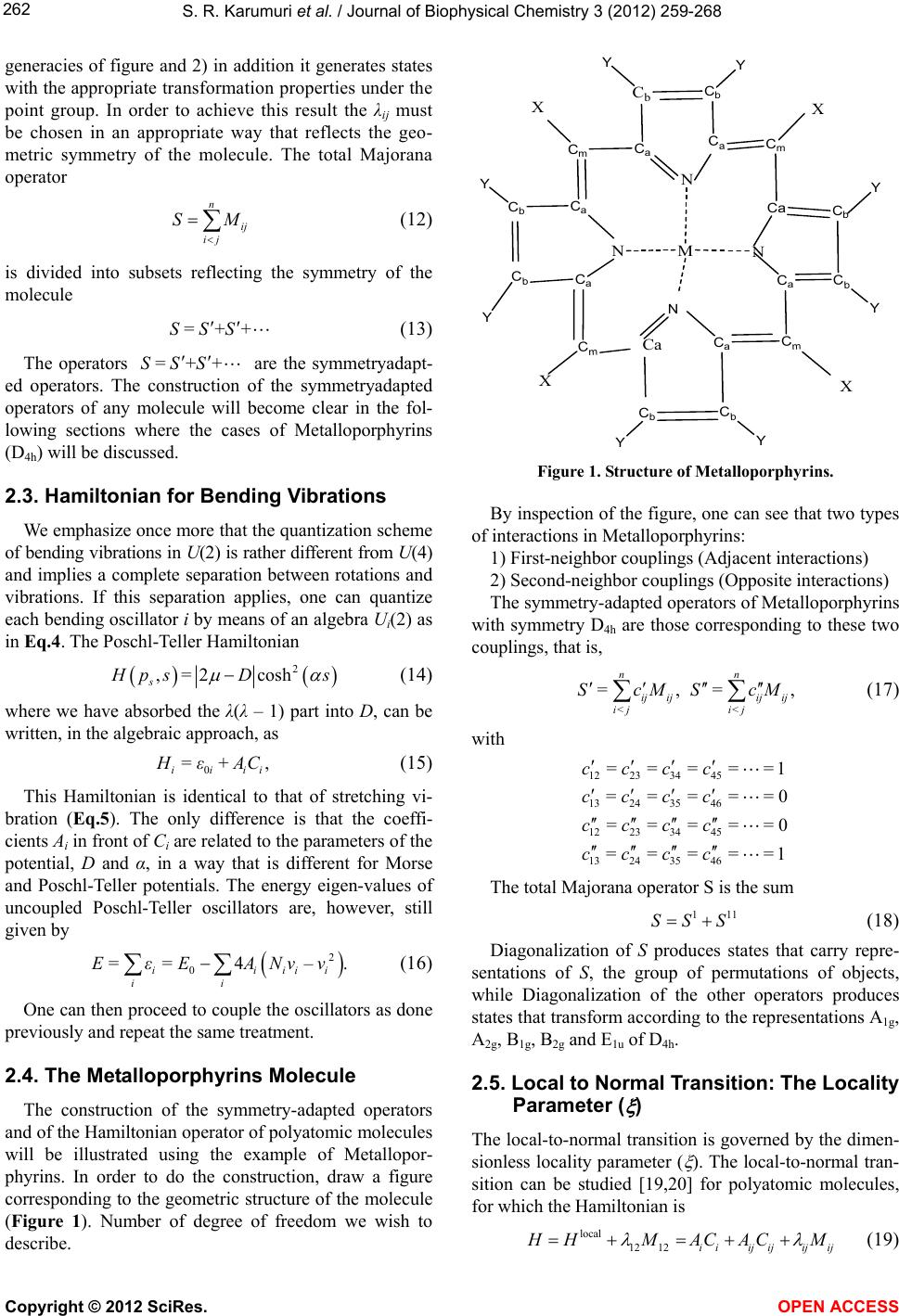
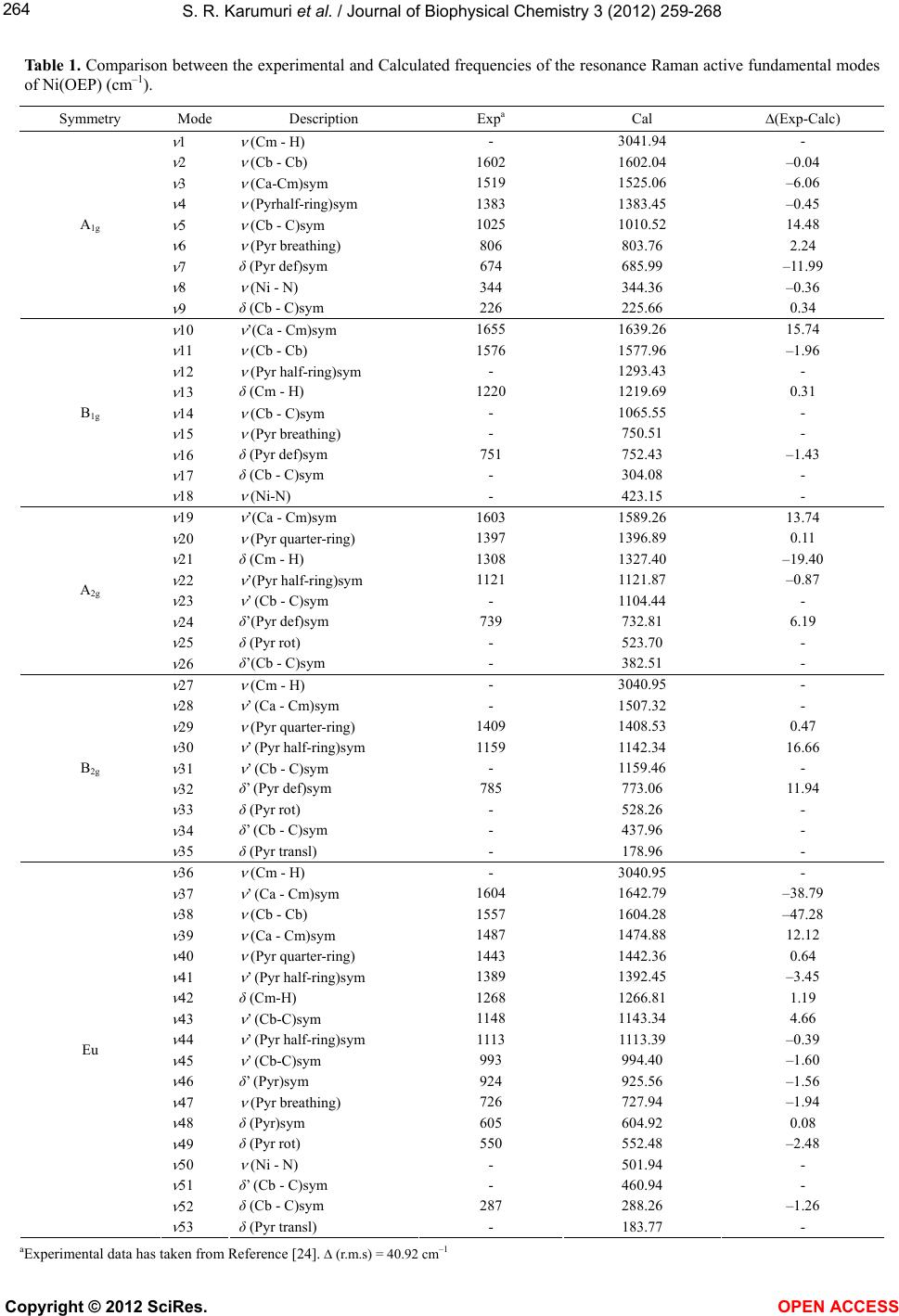
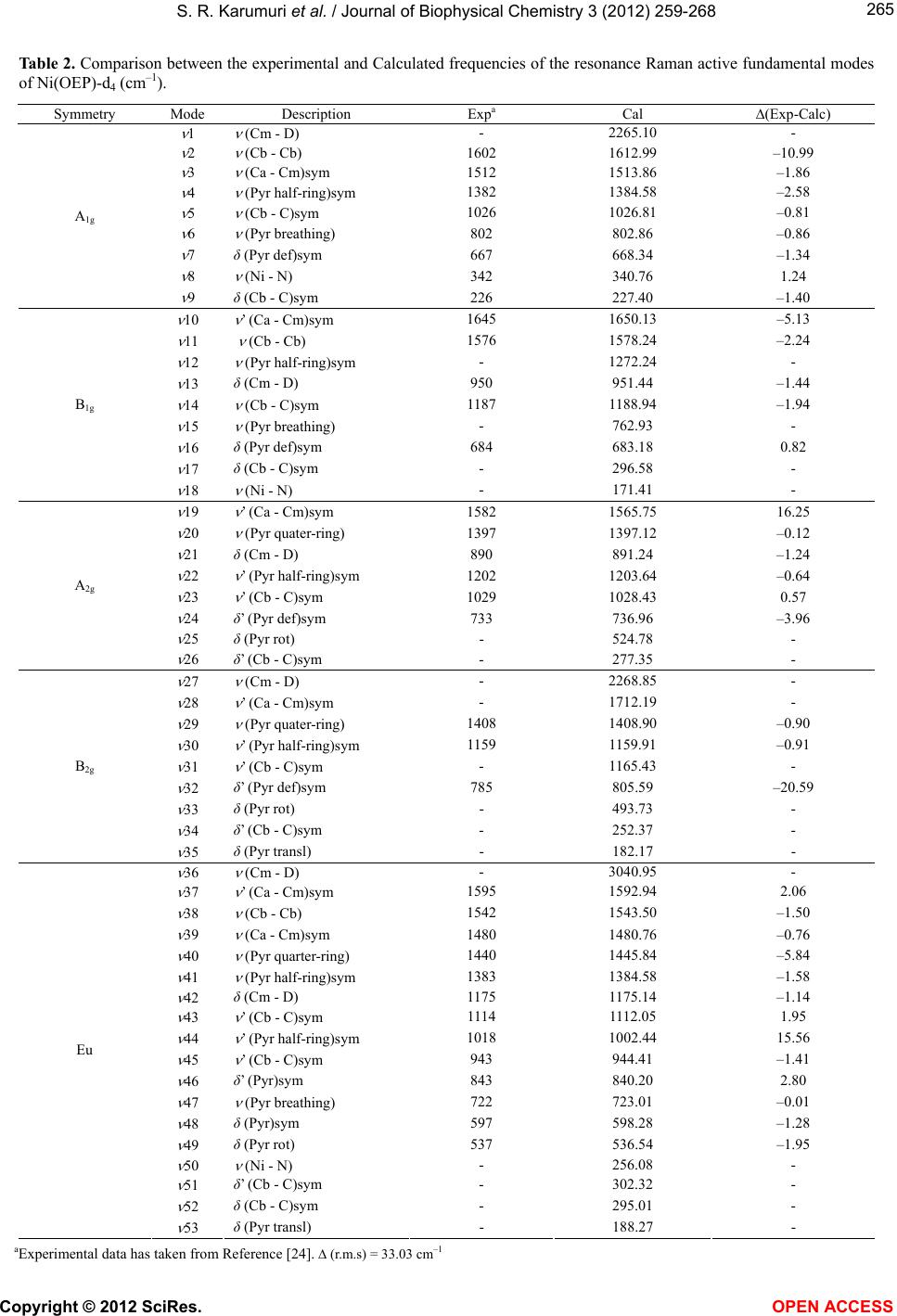
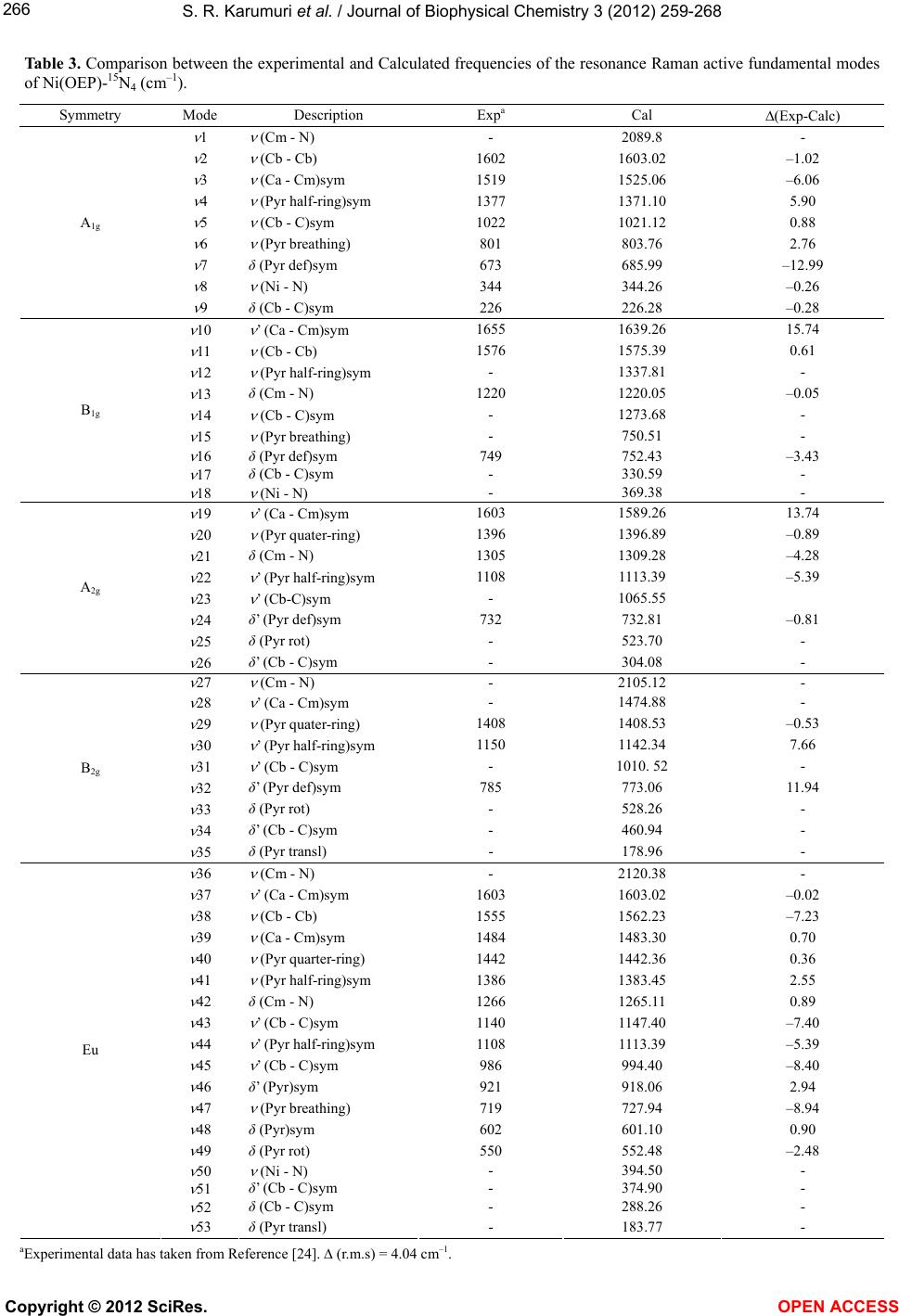
S. R. Karumuri et al. / Journal of Biophysical Chemistry 3 (2012) 259-268
Copyright © 2012 SciRes. OPEN ACCESS
268
5. ACKNOWLEDGEMENTS
The author Srinivasa Rao Karumuri would like to thank Prof. Thom-
son G. Spiro for providing necessary literature for this study.
The author Srinivasa Rao Karumuri also would like to thank Uni-
versity of Grant Commission (UGC), New Delhi, India, for providing
the financial assistance for this study.
REFERENCES
[1] Kroto, H.W., Heath, J.R., Brien, S.C.O., Curl, R.F. and
Smalley, R.E.
(1985) C
60
: Buckminsterfullerene. Nature,
318
, 162-163. doi:10.1038/318162a0
[2]
Treubig Jr., J.M. and Brown, P.R. (2002) Analysis of C
60
and C
70
fullerenes using high-performance liquid chro-
matography—Fourier transform infrared spectroscopy.
Jour-
nal of Chromatography A
, 960, 135-142.
[3]
Levine, R.D. (1982) Representation of one-dimensional
motion in a morse potential by a quadratic Hamiltonian.
Chemical Physics Letters, 95, 87-90.
doi:10.1016/0009-2614(83)85071-4
[4]
Iachello, F. and Levine, R.D. (1982) Algebraic approach
to molecular rotation
—vibration spectra. I. Diatomic mo-
lecules.
Journal of Chemical Physics, 77, 3046-3055.
doi:10.1063/1.444228
[5]
van Roosmalen, O.S., Dieperink, A.E.L. and Iachello, F.
(1982) A unified algebraic model description for inter-
acting vibrational modes in ABA molecules.
Chemical
Physics Letters
, 85, 32-36.
doi:10.1016/0009-2614(82)83455-6
[6]
van Roosmalen, O.S., Iachello, F., Levine, R.D. and Die-
perink, A.E.L. (1983) The geometrical-classical limit of
algebraic Hamiltonians of molecular vibrational spectra.
Chemical Physics Letters, 79, 2515.
doi:10.1063/1.446164
[7]
Sarkar, N.K., Choudhury, J. and Bhattacharjee, R. (2006)
An algebraic approach to the study of the vibrational
spectra of HCN.
Molecular Physics, 104, 3051-3055.
doi:10.1080/00268970600954235
[8]
Sarkar, N.K., Choudhury, J., Karumuri, S.R. and Bhat-
tacharjee, R. (2008) An algebraic approach to the com-
parative study of the vibrational spectra of monofluo-
roacetylene (HCCF) and deuterated acetylene (HCCD).
Molecular Physics, 106, 693-702.
doi:10.1080/00268970801939019
[9]
Sarkar, N.K., Choudhury, J. and Bhattacharjee, R. (2008)
Study of vibrational spectra of some linear triatomic mole-
cules.
Indian Journal of Physics, 82, 767.
[10]
Choudhury, J., Karumuri, S.R., Sarkar, N.K. and Bhat-
tacharjee, R. (2008) Vibrational spectroscopy of CCl
4
and SnBr
4
using lie algebraic approach. Physics and As-
tronomy
, 71, 439-445. doi:10.1007/s12043-008-0123-z
[11]
Choudhury, J., Karumuri, S.R. and Bhattacharjee, R.,
(2008) Algebraic approach to analyze the vibrational spec-
tra of tetrahedral molecules.
Indian Journal of Physics,
82, 561-565.
[12]
Karumuri, S.R., Sarkar, N.K., Choudhury, J. and Bhat-
tacharjee, R.
(2008) Vibrational spectroscopy of C
m
-H,
C
-C
stretching vibrations of Nickel metalloporphyrins.
Molecular Physics, 106, 1733-1738.
doi:10.1080/00268970802248998
[13]
Karumuri, S.R., Choudhury, J., Sarkar, N.K. and Bhat-
tacharjee, R. (2008) Analysis of resonance raman spectra
of nickeloctaethyl porphyrin using lie algebra.
Journal of
Environmental Research and Development
, 3, 250-256.
[14]
Karumuri, S.R., Sarkar, N.K., Choudhury, J. and Bhat-
tacharjee, R. (2009) Study of vibrational spectra of Nickel
metalloporphyrins: An algebraic approach.
Pramana—
Journal of Physics, 72, 517-525.
[15]
Karumuri, S.R., Sarkar, N.K., Choudhury, J. and Bhat-
tacharjee, R. (2009) Vibrational spectroscopy of stretch-
ing and bending modes of nickel tetraphenyl porphyrin:
An algebraic approach.
Chinese Physics Letters, 26, 093-
301. doi:10.1088/0256-307X/26/9/093301
[16]
Karumuri, S.R., Sarkar, N.K., Choudhury, J. and Bhat-
tacharjee, R. (2009
) U(2) algebraic model applied to vi-
brational spectra of Nickel Metalloporphyrins.
Journal of
Molecular Spectroscopy
, 255, 183-188.
doi:10.1016/j.jms.2009.03.014
[17]
Karumuri, S.R., Choudhury, J., Sarkar, N.K. and Bhat-
tacharjee, R. (2010) Vibrational Spectroscopy of C
m
-C/
C
b
-C
b
stretching vibrations of Copper Tetramesityl Por-
phyrin Cu (TMP): An algebraic approach.
Pramana—Jour-
nal of Physics
, 74, 57-66.
doi:10.1007/s12043-010-0007-x
[18]
Karumuri, S.R. (2010) Calculation of vibrational spectra
by an algebraic approach: Applications to Copper Tetra-
mesityl Porphyrins and its Cation radicals.
Journal of Mo-
lecular Spectroscopy
, 259, 86-92.
doi:10.1016/j.jms.2009.11.005
[19]
Iachello, F. and Levine, R.D. (1995) Algebraic theory of
molecules. Oxford University Press, Oxford.
[20]
Iachello, F. and Oss, S. (2002), Algebraic methods in quan-
tum mechanics: From molecules to polymers
. Physics
and Astronomy
, 19, 307-314.
doi:10.1140/epjd/e20020089
[21]
Child, M.S. and Halonen, L.O. (1984) Overtone frequen-
cies and intensities in the local mode picture.
Advances
in Chemical Physics
, 57, 1.
doi:10.1002/9780470142813.ch1
[22]
Wood, B.R., Stoddart, P.R. and McNaughton, D. (2007)
Molecular imaging of red blood cells by raman spec-
troscopy.
Australian Journal of Chemistry, 387, 1691.
[23]
Phuber, K. and Herzberg, G., (1979) Molecular spectra
and molecular structure IV: Constants of diatomic mole-
cules. Van Nostrand Reinhold Co., New York.
[24]
Kitagawa, T., Abe, M. and Ogoshi, H. (1978) Resonance
Raman spectra of octaethylporphyrinato
—Ni(II) and meso—
deuterated and
15
N substituted derivatives. II. A normal
coordinate analysis.
Journal of Chemical Physics, 69,
4526. doi:10.1063/1.436450
[25]
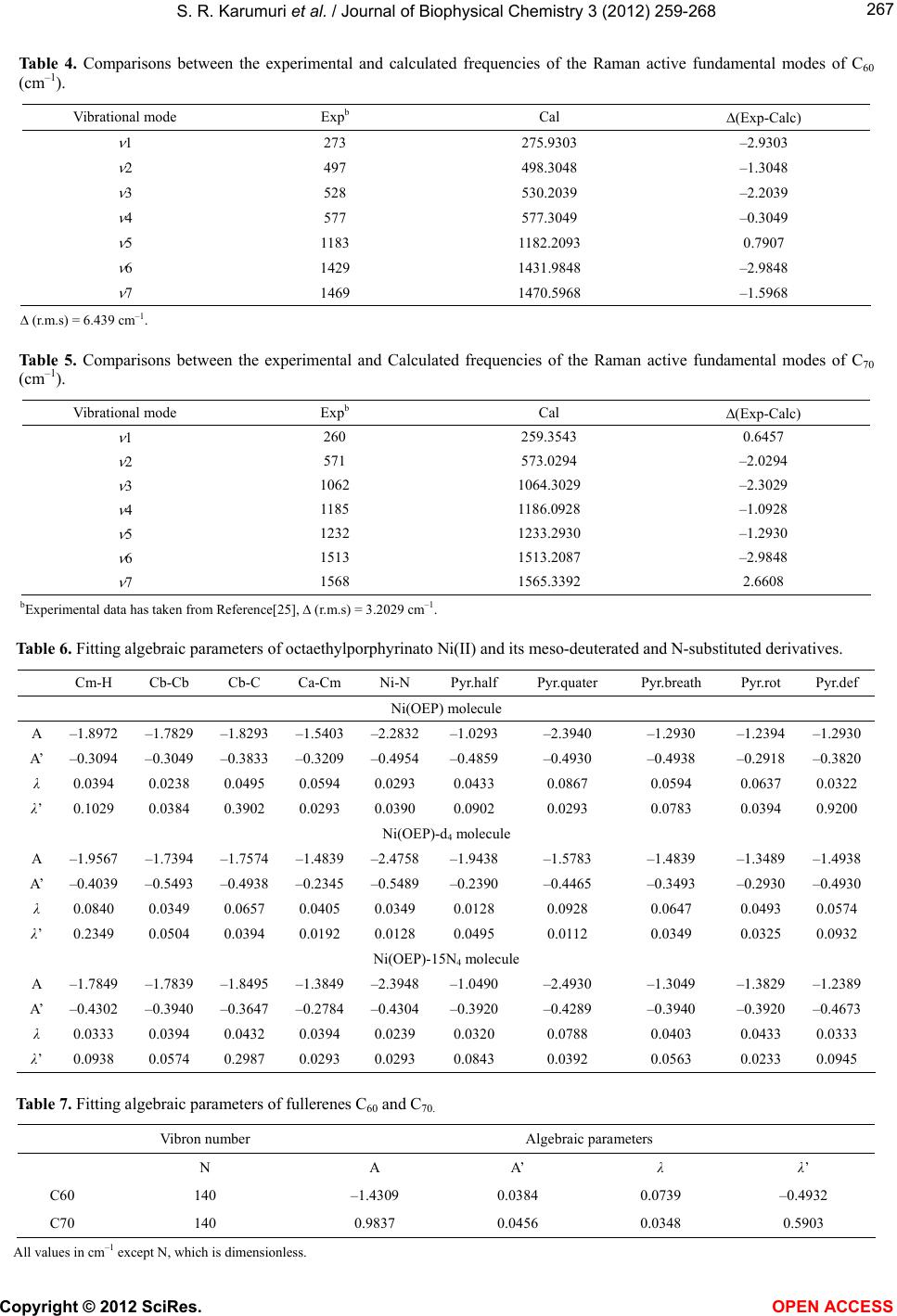
Schettino, V., Pagliai M. and Cardini, G. (2002) The in-
frared and raman spectra of fullerene C
70
. DFT calcula-
tions and correlation with C
60
. The Journal of Physical
Chemistry A
, 106, 1815-1823. doi:10.1021/jp012680d.