S. ABU-BAKER, G. A. LORIGAN
10
H-Met1-Asp-Lys-Val-Gln-Tyr- Arg-Ser10-Ala-Ile-Arg-Ar
g-Ala-Ser-Thr-Ile-Glu-Met20-Pro-Gln-Gln-Ala-Arg-Gln-Asn-Leu-
Gln-Asn30-Leu-Phe-Ile-Asn-Phe-Cys-Leu-Ile-Leu-Ile40-Cys-Leu-
Leu-Leu-Ile-Cys-Ile-Ile-Val-Met50-Leu-Leu-OH
(a)
H-Met1-Asp-Lys-Val-Gln-Tyr- Arg-Ser10-Ala-Ile-Arg-Ar
g-Ala-Ser(PO
3
H
2
)-Thr-Ile-Glu-Met20-Pro-Gln-Gln-Ala-Arg-Gln-
Asn-Leu-Gln-Asn30-Leu-Phe-Ile-Asn-Phe-Cys-Leu-Ile-Leu-Ile40-
Cys-Leu-Leu-Leu-Ile-Cys-Ile-Ile-Val-Met50-Leu-Leu-OH
(b)
Leu-Thr-
Leu-Thr-
Figure 1. Pr imary sequence of (a) PLB and (b) P-PLB. Site s
of pseudoproline substitution are highlighted in red. P-Ser
residue highlighted in blue was introduced using Fmoc-Ser
(PO(OBzl)OH)-OH.
O
O
N
H
O
N
O
COOH
CH3
Figure 2. The pseudoproline dipeptide F moc -Leu-Thr(CM e,
Mepro)-OH. This structure was generated using Chem-
Draw software and it is similar to the structure shown in
the Novabiochem website [13].
To control the progress of the synthesis, the deprotec-
tion and coupling steps can be monitored using a UV
detector. Several approaches including switching to dif-
ferent resins and activating reagents as well as using a
pseudoproline dipeptide has been suggested to improve
the yield of poor synthesis [12]. Figure 2 shows the
pseudoproline dipeptide Fmoc-Leu-Thr(CMe,Mepro)-OH.
In this dipeptide, the Thr residue has been reversibly
protected as proline-like TFA-labile oxazolidine [13 ].
WT-PLB was synthesized according to a new proce-
dure developed in the Lorigan’s lab. Briefly, WT-PLB
was synthesized using modified Fmoc-based solid-phase
methods with an ABI 433A peptide synthesizer (Applied
Biosystems, Foster city, CA). WT-PLB is very hydro-
phobic; thus, the synthesis of this peptide is very chal-
lenging. Nevertheless, by using a combination of ex-
tended coupling and deprotection protocols with a single
pseudoproline dipeptide substitution, we were able to
obtain both purified PLB and P-PLB in a yield of 25%.
Couplings were performed using 10-fold excess of Fmoc-
amino acids activated with HBTU/DIPEA. The synthe-
sizer was programmed to use conditional UV feedback
monitoring; coupling and deprotection reactions are ex-
tended automatically, and a capping step introduced after
the coupling step, based on the kinetic profile of the
Fmoc deprotection reaction. For certain residues addi-
tional extensions to the coupling times were used as in-
dicated in Table 1.
All peptides were cleaved from the resin by treatment
with TFA/EDT/thioanisole/water (10:0.5:0.25:0.5) for
2.5 h, and isolated by centrifugation followed by precipi-
tation with methyl t-butyl ether. PLB consists of a hy-
drophilic N-terminus (residues 1 - 20), a hinge region (21
- 30) and a hydrophobic
-helical transmembrane tail (31
- 52) [1]. From previous work by Lorigan and co-work-
ers [11], it is known that the synthesis of the C-terminal
transmembrane region of PLB is extremely difficult, par-
ticularly the region from Cys36 to Cys45. To overcome
these difficulties, the Lorigan gro up developed a strategy
involving extended double coupling together with cap-
ping and conditional repetition of the Fmoc deprotection
reaction [11]. Using this approach, PLB (24 - 52) seg-
ment was obtained in a purified yield of 37% [11].
Initially, we attempted the synthesis of full length PLB
with standard amino-acid building blocks using the pro-
tocols previously described [11]. A PEG-PS resin (0.22
mmol/g) was selected as the solid support to reduce steric
crowding and aggregation during chain assembly. Using
the conditional feedback monitoring, this synthesis was
completed in 9 days, as compared to 10 days for the
shorter PLB ( 2 4 - 52) prepare d o n p ol ystyrene resi n [ 1 1].
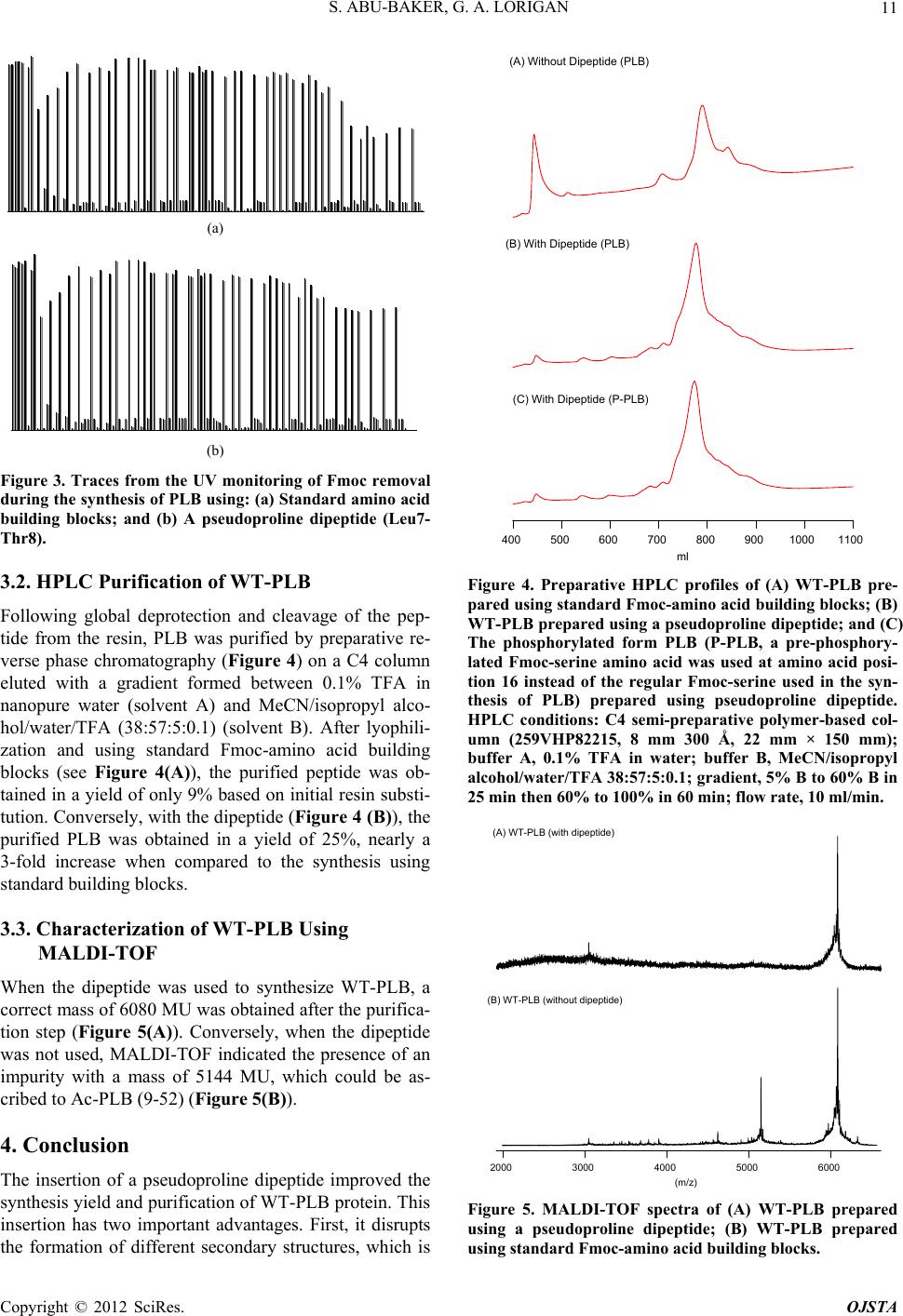
UV monitoring of the Fmoc deprotection reactions in-
dicated that the peptide assembly proceeded smoothly
until Leu-7 (Figure 3(a)). However, following introduc-
tion of this residue, there was a marked decrease in the
height of the Fmoc deprotection peak, indicating difficul-
ties in the coupling of Leu-7 to Thr-8. Attempts to im-
prove this coupling by double coupling or extending the
reaction time to 6 hours had little effect. In view of the
problems with the coupling of Leu-7 to Thr-8, the syn-
thesis was repeated in exactly the same manner, except
that Leu-7 and Thr-8 were introduced simultaneously
using the pseudoproline dipeptide Fmoc-Leu-Thr(CMe,
Mepro)-OH (Figure 2). In the presence of this dipeptide,
UV monitoring of the Fmoc deprotection reactions indi-
cated that the peptide assembly proceeded reasonably
smoothly until the end of t his synthesis (see Figure 3(b)).
Table 1. Coupling protocols used for assembly of PLB pep-
tides.
Cycle Method
2 - 5 Single coupling
6, 20 - 26, 28, 29 Single coupling + 1 h extension
7 - 14 Double coupling + 6 h ex tension
15 - 19, 31, 34 - 39,
41, 42, 47, 49 - 51 Double coupling
27, 30, 33, 40,
43 - 46, 48 Double coupling + 2 h extension
Copyright © 2012 SciRes. OJSTA