Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2010, 3, 672-676 JBiSE doi:10.4236/jbise.2010.37091 Published Online July 2010 (http://www.SciRP.org/journal/jbise/). Published Online July 2010 in SciRes. http://www.scirp.org/journal/jbise Predicting DNA methylation status using word composition Lingyi Lu1,2, Kao Lin2,3*, Ziliang Qian 1,2, Haipeng Li3, Yudong Cai4, Yixue Li1,5,6 1Key Lab of Molecular Systems Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China; 2Graduate School of the Chinese Academy of Sciences, Beijing, China; 3CAS-MPG Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, China; 4Department of Chemistry, College of Sciences, Shanghai University, Shanghai, China; 5Shanghai Center for Bioinformation Technology, Shanghai, China; 6College of Life Science & Biotechnology, Shanghai Jiao Tong University, Shanghai, China. Email: lylu@sibs.ac.cn; cyd@picb.ac.cn Received 1 April 2010; revised 20 April 2010; accepted 21 April 2010. ABSTRACT Background: DNA methylation will influence the gene expression pattern and cause the changes of the genetic functions. Computational analysis of the me- thylation status for nucleotides can help to explore the underlying reasons for developing methylations. Results: We present a DNA sequence based method to analyze the methylation status of CpG dinucleo- tides using 5bp (5-mer) DNA fragments – named as the word composition encoding method. The predic- tion accuracy is 75.16% when all 5bp word composi- tions are used (totally 45 = 1024). Furthermore, 5-bp DNA fragments/words having the most impact on the methylation status are identified by mRMR (Maxi- mum-Relevant-Minimum-Redundancy) feature se- lection method. As a result, 58 words are selected, and they are used to build a compact predictor, which achieves 77.45% prediction accuracy. When the word composition encoding method and the feature selec- tion strategy are coupled together, the meaning of these words can be analyzed through their contribu- tion towards the prediction. The biological evidence in the literature supports that the surrounding DNA sequence of the CpG dinucleotides will affect the methylation of the CpG dinucleotides. Conclusions: The main contribution of this paper is to find out and analyze the key DNA words taken from the neighbor- hood of the CpG dinucleotides that are inducing the DNA methylation. Keywords: Feature Selection; MRMR; 5bp Nucleotide Fragment; Nearest Neighbor Algorithm 1. BACKGROUND DNA methylation is an epigenetic modification that typi- cally occurs on cytosine of CpG dinucleotide, during which the cytosine is transferred to 5-methylcytosine by DNA methyltransferase. In human genome, unmethy- lated CpG dinucleotides are normally clustered together in a region called CpG island which is highly associated with gene promoters [1]. Methylation of CpG inhibits the expression of the downstream gene by firstly pre- venting some DNA-binding factors from recognizing their binding sites [2,3], and secondly by captivating some proteins that recognizing the methyl-CpG [4,5] to elicit the DNA’s repressive capacity. When CpG dinu- cleotides are methylated, it may affect the transcription and cause diseases [6]. DNA methylation patterns are altered in many cancer cells [7-9] since the tumor sup- pression genes are silenced by the DNA methylation. A study in [10] shows that CpG methylation may veil the presence of virus from cytotoxic T-cell immune surveil- lance and cause viral tumorigenesis. Berretta esophagus, Helicobacter pylori gastritis, inflammatory bowl disease and viral hepatitis are also thought to be associated with CpG methylation [11]. It is not possible to predict the DNA methylation status purely from the DNA sequence because the DNA methylation status is affected by many outer factors, i.e., the same sequence may have different methylation status due to these factors. A computational approach will be very hard to be devised to analyze the DNA methylation status affected by the outer factors, e.g. DNA methylation caused by heat stress [12], by over- production of DNA cytosine methyltransferases [13] and hepatitis B virus [14]. However, using computational tools, we are able to analyze what DNA sequence envi- ronment is more feasible for inducing methylation caused by those factors. The last three authors contributed equally to this work.  L. Y. Lu et al. / J. Biomedical Science and Engineering 3 (2010) 672-676 Copyright © 2010 SciRes. JBiSE 673 Machine learning method is used to predict DNA me- thylation status in our study. The DNA sequence, con- taining the CpG dinucleotides, is truncated into a 1000bp DNA sequence with the CpG dinucleotides being the center of the sequence. Instead of raw binary encoding approach [15], a 5bp (5-mer) word composition method, which slices the 1000bp sequence into 996 5bp-words, is used as the input data for the predictor. Nearest neighbor classification algorithm [16] is adopted as the predictor, which achieves an accuracy rate of 75.16%, evaluated by 10-fold cross validation. These 5bp DNA fragments (words) are analyzed by a feature analysis method – the mRMR algorithm [17], and 58 most important words are identified. The literature evidence shows that these 5bp words have other significant roles in their biological functions, e.g. some of them are modifying sites and binding sites of enzymes and some are binding motifs of some transcription factors. Since these 5bp words are important for DNA methylation, they are probably asso- ciated with the binding of DNA methyltransferase. Using these 58 words instead of all the words, the prediction accuracy increases from 75.16% to 77.45%. This indi- cates that the predictor suffers from over-fit problem when all words are used for the prediction. An online web server for the predictor used in this study is freely available at http://p cal.biosino.org/. 2. RESULTS Firstly, all 1024 features were used to distinguish the methylated CpG dinucleotides from unmethylated ones. Based on the 10-fold cross-validation test, we got 75.16% prediction accuracy rate. The accuracy rate is shown in Figure 1 as the red dashed line. The top fea- tures, generated by the MaxRel (Max-Relevance) algo- rithm, are input into the predictor. The prediction accu- racy curve, in increasing number of features, is shown in Figure 1 as the grey curve. Compared with the predic- tion accuracy using the total 1024 features, no signifi- cant improvement is achieved from the mRMR features. Thus a backward sequential feature selection is applied to extract a compact feature subset to improve the pre- diction accuracy. The black curve in Figure 1 shows prediction accuracy curve obtained from the backward feature searching algorithm. The prediction curve is sm- ooth indicating that the prediction performance is stable using backward feature searching method. The highest prediction accuracy occurs when 58 features are inclu- ded, achieving an accuracy rate of 77.45% (see Table 1). The result of mRMR feature selection program con- tains two lists of features (see supplemental material l). The first part lists 500 MaxRel features in a descending order, and the second part lists the mRMR features in the feature selection order. Since the MaxRel features indi- The highest accuracy 77.40% is achieved using backward feature se- lection when 58 features are included. The red dashed line indicates the 75.16% prediction accuracy obtained by using all 1024 features, and the blue dashed line indicates the 73.23% prediction accuracy obtained by the BLAST method. Figure 1. Prediction accuracy curves. cate how well each 5bp word contributes to the predic- tion, these words may have other significant biological functions besides the DNA methylation prediction. By manually searching over the internet, we found that more than half of the top MaxRel features have other significant roles in biological function. This may indi- cate that these 5bp words are important in binding with many enzymes including the methyltransferase. For example, it is reported that both methylases (LlaDII and Bsp6I R/M) have two recognition sites (5’-GCGGC-3’ and 5’-GCCGC-3’) [18]. DNA methyltransferase (me- thylase) FauIA (of the restriction-modification system FauI from Flavobacterium aquatile)’s recognization site is 5’-CCCGC-3’ [19]. Our study offers a perspective to find a connection between the DNA sequence fragments and the methylation mechanism. 3. CONCLUSIONS We introduce a DNA word encoding method for the analysis of the DNA methylation status. The most im- portant words inducing the methylation are found using mRMR and backward feature selection methods. Some of these words are the recognition sites for methylases, while most words’ biological role in methylation still remains unknown. These words should be paid with more attention when the biologists investigate the me- chanism of methylation. The length of the words is set to be 5 as the length is a bit longer than the 3-length DNA words for the translation of the proteins, and the number of the variation of the words can be coped easily with by the feature selection methods. A future research could be 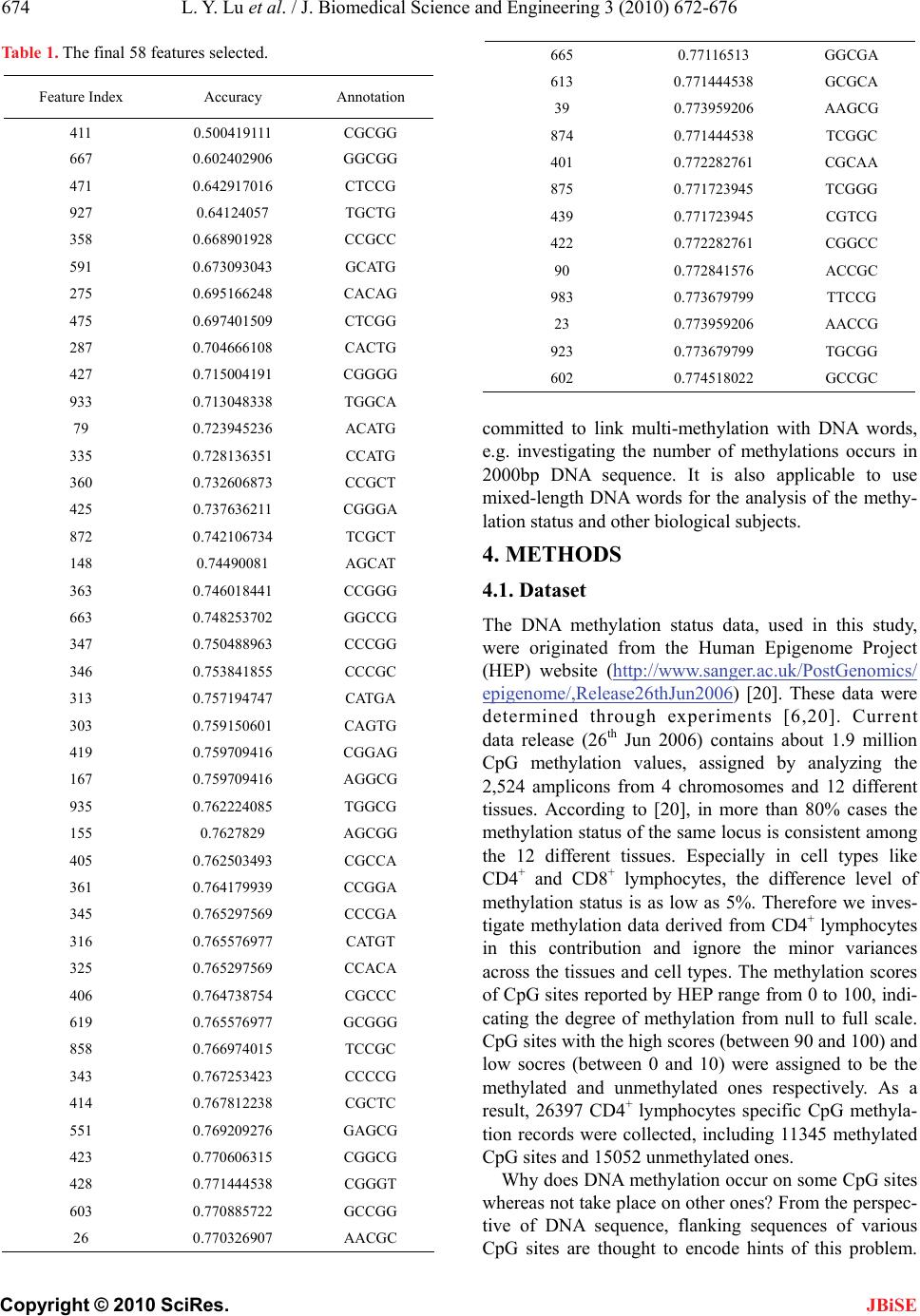 L. Y. Lu et al. / J. Biomedical Science and Engineering 3 (2010) 672-676 Copyright © 2010 SciRes. JBiSE 674 Table 1. The final 58 features selected. Feature Index Accuracy Annotation 411 0.500419111 CGCGG 667 0.602402906 GGCGG 471 0.642917016 CTCCG 927 0.64124057 TGCTG 358 0.668901928 CCGCC 591 0.673093043 GCATG 275 0.695166248 CACAG 475 0.697401509 CTCGG 287 0.704666108 CACTG 427 0.715004191 CGGGG 933 0.713048338 TGGCA 79 0.723945236 ACATG 335 0.728136351 CCATG 360 0.732606873 CCGCT 425 0.737636211 CGGGA 872 0.742106734 TCGCT 148 0.74490081 AGCAT 363 0.746018441 CCGGG 663 0.748253702 GGCCG 347 0.750488963 CCCGG 346 0.753841855 CCCGC 313 0.757194747 CATGA 303 0.759150601 CAGTG 419 0.759709416 CGGAG 167 0.759709416 AGGCG 935 0.762224085 TGGCG 155 0.7627829 AGCGG 405 0.762503493 CGCCA 361 0.764179939 CCGGA 345 0.765297569 CCCGA 316 0.765576977 CATGT 325 0.765297569 CCACA 406 0.764738754 CGCCC 619 0.765576977 GCGGG 858 0.766974015 TCCGC 343 0.767253423 CCCCG 414 0.767812238 CGCTC 551 0.769209276 GAGCG 423 0.770606315 CGGCG 428 0.771444538 CGGGT 603 0.770885722 GCCGG 26 0.770326907 AACGC 665 0.77116513 GGCGA 613 0.771444538 GCGCA 39 0.773959206 AAGCG 874 0.771444538 TCGGC 401 0.772282761 CGCAA 875 0.771723945 TCGGG 439 0.771723945 CGTCG 422 0.772282761 CGGCC 90 0.772841576 ACCGC 983 0.773679799 TTCCG 23 0.773959206 AACCG 923 0.773679799 TGCGG 602 0.774518022 GCCGC committed to link multi-methylation with DNA words, e.g. investigating the number of methylations occurs in 2000bp DNA sequence. It is also applicable to use mixed-length DNA words for the analysis of the methy- lation status and other biological subjects. 4. METHODS 4.1. Dataset The DNA methylation status data, used in this study, were originated from the Human Epigenome Project (HEP) website (http://www.sanger.ac.uk/PostGenomics/ epigenome/,Release26thJun20 06) [20]. These data were determined through experiments [6,20]. Current data release (26th Jun 2006) contains about 1.9 million CpG methylation values, assigned by analyzing the 2,524 amplicons from 4 chromosomes and 12 different tissues. According to [20], in more than 80% cases the methylation status of the same locus is consistent among the 12 different tissues. Especially in cell types like CD4+ and CD8+ lymphocytes, the difference level of methylation status is as low as 5%. Therefore we inves- tigate methylation data derived from CD4+ lymphocytes in this contribution and ignore the minor variances across the tissues and cell types. The methylation scores of CpG sites reported by HEP range from 0 to 100, indi- cating the degree of methylation from null to full scale. CpG sites with the high scores (between 90 and 100) and low socres (between 0 and 10) were assigned to be the methylated and unmethylated ones respectively. As a result, 26397 CD4+ lymphocytes specific CpG methyla- tion records were collected, including 11345 methylated CpG sites and 15052 unmethylated ones. Why does DNA methylation occur on some CpG sites whereas not take place on other ones? From the perspec- tive of DNA sequence, flanking sequences of various CpG sites are thought to encode hints of this problem.  L. Y. Lu et al. / J. Biomedical Science and Engineering 3 (2010) 672-676 Copyright © 2010 SciRes. JBiSE 675 An early study [21] showed that a flanking sequence size of 800bp is optimal for methylation status determination. We focus on flanking sequences around CpG sites with total length 1000bp, exactly 499bp nucleotides upstream and 499bp nucleotides downstream of the CpG site. Comethylation occurs over short distance (≤ 1000bp) [20], which may cause the flanking sequences of the neighboring methylation sites overlapped. To avoid this, similar sequences were filtered using the homologous sequence alignment program CD-HIT [22]. Finally, a sequence set with no more than 80% sequence similarity between any pair of them is obtained. The dataset com- prises 1994 flanking sequences of methylated CpG sites and 1585 unmethylated sequences. The human genome data of chromosome 6, 20 and 22 were downloaded from Ensembl ftp site (ftp://ftp.ensembl.org/pub/release-25/ human-25.34e/data/fasta/dna). 4.2. Word Composition Based Encoding Approach Given a piece of DNA sequence without other transcen- dent knowledge such as the genome location and the gene context, how can the DNA sequence provide in- formation to infer the CpG methylation status? An intui- tive thought would be to use the difference between the DNA sequences to determine the methylation status. However, though the differences can be located rela- tively easy, the analysis afterwards will be a rather com- plex task. In this study, we encode the long sequence into short pieces so as to easily analyze the DNA pieces using feature analysis method. We fix the length of each piece to be 5 bp, thus combining the 4 different nucl- eotides into a length of 5 will give 1024 (= 45) varia- tions/compositions. Notice that a 1000-length sequence will result in 996 words by sliding the sequence from the first nucleotide to the 996th. The prediction data is built by counting the occurrence of each composition among the 996 words. Thus, each DNA sequence will result in a 1024-dimensional vector. The encoding approach can be summarized as follows: 1) each 1000bp DNA sequence is sliced into 5bp nucleotide fragments by the shift-by- one cut; 2) a 1024 dimensional vector is built by count- ing the occurrence of each composition of the 5-length words appearing in the sliced 5bp fragments. For exam- ple, if “AAGTT” is the th icomponent of the 1024D vector and the 996 fragments contain n “AAGTT” fragments, the th icomponent of the corresponding vec- tor is set to be n (ncan be null). The next step is to apply the mRMR (Maximum- Relevant-Minimum-Redundancy) [17] and backward feature selection methods for the searching of the pro- minent words. 4.3. The mRMR and Backward Feature Selection Wrapper The mRMR (minimum-redundancy-maximum-relevance) framework aims at maximizing the relevance between the to-be-selected feature set Sand the target class c and at the same time minimizing the redundancy be- tween the to-be-selected features [23]. The maximum relevance criterion (Max-Relevance) drives to search for features that are maximizing the mutual information between S and c: max ,1/;1,2,..., i i xS DScDSI xcim , (1) where (;) i I xcis the mutual information between feature i x and c, and 1024m is the total number of fea- tures. The minimum redundancy criterion (Min-Redundancy) is formulated as: 2 , min(),1 /, ij ij xx S RR SIxx (2) which is used to minimize the mutual information of all couples of the features. The mRMR method combines the Max-Relevance and Min-Redundancy as: max( , ),DRD R (3) to optimize D and R simultaneously. An incremental search procedure is adopted to im- plement the mRMR algorithm. Suppose we already have a feature set 1k S with 1(2)kkm features, the th k feature can be selected from the rest of the feature set 1k XS as: 11 {} max,1/1, 2 jk jk iji xXS xS IxckIx x km (4) The first feature is selected through the Max-Relevance criterion using Formula 1. The rest features are gained using Formula 4. Since mRMR method only performs pre-selection role in the feature selection, these pre-selected features need to be refined using a more accurate yet more time- consuming feature selection method. Backward sequen- tial selection scheme is adopted here to do the refine- ment. It works by excluding one feature each time from the current feature set k S. Initially, k S is the pre- selected feature set resulted from the mRMR method. Each feature is in turn excluded from k S and the pre- diction accuracy is obtained using the rest 1k fea- tures by the KNN predictor, i.e., there are kdifferent  L. Y. Lu et al. / J. Biomedical Science and Engineering 3 (2010) 672-676 Copyright © 2010 SciRes. JBiSE 676 configurations in obtaining the 1k features. The con- figuration that achieves the highest prediction rate is chosen as the next selected feature set 1k S. If multiple configurations lead to the same accuracy, one of them is chosen randomly. This decremental selection procedure is repeated until one feature is left. The optimal feature subset can be chosen by selecting the feature subset that performs the best prediction. 5. COMPETING INTERESTS No competing of interests. 6. AUTHORS’ CONTRIBUTIONS LL, KL, HL, YC and YL proposed the research topics, developed the methods, designed the experiments, ana- lyzed the data, and wrote the paper. LL and KL imple- mented the experiments. 7. ACKNOWLEDGEMENTS This work was supported by National Technology Research and De- velopment Program (Grant No. 2006AA02Z320, part of a general science and technology plan, known as the Jiu-Liu-San plan, started in 1986) and National Natural Science Foundation of China (Grant No. 30700154). REFERENCES [1] Tost, J., Schatz, P., Schuster, M., Berlin, K. and Gut, I.G. (2003) Analysis and accurate quantification of CpG me- thylation by MALDI mass spectrometry. Nucleic Acids Research, 31(9), e50. [2] Klose, R.J. and Bird, A.P. (2006) Genomic DNA methy- lation: the mark and its mediators. Trends in Biochemical Sciences, 31(2), 89-97. [3] Watt, F. and Molloy, P.L. (1988) Cytosine methylation pre- vents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes & Development, 2(9), 1136-1143. [4] Boyes, J. and Bird, A. (1991) DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell, 64(6), 1123-1134. [5] Hendrich, B. and Bird, A. (1998) Identification and char- acterization of a family of mammalian methyl-CpG binding proteins. Molecular and Cellular Biology, 18(11), 6538-6547. [6] Rakyan, V.K., Hildmann, T., Novik, K.L., Lewin, J., Tost, J., Cox, A.V., Andrews, T.D., Howe, K.L., Otto, T., Olek, A., et al. (2004) DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biology, 2(12), e405. [7] Schulz, W.A. (1998) DNA methylation in urological malignancies (review). International Journal of Oncol- ogy, 13(1), 151-167. [8] Ushijima, T. (2005) Detection and interpretation of al- tered methylation patterns in cancer cells. Nature Re- views, 5(2), 223-231. [9] Agrawal, A., Murphy, R.F. and Agrawal, D.K. (2007) DNA methylation in breast and colorectal cancers. Mod- ern Pathology, 20(7), 711-721. [10] Robertson, K.D., Manns, A., Swinnen, L.J., Zong, J.C., Gulley, M.L. and Ambinder, R.F. (1996) CpG methyla- tion of the major Epstein-Barr virus latency promoter in Burkitt’s lymphoma and Hodgkin’s disease. Blood, 88(8), 3129-3136. [11] Chan, A.O. and Rashid, A. (2006) CpG island methyla- tion in precursors of gastrointestinal malignancies. Cur- rent Molecular Medicine, 6(4), 401-408. [12] Zhu, J.Q., Liu, J.H., Liang, X.W., Xu, B.Z., Hou, Y., Zhao, X.X. and Sun, Q.Y. (2008) Heat stress causes ab- errant DNA methylation of h19 and igf-2r in mouse blastocysts. Molecules and Cells, 25(2), 211-215. [13] Bandaru, B., Gopal, J. and Bhagwat, A.S. (1996) Over- production of DNA cytosine methyltransferases causes methylation and C --> T mutations at non-canonical sites. The Journal of Biological Chemistry, 271, 7851-7859. [14] Zhang, J.C., Lu, J., Li, H.P., Wu, J.M. and Hu, L.H. (2006) High Rate of P16 Methylation Associated with Hepatitis B Virus Infection in Hepatocellular Carcinoma. The Chinese-German Journal of Clinical Oncology, 5, 84-89. [15] Bhasin, M., Zhang, H., Reinherz, E.L. and Reche, P.A. (2005) Prediction of methylated CpGs in DNA sequences using a support vector machine. FEBS Letters, 579(20), 4302-4308. [16] Chou, K.C. and Cai, Y.D. (2006) Predicting protein-pro- tein interactions from sequences in a hybridization space. Journal of Proteome Research, 5(2), 316-322. [17] Peng, H., Long, F. and Ding, C. (2005) Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Transactions on Pattern Analysis and Machine Intelligence, 27(3), 1226-1238. [18] Madsen, A. and Josephsen, J. (1998) Cloning and char- acterization of the lactococcal plasmid-encoded type II restriction/modification system, LlaDII. Applied and En- vironmental Microbiology, 64(7), 2424-2431. [19] Chernukhin, V.A., Kashirina, Y.G., Sukhanova, K.S., Abdurashitov, M.A., Gonchar, D.A. and Degtyarev, S. (2005) Isolation and characterization of biochemical properties of DNA methyltransferase FauIA modifying the second cytosine in the nonpalindromic sequence 5’-CCCGC-3’. Biochemistry, 70(6), 685-691. [20] Eckhardt, F., Lewin, J., Cortese, R., Rakyan, V.K., Att- wood, J., Burger, M., Burton, J., Cox, T.V., Davies, R., Down, T.A., et al. (2006) DNA methylation profiling of human chromosomes 6, 20 and 22. Nature Genetics, 38(12), 1378-1385. [21] Das, R., Dimitrova, N., Xuan, Z., Rollins, R.A., Haghighi, F., Edwards, J.R., Ju, J., Bestor, T.H. and Zhang, M.Q. (2006) Computational prediction of methylation status in human genomic sequences. Proceedings of the National Academy of Sciences of the United States of America, 103(28), 10713-10716. [22] Li, W. and Godzik, A. (2006) Cd-hit: A fast program for clustering and comparing large sets of protein or nucleo- tide sequences. Bioinformatics (Oxford, England), 22(13), 1658-1659. [23] Ding, C. and Peng, H. (2005) Minimum redundancy feature selection from micro array gene expression data. Journal of Bioinformatics and Computational Biology, 3(2), 185-205. |