training set and is unprun ed to obtain low-bias trees. The variables used for splitting the tree nodes are a random subset of the whole variable set. The classification deci- sion of a new instance is made by majority voting over all trees. About one-third of the instances are left of the bootstrap sample and not used in the construction of the tree. These instances in the training set are called “out- of-bag” instances and are used to evaluate the perform- ance of the classifier, which can achieve both low bias and low variance with bagging and randomization. 2.3. Feature Selection Using Random Forests Random Forest calculates several measures of variable importance. The mean decrease in accuracy measure was employed in [18] to rank the importance of the features in prediction. This measure is based on the decrease of classification accuracy when values of a variable in a node of a tree are permuted randomly. In this study, two packages of R, randomForest and varSelRF [18], were utilized to compute the importance of the nucleotides in a given gene sequence dataset. The effectiveness and ro- bustness of this technique as a feature selection method has been demonstrat ed in va rious studies [19-24]. Random Forests produce non-deterministic outcomes. To compensate this bias, the Random Forests algorithm was run multiple times and then the average of the results was taken. The importance of each position in the nu- cleotide sequences was based on the averaged calcula- tions by using the function randomVarImpsRF in var- SelRF repeated 5 times. 3. RESULTS 3.1. Comparison of Ten Genes of Influenza Viruses Based on their Consensus Nucleotide Sequences To explore the relationship among the genes of influenza viruses, the Hamming distance, defined as the number of positions at which the corresponding nucleotides of two sequences are different, of any tw o consensus nucleotid e sequences of avian, human, 2009 pandemic H1N1, and swine viruses was calculated. The distance information in Ta b l e 1 provided insight into the sequence similarity between the genes of different viruses. In particular, the distances between 2009 pandemic H1N1 and avian, hu- man, and swine viruses reflected the origin of 2009 pandemic H1N1 with its genes derived from avian (PB2 and PA), human H3N2 (PB1), classical swine (HA, NP, and NS), and Eurasian avian-like swine H1N1 (NA and M) lineages [25]. 3.2. Important Nucleotide Host Markers in Ten Genes of Influenza Viruses In [8], important amino acid host markers in ten proteins of influenza viruses were uncovered, based on which the novel host markers in 20 09 pande mic H1N1 wer e identi- fied. The main task here was to find the nucleotide host markers in the ten corresponding genes of 2009 pan-  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 686 Table 1. This table contains the Hamming distances of ten genes of avian, human, 2009 pandemic H1N1, and swine viruses based on their consensus nucleotide sequences. Genes HA NA NP M1 M2 NS1 NS2 PA PB1 PB2 Dist (Avian, 2009_pandemic) 389 249 242 51 15 108 40 160 2 56 231 Dist (Human, 2009_pandemic) 389 298 250 98 35 115 55 352 118 354 Dist (Swine, 2009_pandemic) 135 263 78 83 15 47 19 192 1 84 211 Dist (Avian, Human) 390 339 254 79 28 61 40 332 215 329 Dist (Avian, Swine) 337 316 212 64 12 77 28 161 158 177 Dist (Human, Swine) 3 42 244 222 64 30 89 42 269 152 286 demic H1N1, avian, and swine viruses, thus offering further information about the adaptation of these viruses to humans. In the following sections, each of the ten genes of human viruses was compared to that of 2009 pandemic H1N1, avian, and swine viruses. Random Forests were employed to locate the top 20 important codons, served as host markers, in the genes of influenza that could separate human from 2009 pandemic H1N1, avian, and swine viruses. In different genes there were several codons that contained two or even three impor- tant nucleotide markers selected by Random Forests, a remarkable feature that amino acid markers lack. The top important codons in each gene for differenti- ating human from 2009 pandemic H1N1, avian, and swine viruses were displayed in single figure (Figures 1-10). The comparison of amino acid markers in [8] and nucleotide markers found in this study revealed several shared sites in each protein/gene, illustrating their sig- nificance as host markers. The consensus nucleotides (codons) comprising these sites in each gene were pre- sented in Tables 2-11, which could also serve as a con- firmation and refinement of the results in [8]. Due to high genetic variation of the HA and NA genes, only the HA nucleotide sequences of H1 subtype and the NA nucleotide sequences of N1 subtype of 2009 pan- demic H1N1, avian, human, and swine viruses were utilized in the current analysis. Therefore, the important codons in HA and NA found in this study were sub- type-specific. Because all the PB1-F2 proteins of 2009 pandemic H1N1 were truncated and nonfunctional, the genes encoding these proteins were excluded in this study. 3.2.1. HA Gene One of the advantages of the nucleotide markers over amino acid markers is their ability to represent the syn- onymous mutations that might be significant for host shifts. In comparison of human with avian, 2009 pan- demic H1N1, and swine viruses, there were several sy- nonymous mutation positions in HA with high impor- tance. They were 197(3) (cac(H), cat(H)) and 230(3) (gag(E), gaa(E)) in avian and 197(3) (cac(H), cat(H)) and 254(3) (gga(G), ggg(G)) in 2009 pandemic H1N1. Codon 197(3) had a very high importance in both avian and 2009 pandemic H1N1, although it contained a syn- onymous mutation in both cases. The codons in 2009 pandemic H1N1 (Figure 1) including 184, 258, and 314 had significant effects on the receptor binding specificity of HA of 2009 pa ndemic H1N1 [26]. The HA activ e site located in a cleft is composed of the codons 91, 150, 152, 180, 187, 191, and 192, and the active site cleft o f HA is formed by its right edge (131_GVTAA) and left edge (221_RGQAGR) [2 7]. Three codons 127( 2), 128(1) , and 129(2) in Table 2 were near the right edge and codon 225(3) in avian (Figure 1) was on the left edge of the active site. The importance values of top codons in avian were more homogenous than those in the 2009 pandemic H1N1 and swine. As in case of the amino acid markers [8], the HA1 domain of HA contained more significant codons than the HA2 domain (Figure 1). 3.2.2. N A Gene In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in NA with high importance. They were 263(3) (gtg(V), gtt(V)) and 410(3) (cca(P), cct(P)) in avian, 156(1) (aga(R), agg(R)), 339(3) (act(T), tcg(S)), and 440(3) (agt(S), agc(S)) in 2009 pandemic H1N1, and 89(3) (tcc(S), tca(S)) and 267(3) (gtt(V), ata(I)) in swine. Furthermore, sequence alignment re- vealed a deletion at codon 435 in the NA nucleotide se- quences of 2009 pandemic H1N1, avian, and swine vi- ruses, causing a very high importance at that codon in avian and swine (Figure 2). The NA active site is a shallow pocket constructed from conserved residues, some of which contact the sub- strate directly and participate in catalysis, while others  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 687 Tabl e 2. This table contains the consensus nucleotides (codons) at positions in HA that have high importance in separating 2009 pandemic H1N1, avian H1, and swine H1 from human H1 viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 45(3) (a) 71(2)(p) 72(1)(s) 74(1)(s) 94(1)(s) 127(2)(s)128(1)(p)129(2)(s)139(1)(a)141(2)(s) 152(2)(s) 157(2)(s)168(1)(p) Avian aat(N) ctc(L) act(T) aac(N) gaa(E)gag(E)aca(T)act(T)tct(S) gcc(A) aca(T) tca(S)aat(N) Human aaa(K) att(I) tcc(S) gaa(E) tat(Y)acc(T)gta(V)acc(T)aat(N)aaa(K) acg(T) ttg(L)aac(N) 2009 H1N1 aga(R) tcc(S) aca(T) agc(S) gat(D)gac(D)tcg(S)aac(N)gct(A)gca(A) gtt(V) tca(S)gat(D) Swine agg(R) ttc(F) aca(T) agc(S) gat(D)gaa(E)aca(T)aac(N)gct(A)gca(A) gta(V) tca(S)aat(N) Position 205(3)(s) 216(2)(p) 235(3)(a) 236(2)(a) 259(2)(a)275(3)(p)298(1)(p)302(1)(p)314(1)(p)365(2)(p) 374(2)(p) 404(3)(a)472(1)(s) Avian aag(K) gct(A) gac(D) caa(Q) aag(K)tgc(C)atc(I) gaa(E)atg(M)cag(Q) gga(G) att(I) gat(D) Human cat(H) aaa(K) gaa(E) ccc(P) aga(R) tgt(C)gtc(V)gag(E)atg(M)caa(Q) ggg(G) atg(M)aac(N) 2009 H1N1 aga(R) ata(I) gag(E) ccg(P) aga(R)tgc(C)atc(I) aaa(K)ctg(L)ctg(L) gag(E) ata(I) gat(D) Swine aaa(K) gca(A) gag(E) cct(P) aga(R)tgt(C)gtc(V)gaa(E) atg(M) caa(Q) ggg(G) ata(I) gat(D) Tabl e 3. This table contains the consensus nucleotides (codons) at positions in NA that have high importance in separating 2009 pandemic H1N1, avian N1, and swine N1 from human N1 viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 126(2)(p) 157(1)(s) 163(3)(s) 166(2)(p) 189(2)(p)214(3)(a,s)221(3)(a)222(3)(a)257(2)(p)269(1)(p) 285(1)(p) 329(3)(a,s)331(2)(p) Avian cac(H) aca(T) gtg(V) gct(A) agt(S) gac(D)aac(N)aac(N)aaa(K)ttg(L) gcc(A) aat(N) gga(G) Human cac(H) gcc(A) cta(L) gct(A) ggc(G)gaa(E)aag(K)caa(Q)aag(K)ttg(L) act(T) aaa(K)gga(G) 2009 H1N1 ccc(P) acc(T) att(I) gtt(V) aat(N) gac(D)aac(N)aat(N)aga(R)atg(M) tct(S) aat(N)aag(K) Swine cac(H) acc(T) att(I) gct(A) gga(G)gat(D)aac(N)aaa(K)aaa(K)ctg(L) aca(T) aat(N)ggg(G) Position 336(1)(s) 340(1)(a,s) 344(1)(a) 351(2)(a) 365(2)(p,s)369(2)(a)395(2)(p)397(2)(p)398(3)(p)435(1)(a,s) 435(2)(a,s) 435(3)(a,s) Avian ggt(G) cct(P) tat(Y) ttt(F) act(T) agc(S)gca(A)act(T)gat(D)--- --- --- Human aat(N) gtt(V) aac(N) tac(Y) aac(N)aag(K)gca(A)act(T) gat(D)aca(T) aca(T) aca(T) 2009 H1N1 ggt(G) tct(S) aat(N) ttc(F) att(I) aac(N)gga(G)aat(N)gag(E)--- --- --- Swine ggc(G) tct(S) aat(N) ttt(F) atc(I) agt(S) gca(A)act(T) gat(D)--- --- --- provide a structural framework [28]. According to the numbering in [29], these residues of N1 are 118, 119, 151, 152, 156, 179, 180, 223, 225, 228, 247, 277, 278, 293, 295, 368, and 402. The important codon s in Figure 2 including 157(1), 221( 3), 222(3 ), and 369( 2) were n ear these residue positions, and codon 156(1) carrying a sy- nonymous mutation in 2009 pandemic H1N1 is at one of these positions. 3.2.3. M1 Gen e Residue positions 115, 121, and 137 were avian-human host shift markers in [5]. Codons 103(3), 115(1), 121(1), 137(1), 218(1), 218(3), and 239(1) were identified as avian-human markers in this study and in [8], with codon 218 being select ed twice, 218(1) and 218(3 ). Remarkably, codons 149 and 180 carry ing a synonymous mutation had a very higher i m portance t han resi due posit ions 11 5, 1 21, and 137. Residue position 137 was a swine-human marker in [2]. There were codons 115(1), 115(3), 137(1), 218(1), and 218(3) selected as swine-human markers in this study and in [8], and two codons 115 and 218 were chosen  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 688 Figure 1. Top important HA codon positions in distinguishing avian H1, human H1, 2009 pandemic H1N1, and swine H1 viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an asterisk are the important residue positions identified in [8]. twice, i.e., 115(1), 115(3), and 218(1), 218(3). Even though the previously discovered residue position 137 received the highest importance, the two newly found codons 17 3 and 180 ha d a very hi gh importance as well. It was noteworthy that codon 180 was significant in both avian an d sw in e and wa s loca ted in the C-terminal part of M1 (codons 165-252) that bind to vRNP (viral ribonu- Figure 2. Top important NA codon positions in distinguishing avian N1, human N1, 2009 pandemic H1N1, and swine N1 viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an asterisk are the important residue positions identified in [8]. cleoproteins) [30] (Figure 3). This study and [8] found that codons 30(1), 30(2), 115(3), 142(3), 166(3), 209(1), 214(3), and 218(3 ) were impor tant host markers in 2009 pandemic H1N1 with codon 30 being chosen twice. In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in M1 with high importance. They were 149(3) (gcc(A), gca(A)) and 180(3) (gtg(V),  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 689 Figure 3. Top important M1 codon positions in distinguishing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an aster- isk are the important residue positions identified in [8]. gtt(V)) in avian, 117(3) (cta(L), ctc(L)), 186(3) (gct(A), gca(A)), and 242(3) (aaa(K), aag(K)) in 2009 pandemic H1N1, and 90(3) (ccg(P), cca(P)), 173(3) (atc(I), ata(I)) and 180(3) (gta(V), gtt(V)) in swine (Figure 3). 3.2.4. M2 Gen e This gene has three domains, one N-terminal extracellu- lar domain (24 codon s) recognized by host immune sys- tem, one transmembrane domain (19 codons) responsi- ble for ion channe l activity, and one cytoplasmic tail (54 Figure 4. Top important M2 codon positions in distinguishing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an aster- isk are the important residue positions identified in [8]. codons) interacting with M1 and required for genome packing and formation of virus particles [3 1]. Residue positions 11, 14, 20, 28, 54, 55, 57, 78, and 86 were avian-human host shift sites found in [5]. Co- dons 11(2), 14(2), 18(2), 20(2), 43(1), 54(2), 55(3), 57(1), 78(1), 86(2), and 93(2) were important avian- human markers in this study and in [8], plus codons 18(2), 43(1), and 93(2) were new markers, with codon 93(2) carrying a high importance.  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 690 Tabl e 4. This table contains the consensus nucleotides (codons) at positions in M1 that have high importance in separating 2009 pandemic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was se- lected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 30(1)(p) 30(2)(p) 115(1)(a,s) 115(3)(a) 115(3)(p,s) 121(1)(a) 137(1)(a,s) Avian gat(D) gat(D) gtt(V) gtt(V) gtt(V) act(T) acg(T) Human gat(D) gat(D) ata(I) ata(I) ata(I) gct(A) gct(A) 2009 H1N1 agt(S) agt(S) gtg(V) gtg(V) gtg(V) act(T) aca(T) Swine gat(D) gat(D) gta(V) gta(V) gta(V) gct(A) act(T) Position 142(3)(p) 166(3)(p) 209(1)(p) 214(3)(p) 218(1)(a,s) 218(3)(a,p,s) 239(1)(a) Avian gtg(V) gtg(V) gct(A) cag(Q) aca(T) aca(T) gcc(A) Human gtg(V) gtg(V) gcc(A) cag(Q) gcc(A) gcc(A) acc(T) 2009 H1N1 gct(A) gct(A) act(T) cat(H) act(T) act(T) gcc(A) Swine gtg(V) gtg(V) gct(A) cag(Q) aca(T) aca(T) gcc(A) Tabl e 5. This table contains the consensus nucleotides (codons) at positions in M2 that have high importance in separating 2009 pandemic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was se- lected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 11(2)(a,p) 13(2)(p) 14(2)(a) 16(2)(p) 18(2)(a)20(2)(a,p,s)28(1)(p)28(2)(s)31(2)(p) 31(3)(s) 43(1)(a,p)43(2)(p) Avian acc(T) aac(N) gga(G) gag(E) aga(R)agc(S) att(I) att(I)agt(S) agt(S) ctt(L) ctt(L) Human atc(I) aac(N) gaa(E) ggg(G) aga(R)aac(N) gtt(V)gtt(V)agt(S) agt(S) ctt(L) ctt(L) 2009 H1N1 acc(T) agc(S) gaa(E) gag(E) aga(R)agc(S) att(I) att(I)aat(N) aat(N) act(T) act(T) Swine acc(T) aac(N) gga(G) gag(E) aga(R)aac(N) gtt(V)gtt(V)agc(S) agc(S) ctt(L) ctt(L) Position 54(2)(a,s) 55(1)(s) 55(3)(a) 57(1)(a,s) 77(2)(p)78(1)(a,p,s)79(1)(s)82(2)(s)86(2)(a,p,s) 89(1)(s) 93(2)(a,s)95(2)(s) Avian cgc(R) ctt(L) ctt(L) tac(Y) cgg(R)cag(Q) gaa(E)agt(S)gtt(V) ggt(G) aac(N) gag(E) Human ctc(L) ttc(F) ttc(F) cac(H) cga(R)aag(K) gaa(E)aat(N)gct(A) agt(S) agc(S) gag(E) 2009 H1N1 cgc(R) ttt(F) ttt(F) tac(Y) caa(Q)cag(Q) gaa(E)agt(S)gtt(V) ggt(G) aac(N) gag(E) Swine cgc(R) ttt(F) ttt(F) tac(Y) cga(R)cag(Q) aaa(K)agt(S)gtt(V) ggt(G) aac(N) gag(E) Residue positions 57, 86, and 93 were swine-human shift markers in [32]. Codons 20(2), 28(2), 31(3), 54(2), 55(1), 57(1 ), 78 (1), 79 (1), 82(2) , 86(2) , 89(1 ), 93(2 ), and 95(2) were primary swine-human markers in this study and in [8], a nd in pa rticular codon 78( 1) was new and had a much higher importance than the residue positions 57, 86, and 93 discovered in [32]. Similarly, codons 11(2), 13(2), 16(2), 20(2), 28(1), 31(2), 43(1), 43(2), 77(2), 78(1), and 86(2) were major host markers in 2009 pan- demic H1N1 in this study and in [8]. In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in M2 with high importance. They were 53(3) (cg t(R), cga(R) ) in avian, 27(3) (gtc(V) , gtt(V)) and 50(3) (tgt(C), tgc(C)) in 2009 pandemic H1N1, and 26(3) (ctc(L), ctt(L)) and 68(3) (gtg(V), gta(V)) in swine (Figure 4). Figur e 4 indicated that codons 20(2), 78(1), 86(2) were significant in all three categories: 2009 pandemic H1N1, avian, an d swi ne, als o co don 2 0(2) was in the N- terminal  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 691 Figure 5. Top important NS1 codon positions in distinguishing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an aster- isk are the important residue positions identified in [8]. extracellular domain and codons 78(1) and 86(2) in the cytoplasmic tail. 3.2.5. NS1 Gene NS1 is a multifunc tional g ene [33]. Its N-termin al reg ion has an RNA-binding domain (codons 1-73) and its C-terminal region (codons 74-237) contains the effector domain that inhibits the maturation and exportation of the host cellular antiviral mRNAs [34]. Residue positions 22, 60, 81, 84, 215, and 227 were avian-human host shift markers in [4]. Codons 22(1), Figure 6. Top important NS2 codon positions in distinguishing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an aster- isk are the important residue positions identified in [8]. 70(1), 79(2), 79(3), 80(3), 81(3), 84(2), 84(3), 98(1), 114(1), 171(2), and 215(1) were sign ificant av ian-human markers in this study and in [8] ( Figure 5). Furthermore, our analysis selected two position s 2 and 3 within codon 84 with a much higher importance than the previous residue positions 22, 60, 81, 215, and 227 discovered in [4]. Another codon had two positions selected at 79(2)  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 692 Ta b le 6 . This table contains the consensus nucleotides (codons) at positions in NS1 that have high importance in separating 2009 pandemic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was se- lected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 6(1)(p) 22(1)(a,s) 25(1)(s) 25(3)(s) 41(2)(s)67(1)(s)70(1)(a)79(2)(a)79(3)(a)80(3)(a) 81(3)(a,s)82(3)(a) Avian gtg(V) ttt(F) caa(Q) caa(Q) aag(K)cgg(R)gag(E)atg(M)atg(M)act(T) att(I) gct(A) Human gtg(V) gtt(V) caa(Q) caa(Q) aag(K)agg(R)aag(K)atg(M)atg(M)acc(T) atg(M) gcc(A) 2009 H1N1 atg(M) ttt(F) aat(N) aat(N) aag(K)tgg(W)aaa(K)atg(M)atg(M)aca(T) att(I) gca(A) Swine gtg(V) ttt(F) aat(N) aat(N) aag(K)tgg(W)aaa(K)atg(M)atg(M)acc(T) att(I) gca(A) Position 84(2)(a,s) 84(3)(a) 91(1)(p) 98(1)(a) 114(1)(a)119(1)(p)129(1)(p)171(1)(p)171(2)(a)205(2)(p) 206(1)(p,s)209(1)(s) Avian gtg(V) gtg(V) act(T) atg(M) tcc(S) atg(M)ata(I) gat(D) gat(D) agc(S) agt(S) gat(D) Human aca(T) aca(T) act(T) ttg(L) cct(P) atg(M)atg(M)att(I) att(I) agc(S) agt(S) aat(N) 2009 H1N1 gta(V) gta(V) tct(S) atg(M) cct(P) ttg(L) gta(V) tat(Y) tat(Y) aac(N) tgt(C) aat(N) Swine gta(V) gta(V) gct(A) atg(M) tct(S) atg(M)ata(I) gat(D) gat(D)agc(S) cgt(R) gat(D) Ta b le 7 . This table contains the consensus nucleotides (codons) at positions in NS2 that have high importance in separating 2009 pandemic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was se- lected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 6(1)(p) 14(1)(a,s)26(1)(s) 32(1)(s) 34(2)(p)40(3)(p)48(1)(p)48(3)(p)49(1)(s) 57(2)(a,p,s) 57(3)(a,s)60(1)(a) Avian gtg(V) atg(M) gag(E) ata(I) cag(Q)ctc(L)gca(A)gca(A) gtg(V) tcc(S) tcc(S) agc(S) Human gtg(V) ttg(L) gag(E) ata(I) cag(Q)atc(I)gca(A)gca(A) gta(V) tta(L) tta(L) aac(N) 2009 H1N1 atg(M) atg(M) gag(E) gta(V) cgg(R)ata(I)act(T)act(T) gtg(V) tac(Y) tac(Y) agc(S) Swine gtg(V) atg(M) gag(E) gta(V) cag(Q)atc(I)gcc(A)gcc(A) gta(V) tac(Y) tac(Y) aac(N) Position 60(2)(a,p) 63(3)(a) 70(1)(a) 70(3)(s) 83(1)(p)85(3)(a)89(2)(a)89(3)(a,s)107(1)(a,s)115(1)(p) 115(3)(a) Avian agc(S) gga(G) agt(S) agt(S) gtg(V)cat(H)att(I) att(I) ctt(L) act(T) act(T) Human aac(N) gga(G) ggt(G) ggt(G) gtg(V)cac(H)aca(T)aca(T) ttt(F) act(T) act(T) 2009 H1N1 agc(S) gaa(E) gga(G) gga(G) atg(M)cac(H) gcg(A)gcg(A)ctt(L) gct(A) gct(A) Swine aac(N) gaa(E) ggt(G) ggt(G) gtg(V)cac(H)atg(M)atg(M) ctt(L) act(T) act(T) and 79(3) as well. Both of these double selected codons were located in the C-terminal region. The results of this study and [8] suggested that codons 22(1), 25(1), 25(3), 41(2), 67(1), 81(3), 84(2), 206(1), 209(1), and 215(1) were key swine-human markers and codons 6(1), 91(1), 100(1), 119(1), 128(3), 129(1), 171(1), 205(2), and 206(1) were essential host markers in 2009 pandemic (Figure 5). In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in NS1 with high importance. They were 157(3) (gtg(V)), (gtt(V)) in avian, 5(3) (acc(T)), (act(T)), 68(3) (atc(I)), (att(I)), 90(3) (ctt(L)), cta(L)), and 128(3) (ata(I)), (atc(I)) in 2009 pandemic H1N1, and 146(1) (cta(L)), (tta(L)) in swine (Figure 5). 3.2.6. NS2 Gene Residue positions 60, 70, and 107 were avian-human host shift markers in [4]. Codons 14(1), 57(2), 57(3), 60(1), 60(2), 63(3), 70(1), 85(3), 89(2), 89(3), 107(1), and 115(3) were avian-human markers in this study and  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 693 in [8] with codons 57, 60, and 89 being selected twice. Codons 57(3) and 89(3) were not only new markers but also had a very high importance, comparable to that of positions 70 and 107 in [4](Figure 6). Codons 14(1), 26(1), 32(1), 49(1 ), 57(2 ), 57( 3), 70 (3) , 89(3), and 107(1) were important swine-human markers in this study and in [8] with codon 57 being selected twice. The same analysis also identified codons 16(1), 34(2), 40(3), 48(1), 48(3), 57(2), 60(2), 83(1), and 115(1) as important host markers in 2009 pandemic H1N1, with codon 48 being selected twice. Notably, there was one codon position 57(2) that was important in all three categories: avian, 2009 pandemic H1N1, and swine, and it was in the M1 binding domain (codons 59-116) [5]. In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in NS2 with high importance. They were 69(3) (ttg(L), cta(L)) and 84(1) (cga(R), aga(R)) in avian, 5(3) (acc(T), act(T)) and 13(3) (ctt(L), cta(L)) in 2009 pandemic H1N1, and 48(3) (gcc(A), gca(A)) and 84(1) (cgg(R), aga(R)) in swine (Figure 6). Further more, codons 8 4(1) and 13(3) r eceived a very high importance in both avian and swine and in 2009 pan- demic H1N1, respectively. Codon 84(1) was in the M1 binding domain. 3.2.7. NP Gene Residue positions 16, 33, 61, 100, 136, 214, 283, 305, 313, 357, 375, and 423 were avian-human host shift markers in [4]. Codons 16(2), 61(1), 100(1), 100(2), 283(2), 305(1), 305(3), 313(2), 357(1), and 455(3) were significant for discriminating avian and human viruses in this study and in [8] with codons 100 and 305 being chosen twice (Figure 7). In this study and in [8], codons 16(2), 61(1), 214(2), 283(2), 289(1), 313(2), 372(3), 442(1), and 455(3) were main swine-human marker s, and similarly codons 53(3), 289(1), 313(1), 316(3), 430(2), and 444(1) were vital host markers in 2009 pandemic H1N1. In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in NP with high importance. They were 108(3) (ctg(L), ctc(L)) and 155(3) (gtg(V), gtt(V)) in avian, 177(3) (ggt(G), gga(G)), 182(3) (gcg(A), gca(A)), and 363(3) (gtc(V), gta(V)) in 2009 pandemic H1N1, and 3(3) (tct(S), tcc(S)), 94(3) (gga(G), ggg(G)), and 376(3) (tcc(S), tca(S)) in swine (Figure 7). NP has three regions (codons 1-160, 256-340 and 340–498) that bind to PB1 and PB2 [35], and codons 108(3) and 155(3) in avian and codons 3(3), 94(3), and 376(3) in swine were in two of these three regions. One region, codons 360-374, in NP of 2009 pandemic H1N1 was deemed extremely important for host range restric- tion and is a common feature of pandemic viruses [36], and codon 363(3) carrying a synonymous mutation in 2009 pandemic H1N1 was in this region . 3.2.8. PA Gen e Residue positions 28, 55 , 57, 65, 66, 100, 225, 268, 321, 337, 356, 382, 400, 404, 409, 421, and 552 were avian-human host shift markers in [4], whereas this study and [8] indicated that codons 55(1), 56(1), 225(1), 337(1), 337(3), 421(2), and 552(2) were important avian-human markers, with codon 337 being chosen twice (Figure 8). Residue positions 268 and 552 were swine-human markers uncovered in [32]. Codons 225(1), 268(1), 272(1), 337(1), 421(2), and 552(2) were key swine- human markers in our analysis and in [8] with codon 337(1) having a similar importance as position 268. Also codons 85(2), 186(1), 275(2), 277(1), and 388(3) were crucial for classifying 2009 pandemic H1N1 and human viruses in this study and in [8]. The N-terminal domain of PA (codons 1-256) harbors several functional domains, including an endonuclease active site with a putative active site motif, two putative nuclear transport motifs (codons 124-139 (NLS1) and codons 186-247 (NLS2)), and a proteolytic domain that can induce generalized proteolysis of both viral and host proteins. The C-ter minal domain of PA (codons 257-716) binds to PB1 for complex formation and nuclear trans- port [37]. In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in PA with high importance. They were 106(3) (ctc(L), cta(L)), 138(3) (ata(I), att(I)), and 270(3) (tta(L), ctt(L)) in avian, 44(3) (gtt(V) , gta(V)), 173(3) (act(T), acc(T)), and 526(3) (tca(S), tct(S)) in 2009 pandem ic H1N 1, and 106(3) (ct t(L), ct a(L)), 290(3) (tta(L), ttg(L)), and 345(3) (cta(L), ttg(L)) in swine (Figur e 8). Codon 138(3) was in the first putative nuclear localization signal (NLS1) region, and codons 270(3) and 526(3) were in the C-terminal domain of PA. 3.2.9. PB 1 G ene Residue position 336 was the only avian-human host shift markers in [4]. Codons 327(2), 336(1), 361(3), 401(3), and 576 (3) were importan t host markers in av ian in this study and in [8] with codons 361(1), 401(3), and 576(3) having a much higher importance than position 336 (Figure 9). The analysis of this study and [8] suggested that co- dons 327(2), 339(3), and 638(3) were important swine- human markers, and codons 12(1), 175(1), 212(1), 339(3), 364(1), 435(2), 618(3), 638(3), 728(1), and 728(3) were vital for classifying 2009 pandemic H1N1 and human viruses with codon 728 being sel ected twice.  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 694 Table 8. This table contains the consensus nucleotides (codons) at positions in NP that have high importance in separating 2009 pan- demic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position num- ber indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 16(2)(a,s) 53(3)(p) 61(1)(a,s) 100(1)(a)100(2)(a)214(2)(s)283(2)(a,s)289(1)(p,s) 305(1)(a) 305(3)(a) Avian ggt(G) gaa(E) ata(I) aga(R) aga(R) aga(R) ctt(L) tat(Y) cgt(R) cgt(R) Human gat(D) gaa(E) ttg(L) gta(V) gta(V) aaa(K) cct(P) tac(Y) aaa(K) aaa(K) 2009 H1N1 ggt(G) gat(D) ata(I) ata(I) ata(I) agg(R) ctt(L) cat(H) aaa(K) aaa(K) Swine ggt(G) gag(E) ata(I) gta(V) gta(V) agg(R) ctt(L) cat(H) aaa(K) aaa(K) Position 313(1)(p) 313(2)(a,s) 316(3)(p) 355(3)(a)357(1)(a)372(3)(s)430(2)(p) 442(1)(s) 444(1)(p) 455(3)(s) Avian ttc(F) ttc(F) att(I) aga(R) caa(Q) gaa(E) aca(T) act(T) atc(I) gat(D) Human tac(Y) tac(Y) atc(I) cgg(R) aaa(K) gat(D) act(T) gca(A) atc(I) gaa(E) 2009 H1N1 gtc(V) gtc(V) atg(M) aga(R) aag(K) gaa(E) agc(S) aca(T) gtt(V) gat(D) Swine ttc(F) ttc(F) atc(I) aga(R) aag(K) gaa(E) act(T) act(T) att(I) gat(D) Table 9. This table contains the consensus nucleotides (codons) at positions in PA that have high importance in separating 2009 pan- demic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position num- ber indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 55(1)(a) 65(1)(a) 85(2)(p) 186(1)(p) 225(1)(a,s) 268(1)(s) 272(1)(s) Avian gat(D) tct(S) aca(T) ggt(G) agc(S) ctc(L) gat(D) Human aat(N) ctt(L) aca(T) ggc(G) tgc(C) atc(I) aat(N) 2009 H1N1 gac(D) tct(S) atc(I) agt(S) agc(S) ctc(L) gat(D) Swine gat(D) tct(S) aca(T) ggt(G) agc(S) ctc(L) gat(D) Position 275(2)(p) 277(1)(p) 337(1)(a,s) 337(3)(a) 388(3)(p) 421(2)(a,s) 552(2)(a,s) Avian cct(P) tct(S) gct(A) gct(A) agc(S) agt(S) act(T) Human cct(P) tat(Y) tca(S) tca(S) agc(S) atc(I) agt(S) 2009 H1N1 ctt(L) cat(H) gct(A) gct(A) gga(G) agc(S) act(T) Swine ccc(P) tct(S) gct(A) gct(A) agt(S) agc(S) act(T) PB1 contains several functional domains, including cRNA binding domain (codons 1-139 and 267-493), vRNA binding domain (codons 1-83 and 233-249 and 494-758), NLS region (codons 180-195 and 202-252), PA binding domain (codons 1-25), and PB2 binding domain (codons 600-757 ) [5]. In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in PB1 with high importance. They were 148(3) (gag(E), gaa(E)), 322(3) (ata(I) , att(I)), and 454(3) (ccg(P), cca(P)) in avian, 62(3) (ggt(G) , ggg(G)), 167(1) (tta(L), ctc(L)), 543(3) (acg(T), aca(T)), and 601(3) (ata(I), atc(I)) in 2009 pandemic H1N1, 60(3) (gag(E), gaa(E)) and 726(3) (gca(A), gcc(A)) in swine (Figur e 9). It was striking that codons 322(3), 167(1), and 60(3), each carrying a synonymous mutation, had the highest importance in 2009 pandemic H1N1, avian, and swine, respectively. Many of these significant codons carrying synonymous mutations were located in the functional domains of PB1. 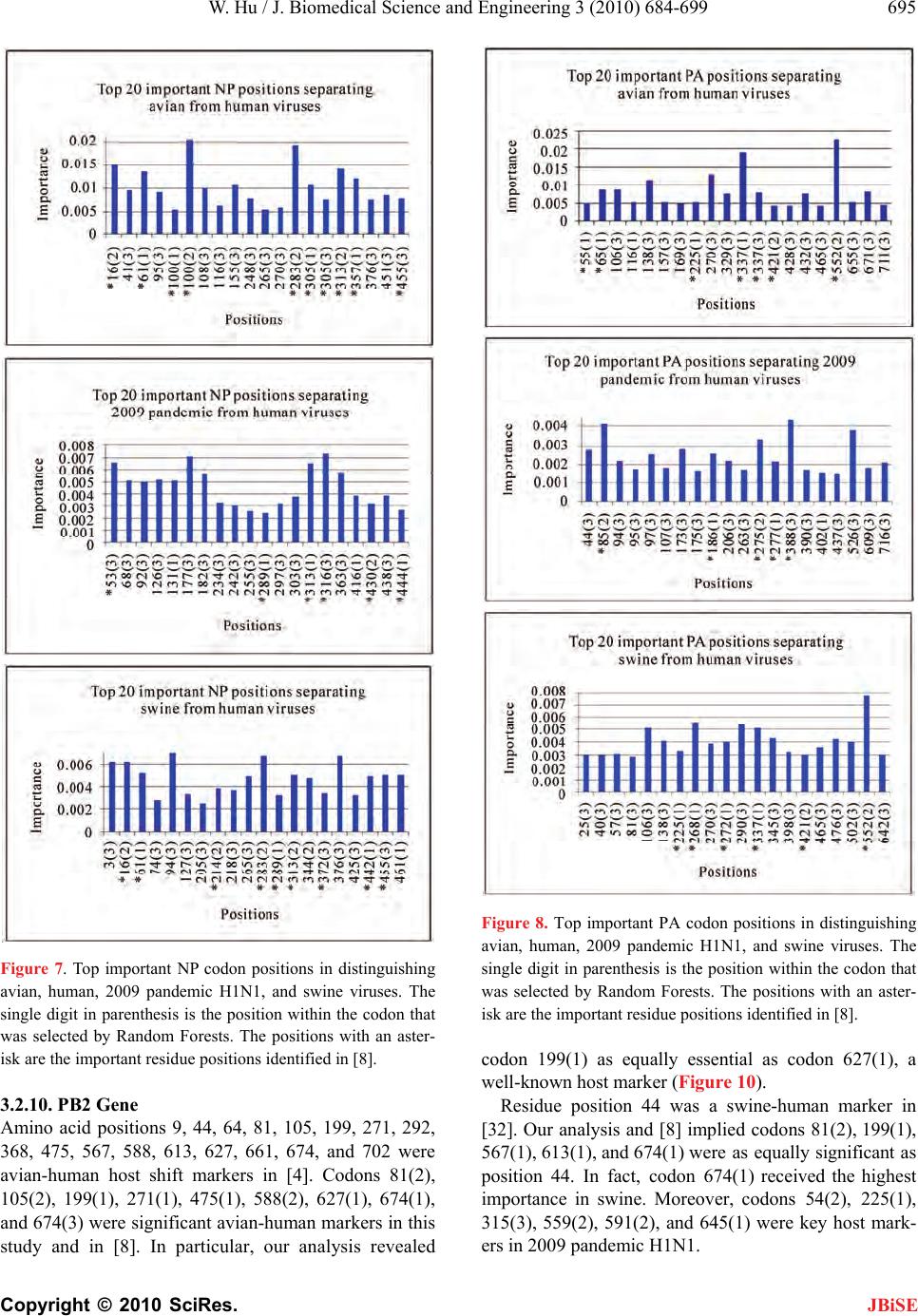 W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 695 Figure 7. Top important NP codon positions in distinguishing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an aster- isk are the important residue positions identified in [8]. 3.2.10. PB2 Gene Amino acid positions 9, 44, 64, 81, 105, 199, 271, 292, 368, 475, 567, 588, 613, 627, 661, 674, and 702 were avian-human host shift markers in [4]. Codons 81(2), 105(2), 199(1), 271(1), 475(1), 588(2), 627(1), 674(1), and 674(3) were significant avian-human markers in this study and in [8]. In particular, our analysis revealed Figure 8. Top important PA codon positions in distinguishing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an aster- isk are the important residue positions identified in [8]. codon 199(1) as equally essential as codon 627(1), a well-known host marker ( Figure 10). Residue position 44 was a swine-human marker in [32]. Our analysis and [8] implied codons 81(2), 199(1), 567(1), 613(1), and 674(1) were as equally significant as position 44. In fact, codon 674(1) received the highest importance in swine. Moreover, codons 54(2), 225(1), 315(3), 559(2), 591(2), and 645(1) were key host mark- ers in 2009 pandemic H1N1. 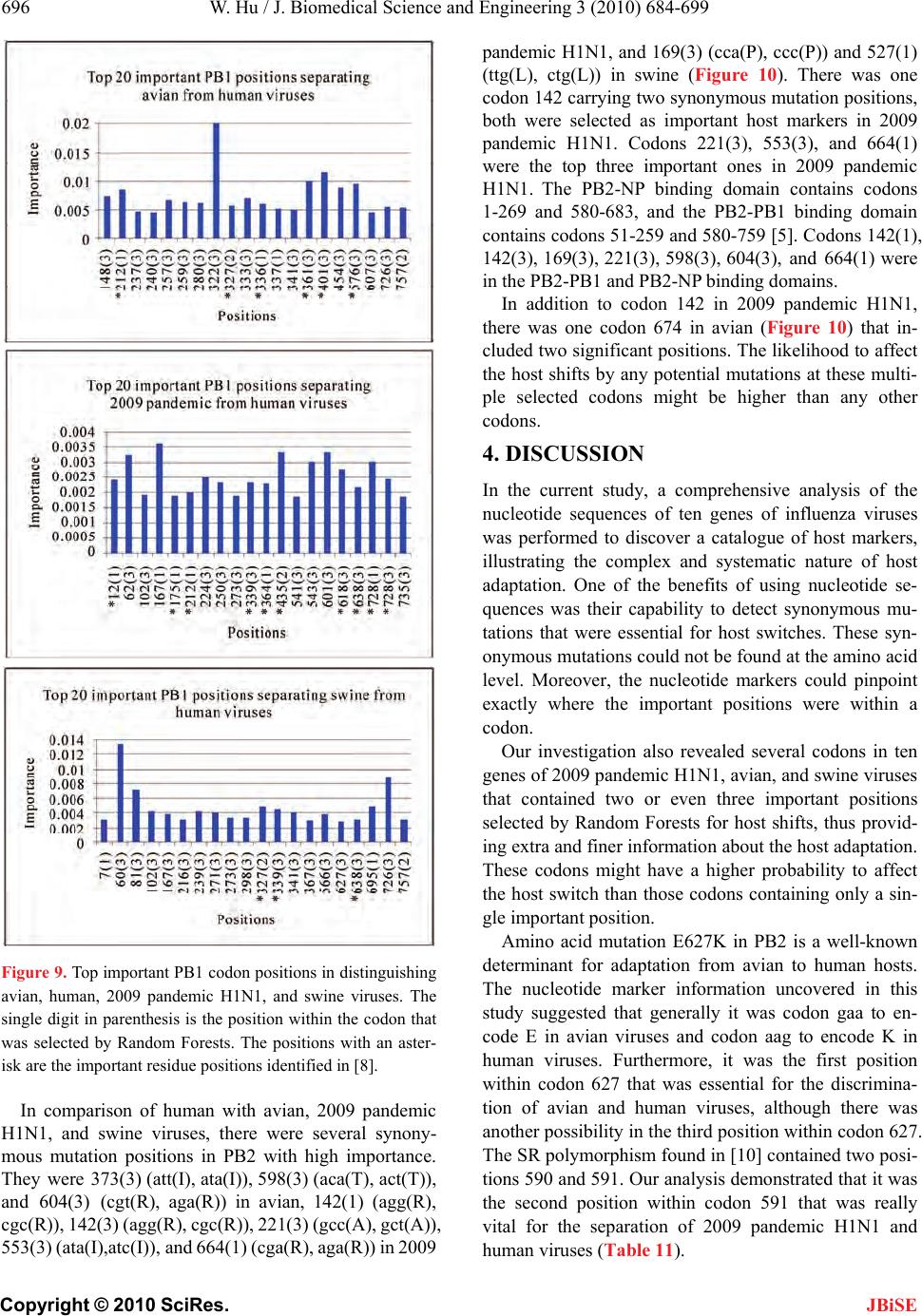 W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 696 Figure 9. Top important PB1 codon positions in distinguishing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an aster- isk are the important residue positions identified in [8]. In comparison of human with avian, 2009 pandemic H1N1, and swine viruses, there were several synony- mous mutation positions in PB2 with high importance. They were 373(3) (att(I), ata(I)), 598(3) (aca(T), act(T)), and 604(3) (cgt(R), aga(R)) in avian, 142(1) (agg(R), cgc(R)), 142(3) ( agg(R), c gc(R)), 221 (3) (gc c(A), gct( A)), 553(3) (ata(I) ,atc(I)), and 664(1) (c ga(R), aga(R)) in 2009 pandemic H1N1, and 169(3) (cca(P), ccc(P)) and 527(1) (ttg(L), ctg(L)) in swine (Figure 10). There was one codon 142 carrying two synonymou s mutation positions, both were selected as important host markers in 2009 pandemic H1N1. Codons 221(3), 553(3), and 664(1) were the top three important ones in 2009 pandemic H1N1. The PB2-NP binding domain contains codons 1-269 and 580-683, and the PB2-PB1 binding domain contains codons 51-2 59 and 580-759 [5]. Codons 142(1) , 142(3), 169(3), 2 21(3), 598(3), 604(3), and 664( 1) were in the PB2-PB1 and PB2-NP binding domains. In addition to codon 142 in 2009 pandemic H1N1, there was one codon 674 in avian (Figure 10) that in- cluded two significant positions. The lik elihood to affect the host shifts by any potential mutations at these multi- ple selected codons might be higher than any other codons. 4. DISCUSSION In the current study, a comprehensive analysis of the nucleotide sequences of ten genes of influenza viruses was performed to discover a catalogue of host markers, illustrating the complex and systematic nature of host adaptation. One of the benefits of using nucleotide se- quences was their capability to detect synonymous mu- tations that were essential for host switches. These syn- onymous mutations could not be found at the amino acid level. Moreover, the nucleotide markers could pinpoint exactly where the important positions were within a codon. Our investigation also revealed several codons in ten genes of 2009 pandemic H1N1, avian, and swine viruses that contained two or even three important positions selected by Random Forests for host shifts, thus provid- ing extra and finer information abo ut the ho st ad aptatio n . These codons might have a higher probability to affect the host switch than those codons containing only a sin- gle important position. Amino acid mutation E627K in PB2 is a well-known determinant for adaptation from avian to human hosts. The nucleotide marker information uncovered in this study suggested that generally it was codon gaa to en- code E in avian viruses and codon aag to encode K in human viruses. Furthermore, it was the first position within codon 627 that was essential for the discrimina- tion of avian and human viruses, although there was another possibility in the th ird pos ition within codo n 627. The SR polymorphism found in [10] contained two posi- tions 590 and 591. Our analysis demonstrated that it was the second position within codon 591 that was really vital for the separation of 2009 pandemic H1N1 and human viruses (Table 11).  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 697 Table 10. This table contains the consensus nucleotides (codons) at positions in PB1 that have high importance in separating 2009 pandemic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was se- lected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 12(1)(p) 175(1)(p) 212(1)(a,p) 327(2)(a,s) 336(1)(a) 339(3)(p,s) 361(3)(a) 364(1)(p) Avian gtt(V) gat(D) ctg(L) aga(R) gtc(V) att(I) agc(S) ctt(L) Human gtt(V) gat(D) gtg(V) aaa(K) atc(I) atc(I) aga(R) ctc(L) 2009 H1N1 att(I) aac(N) ctg(L) aga(R) atc(I) atg(M) aga(R) att(I) Swine gtg(V) gat(D) ttg(L) aga(R) gtt(V) att(I) agc(S) ctc(L) Position 401(3)(a) 435(2)(p) 576(3)(a) 618(3)(p) 638(3)(p,s) 728(1)(p) 728(3)(p) Avian gcc(A) aca(T) ctg(L) gaa(E) gag(E) att(I) att(I) Human gca(A) aca(T) cta(L) gag(E) gag(E) att(I) att(I) 2009 H1N1 gca(A) ata(I) tta(L) gat(D) gat(D) gtc(V) gtc(V) Swine gca(A) aca(T) cta(L) gaa(E) gaa(E) att(I) att(I) Table 11. This table contains the consensus nucleotides (codons) at positions in M1 that have high importance in separating 2009 pandemic H1N1, avian, and swine from human viruses. The single digit in parenthesis is the position within the codon that was se- lected by Random Forests. The single letter ‘a’ (for avian) or ‘p’ (for pandemic 2009) or ‘s’ (for swine) in parenthesis after a position number indicates whether the position is important for avian or 2009 pandemic H1N1 or swine viruses. Position 44(1)(s) 54(2)(p) 81(2)(a,s) 105(2)(a) 199(1)(a,s) 225(1)(p) 271(1)(a) 315(3)(p) 475(1)(a) Avian gca(A) aaa(K) aca(T) aca(T) gct(A) agc(S) aca(T) atg(M) ctg(L) Human tca(S) aaa(K) atg(M) gtg(V) tct(S) agc(S) gca(A) atg(M) atg(M) 2009 H1N1 gca(A) aga(R) aca(T) aca(T) gct(A) ggc(G) gca(A) ata(I) ctg(L) Swine gca(A) aaa(K) aca(T) aca(T) gct(A) agc(S) aca(T) atg(M) ctg(L) Position 559(2)(p) 567(1)(s) 588(2)(a) 591(2)(p) 613(1)(s) 627(1)(a) 645(1)(p) 674(1)(a,s) 674(3)(a) Avian act(T) gac(D) gcc(A) caa(Q) gtt(V) gaa(E) atg(M) gca(A) gca(A) Human gct(A) aat(N) att(I) caa(Q) acc(T) aag(K) atg(M) act(T) act(T) 2009 H1N1 att(I) gat(D) acc(T) cgg(R) gtc(V) gaa(E) ttg(L) gca(A) gca(A) Swine act(T) gac(D) gcc(A) caa(Q) gtc(V) gaa(E) atg(M) gca(A) gca(A) In [8], a set of residue positions in the PB2 protein in- cluding the SR polymorphism found in [10 ] were identi- fied as host markers in 2009 pandemic H1N1. Codons 54(2), 225(1), 315( 3), 559(2), 591(2) , and 645(1) in PB2 of 2009 pandemic H1N1 were concluded as essential host markers both in this study and in [8]. Furthermore, the current study fo und thr ee new codo ns 221(3), 553(3), and 664(1) that were the top three significant codons in 2009 pa ndemic H1N1. They coul d augm ent the re pertoire of existing host markers in PB2 that might play com- pensatory roles, as the SR polymorphism, in the viral replication and transmission of 2009 pandemic H1N1. Additionally, the new nucleotide markers carrying syn- onymous m utations fo und in NP, PA, and PB 1, along with those in PB2 would provide further information for the life cycle of 2009 pandemic H1N1. Also, co dons 728(1), 728(3) in PB1, carrying a non-synonymous mutation, and codons 142(1), 142(3) in PB2, carrying a synonymous mutation, were of special in terest in this regard as multi- ple selected markers within the same codon.  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 698 Figure 10. Top important PB2 codon positions in distinguish- ing avian, human, 2009 pandemic H1N1, and swine viruses. The single digit in parenthesis is the position within the codon that was selected by Random Forests. The positions with an asterisk are the important residue positions identified in [8]. 5. CONCLUSION As an exten sion of our earlier work in [8], Random For- ests were employed to discover a collection of nucleo- tide positions, served as host markers, in ten genes of influenza that could separate 2009 pandemic H1N1, avian, and swine viruses from human viruses with high confidence. Our results indicated that two or even three important positions could coexist within a single codon, i.e., multiple nucleotide markers might be present within one codon, and the different importance values of these positions could further differentiate these multiple mar- kers within a codon. The nucleotide markers uncovered in the current study provided a complete genomic view of host adaptation of influenza viruses. They verified and enriched the known amino acid host markers and generated new information about the adaptation of zoo- notic viruses to humans, thus offering a larger set of fin- er potential sites for further experimental verification to elucidate their biological functions in cellular processes. 6. ACKNOWLEDGEMENTS We thank Houghton College for its financial support. REFERENCES [1] Chen, G.W., Chang, S.C., Mok, C.K., Lo, Y.L., Kung, Y.N., et al. (2006) Genomic signatures of human versus avian influenza A viruses. Emerging Infectious Diseases, 12(9), 1353-1360. [2] Chen, G.W. and Shih, S.R. (2009) Genomic signatures of influenza A pandemic (H1N1) 2009. Emerging Infectious Diseases, 15(12), 1897-1903. [3] Pan, C., Cheung, B., Tan, S., Li, C., Li, L., et al. (2010) Genomic signature and mutation trend analysis of pan- demic (H1N1) 2009. Influenza A Virusus PLoS One, 5(3), e9549. [4] Miotto, O., Heiny, A., Tan, T. W., August, J.T. and Brusic, V. (2008) Identification of human-to-human transmissi- bility factors in PB2 proteins of influenza A by large- scale mutual information analysis. BMC Bioinformatics., 9(Suppl 1), S18. [5] Miotto, O., Heiny, A.T., Albrecht, R., García-Sastre, A., Tan, T.W., August, J.T. and Brusic, V. (2010) Com- plete-proteome mapping of human influenza A adaptive mutations: Implications for human transmissibility of zoonotic strains. PLoS One, 5(2), e9025. [6] Finkelstein, D.B., Mukatira, S., Mehta, P.K., Obenauer, J.C., Su, X., Webster, R.G. and Naeve, C.W. (2007) Per- sistent host markers in pandemic and H5N1 influenza viruses. Journal of Virology, 81(19), 10292-10299. [7] Allen, J.E., Gardner, S.N., Vitalis, E.A. and Slezak, T.R. (2009) Conserved amino acid markers from past influ- enza pandemic strains. BMC Microbiology, 9(1), 77. [8] Hu, W. (2010) Novel host markers in the 2009 pandemic H1N1 influenza A virus. Journal of Biomedical Science and Engineering, 3(6), 584-601. [9] Herfst, S., Chutinimitkul, S., Ye, J., de Wit, E., Munster, V.J., Schrauwen, E.J., Bestebroer, T.M., Jonges, M., Meijer, A., Koopmans, M., Rimmelzwaan, G.F., Oster- haus, A.D., Perez, D.R. and Fouchier, R.A. (2010) Intro- duction of virulence markers in PB2 of pandemic swine-origin influenza virus does not result in enhanced virulence or transmission, Journal of Virology, 84(8), 3752-3758. [10] Mehle, A. and Doudna, J.A. (2009) Adaptive strategies of the influenza virus polymerase for replication in humans. Proceedings of National Academic Science in USA., 106(50), 21312-21316. [11] Alexande,r S., Benjamin, G., Gustavo, P., Ian Lipkin, W. and Raul R. (2010) Host dependent evolutionary patterns  W. Hu / J. Biomedical Science and Engineering 3 (2010) 684-699 Copyright © 2010 SciRes. JBiSE 699 and the origin of 2009 H1N1 pandemic influenza. PLoS Current Influenza, RRN1147. [12] Rabadan, R., Levine, A.J. and Robins, H. (2006) Com- parison of avian and human influenza A viruses reveals a mutational bias on the viral genomes. Journal of Virology, 80(23), 11887-11891. [13] Microbiol Biotechnol, J. (2010) Comparative study of the nucleotide bias between the novel H1N1 and H5N1 sub- types of influenza A viruses using bioinformatics tech- niques. Ahn I, Son HS. Bioinformatics Team, 20(1), 63- 70. [14] Valli, M.B., Meschi, S., Selleri, M., Zaccaro, P., Ippolito, G., Capobianchi, M.R. and Menzo, S. (2010) Evolution- ary pattern of pandemic influenza (H1N1) 2009 virus in the late phases of the 2009 pandemic. PLoS Current In- fluenza, RRN1149. [15] Ramakrishnan, M.A., Gramer, M.R., Goyal, S.M. and Sreevatsan, S. (2009) A Serine12Stop mutation in PB1- F2 of the 2009 pandemic (H1N1) influenza A: a possible reason for its enhanced transmission and pathogenicity to humans. Journal of Veterinary Science, 10(4), 349-351. [16] Katoh, K., Kuma, K., Toh, H. and Miyata, T. (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acid Research, 33, 511-518. [17] Breiman, L. (2001) Random Forests, Machine Learning, 45(1), 5-32. [18] Díaz-Uriarte, R. and Alvarez de Andrés, S. (2006) Gene selection and classification of microarray data using random forest. BMC Bioinformatics, 7(3), 3-16. [19] Archer, K.J. and Kimes, R.V. (2008) Empirical charac- terization of random forest variable importance measures. Computational Statistics and Data Analysis, 52(4), 2249- 2260. [20] Reif, D.M., Motsinger, A.A., McKinney, B.A., Crowe, J.E. and Moore, J.H. (2006) Feature selection using a random forests classifier for the integrated analysis of multiple data types. Proceedings of 2006 IEEE Symposium on Computa- tional Intelligence and Bioinformatics and Computational Biology, CIBCB ’06. [21] Granittoa, P.M., Furlanellob, C., Biasiolia, F. and Gas- peria, F. (2006) Recursive feature elimination with ran- dom forest for PTR-MS analysis of agroindustrial prod- ucts. Chemometrics and Intelligent Laboratory Systems, 83(2), 83-90. [22] Menze1, B.H., Kelm, B.M., Masuch, R., Himmelreich, U., Bachert, P., Petrich, W. and Hamprecht, F.A. (2009) A comparison of random forest and its Gini importance with standard chemometric methods for the feature se- lection and classification of spectral data. BMC Bioin- formatics, 10, 213. [23] Gao, D., Zhang, Y.-X. and Zhao, Y.-H. (2009) Random forest algorithm for classification of multi-wavelength data. Research in Astronomy and Astrophysics, 9(2), 220- 226. [24] Hu, W. (2009) Identifying predictive markers of chemo- sensitivity of breast cancer with random forests. Journal of Biomedical Science and Engineering, 3(1), 59-64. [25] Gavin, J.D., Smith, D.V., Justin, B., Samantha, J.L., et al. (2009) Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature, 459(7250), 1122-1125. [26] Hu, W. (2010) Quantifying the effects of mutations on receptor binding specificity of influenza viruses. Journal of Biomedical Science and Engineering, 3(3), 227-240. [27] KováccaronOVá, A., Ruttkay-Nedecký, G., HaverlíK1, I.K. and Janecccaronek, S. (2002) Sequence similarities and evolutionary relationships of influenza virus A he- magglutinins. Virus Genes, 24(1), 57-63. [28] Colman, P.M., Hoyne, P.A. and Lawrence, M.C. (1993) Sequence and structure alignment of paramyxovirus he- magglutinin-neuraminidase with influenza virus neura- minidase. Journal of Virology, 67(6), 2972-2980. [29] Maurer-Stroh, S., Ma, J.M., Lee, R.T.C., Sirota, F.L. and Eisenhaber, F. (2009) Mapping the sequence mutations of the 2009 H1N1 influenza A virus neuraminidase relative to drug and antibody binding sites. Biology Direct, 4, 18. [30] Baudin, F., Petit, I., Weissenhorn, W. and Ruigrok, R.W.H. (2001) In vitro dissection of the membrane bind- ing and RNP binding activities of influenza virus M1 protein. Virology, 281(1), 102-108. [31] Furuse, Y., Suzuki, A., Kamigaki, T. and Oshitani, H. (2009) Evolution of the M gene of the influenza A virus in different host species: Large-scale sequence analysis. Journal of Virology, 6(1), 67. [32] Yang, H., Carney, P. and Stevens, J. (2010) Structure and Receptor binding properties of a pandemic H1N1 virus hemagglutinin. PLoS Current Influenza, RRN1152. [33] Dundon, W.G. and Capua, I. (2009) A closer look at the NS1 of influenza virus. Viruses, 1(3), 1057-1072. [34] Lin, D., Lan, J. and Zhang, Z. (2007) Structure and func- tion of the NS1 protein of influenza A virus. Acta Bio- chim Biophys Sin (Shanghai), 39(3), 155-162. [35] Ye, Q., Krug, R.M. and Tao, Y.J. (2006) The mechanism by which influenza A virus nucleoprotein forms oli- gomers and binds RNA. Nature, 444(7122), 1078-1082. [36] Liu, X. and Zhao, Y.P. (2010) Switch region for patho- genic structural change in conformational disease and its prediction. PLoS One, 5(1), e8441. [37] Yuan, P.W., Bartlam, M., Lou, Z.Y., Chen, S.D., Zhou, J., He, X.J., Lv, Z.Y., Ge, R.W., Li, X.M., Deng, T., Fodor, E., Rao, Z.H. and Liu, Y.F. (2009) Crystal structure of an avian influenza polymerase PAN reveals an endonuclease active site. Nature, 458(7240), 909-913. |