 Journal of Minerals and Materials Characterization and Engineering, 2012, 11, 709-718 Published Online July 2012 (http://www.SciRP.org/journal/jmmce) Characterization and Comparison of Saprist and Fibrist Newfoundland Sphagnum Peat Soils Emmanuel S. Asapo1,2*, Cynthia A. Coles1 1Faculty of Engineering and Applied Science, Memorial University of Newfoundland, St. John’s, Canada 2Chemical and Polymer Engineering Department, Faculty of Engineering, Epe Campus, Lagos State University, Lagos, Nigeria Email: *esasapo@mun.ca Received April 5, 2012; revised May 21, 2012; accepted June 9, 2012 ABSTRACT Saprist and fibrist sphagnum peat soils obtained from the same natural peat bog owned by Traverse Nurseries, Torbay, Newfoundland, Canada were characterized to study their potential for adsorbing metals. Both peat soils had a pH of 4.2. The saprist peat had the lower fiber content (68.6% versus 75%), higher cation exchange capacity (70 meq/100g versus 45 meq/100g), higher moisture content (86% versus 82%), higher organic matter content (91% versus 84%), higher wet bulk density (0.65 g/cm3 versus 0.60 g/cm3) and higher dry bulk density (0.28 g/cm3 versus 0.20 g/cm3). A crystallog- raphy study showed that the saprist peat was completely amorphous and the metal content analysis showed high cal- cium and iron concentrations in both types of peat with higher values in the fibrist peat. Carboxylic acid, alcoholic hy- droxyl, phenolic hydroxyl, amine and amide functional groups were present and these could be responsible for binding metal ions via ion exchange and or complexation reactions. Keywords: XRD; SEM; FTIR; NMR; ICP-MS; Functional Groups 1. Introduction Mining is a major contributor of soil and water pollution [1] and a high proportion of dissolved metals exist in areas surrounding mining sites [2] and have been ana- lyzed in water samples long after the mines have been closed or abandoned [1]. Aqueous phase metals are highly mobile and aquatic life around any mine site could be perpetually endangered if improperly treated waste- waters are discharged. Adsorption using low cost adsorbents such as agricul- tural wastes (orange skins, banana peels, [3]) saw dust [4], peat [5,6], clay and zeolites [7], is an effective alter- native to precipitation, membrane technology and floata- tion for metal removal from wastewater. An adsorbent is “low cost” if it is readily available, requires minimal or no processing [8] and is inexpensive or of zero cost. Peat soils are promising adsorbents for heavy metal removal [5,6,9], easily harvested and are economical as can be seen from Table 1. Peat is partially fossilized plant matter that is formed in poorly oxygenated waters of marshes, bogs, and swamps where the rate of plant matterproduction and accumulation exceeds the rate of microbial oxidation [15]. Though peat is most common in the northern hemisphere, large deposits exist in Brazil, Indonesia and South Africa [16]. Peatlands or peat bogs record environmental and paleoenvironmental evolution and provide a reference for measuring past and present global climate change [17]. Since 1922 the von Post scale has been used to classify peat from poorly decomposed (1 H) to completely de- composed (10 H) and peat has also been classified as being highly decomposed (saprist), moderately decom- posed (hemic) or poorly decomposed (fibrist) [18,19]. Peat characterization has remained a difficult task since peat soils may form under a variety of conditions of vegetation and environment [20]. In the past characteri- zation has provided details on the organic and inorganic compounds present in the peat, usually obtained through partial or complete destruction of the peat matrix, and details on the morphology and particle size. Previous studies have focused on the humic and fulvic acid frac- tions extracted from the peat [21-25] but the determina- tion of the constituents or composition of peat, with minimal destruction is preferable and may be more real- istic [26]. Poorly humified or fibrist peat (horticultural peat) has been well studied as an adsorbent for heavy metals [27-29] although the highly humified or saprist peat has not been studied to the same extent and the peat-metal bonding mechanisms have not been fully understood or *Corresponding author. Copyright © 2012 SciRes. JMMCE  E. S. ASAPO, C. A. COLES 710 Table 1. Relative cost of some adsorbent materials. Adsorbent material Cost (US $/kg) Source Chitosan 15.43 [10] Activated Carbon ~2.54 (production, quality and source dependent)[11] Zeolites 0.03 - 0.14 (quality and end-use dependent) [12] Clay 0.03 - 0.375 (quality and type dependent) [13] Peat 0.024 - 0.052 (peat type and processing level dependent)[14] established [30], possibly due in part to the destructive characterization procedures employed. Saprist peat can be used as an agricultural enhancement for improving the water holding capacity of sandy soils [23] and possess a higher metal adsorptive capacity than the widely studied fibrist peat [31]. This paper describes and compares the properties of untreated saprist and fibrist peat soils from the same peat bog. Physico-chemical characterization of the two peat soils was undertaken using standard laboratory proce- dures and in addition, was supplemented with non-de- structive characterization of the two peat soils employing five types of equipment including an X-Ray Diffracto- meter (XRD), a Scanning Electron Microscope (SEM), a Fourier Infrared Spectroscope (FTIR), a Solid State 13Carbon Nuclear Magnetic Resonance (NMR) and an Inductively Coupled Plasma-Mass Spectrometer (ICP- MS). The saprist and fibrist peat soils are compared and the results are discussed in terms of the potential of the saprist peat to remove metals from wastewater, though metal removal studies were not a part of this research. 2. Methods The two peat soils were obtained, courtesy of the Trav- erse Nursery, from a natural peat bog located between Torbay and Flat Rock (15 km north of St. John’s), and an area, which is part of the largest peatland on the Avalon Peninsula of Newfoundland [32]. The peats were har- vested at about 0.4 m depth (fibrist) and at about 1.6 m depth (saprist), transferred in flexi bags to the lab, weighed, spread on a plastic tray, and air dried at room temperature (about 23˚C) to remove about 70% of the original moisture content. Each was then homogenized by manually mixing after removing pebbles and unde- composed woody materials. The physico-chemical prop- erties that could influence metal adsorption of the peat soils were determined by standard methods [33] as sum- marized in Table 2. All of the tests were conducted on the air-dried peat soils except for the wet bulk density tests that were conducted on the peat fresh from the field, Table 2. Physico-chemical parameters of the saprist and fibristpeat soils. Values Parameter Method Used Saprist Fibrist Degree of decompositionvon Post 8H 3H pH (in de-ionized water)ASTM D2976-71 4.2 4.2 Fiber content (%) ASTM D1997-91 68.8 75 CEC at 7.0 pH (meq/100g) Calcium acetate/chloridea 70 45 Moisture content (%) ASTM D2974-07A 86 82 Organic matter (%) ASTM D2974-07A 91 84 Ash content (%) ASTM D2974-07A 9 16 Wet bulk density (g/cm3 )ASTM D4531-86 0.65 0.60 Dry bulk density (g/cm3)ASTM D4531-86 0.28 0.21 aCalcium acetate/chloride method [34]. and which was homogenized after removing the pebbles and woody materials. Grain size determination of the air- dried peats was obtained by sieving triplicate dried peat samples over a series of mechanically stacked sieves. To determine the total metallic contents of the air- dried homogenized saprist and fibric peat soils, each soil was crushed in a mortar, acidified with 14.5 N HF and 8 N HNO3 and left on a hot plate for several days until completely digested so that all the organic components were released. Then 6 N HCl and 8 N HNO3 were added to dissolve the samples further and this step released the inorganic components. Finally 8 N HNO3 was added and diluted with nano-pure water according to the rock dis- solution procedure [33] in the Earth Sciences Department at Memorial University of Newfoundland (MUN) where the samples were finally analyzed with a model ELAN DRC-2 ICP-MS. Micrographs of the peat pore orientation and surface morphology were obtained using the Hitachi S-570 SEM (Biology Department, MUN). To prepare the soil sam- ples for each of the size fractions obtained from the dry granulometry test but excluding the dust fraction (Table 3) for the saprist and fibrist peat (for a total of twelve samples), each was spread over a carbon taped stud and coated with 550× Sputter Coater for gold operated at 20 mA in a vacuum of 0.2 mbar for 2.5 mins resulting in a 15 nm thick coating on the peat. The mineral content was analyzed for each of the same twelve size fractions of the saprist and fibrist peat sam- ples that micrographs had been taken of. Samples were packed on a vertically placed stud of the Rigaku Rotaflex D/Max 1400 rotating anode powdered XRD (Earth Sci- ences Department, MUN) with Cu-Kα radiation source Copyright © 2012 SciRes. JMMCE  E. S. ASAPO, C. A. COLES 711 Table 3. Dry granulometry resultsfor the two peat types. Average % retained by weight Sieve No. Sieve size (µm) Saprist Fibrist 4 4750 13 15 8 2000 - 19 20 850 52 - 40 425 15 45 50 300 5 - 60 250 - 9 100 150 6 4 200 75 3 6 Smaller dust 6 2 operated at 40 kV and 100 mA from Rigaku/MSC—Ja- pan equipped with an X-ray stream 2000 low tempera- ture system. The spectra obtained were matched through the JADE data software (Earth Sciences Department, MUN). The functional group content of the same twelve frac- tions of the saprist and fibrist peat soils that were ana- lyzed with a SEM and an XRD was identified using the Bruker TENSOR 27 FTIR (Chemistry Department, MUN) equipped with a MIRacle ATR accessory coated with crystallized ZnSe with absorbance range from 4000 and 650 cm−1. A few grains of each air dried homogenized peat sample was placed on the pressure tip, compressed onto the sampling area at the center of the ZnSe crystal plate, and was scanned for one minute in transmission mode double sided forward/backward at a spectral reso- lution of four wavenumbers. The equipment incorporates a KBr beam splitter, aperture (6 mm setting) and detector. Solid state 13C NMR of a peat samples were taken from fraction < 425 µm to identify the dominant functional groups. The spectra were obtained at 298 K using a Bruker Avance II 600 spectrometer, equipped with a SB Bruker 3.2 mm MAS triple-tuned probe operating at 600.33 MHz for 1 H and 150.97 MHz for 13C. Chemical shifts are referenced to tetramethylsilane (TMS) using adamantane as an intermediate standard for 13C. The sam- ples were spun at 20 kHz for 13C NMR spectra. Cross- polarization spectra were collected with a Hartmann- Hahn match at 62.5 kHz and 100 kHz 1 H decoupling. The recycle delay was 2 s. The contact time was 2000 ms for 13C NMR. 3. Results and Discussion 3.1. Physico-Chemical Properties The physico-chemical properties of the two peat types are summarized in Table 2. On the von Post scale of classification, the saprist peat was 8H while the fibrist peat was 3 H. Both peat samples were acidic and had relatively high fiber contents, with the fiber content being greater in the fibrist peat (as would be expected). The cation exchange capacity (CEC) was 70 meq/100g for the saprist peat compared to 45 meq/100 for the fi- brist peat. The higher CEC of the saprist peat suggests that it could be a better adsorbent for metal removal though it is the fibrist peat that has been the most widely tested as an adsorbent for metals. Both peats had high moisture holding capacities with the saprist peat having the higher value. Since CEC influences uptake of hy- drated cations, CEC is also related to moisture holding capacity and so the results are reasonable. The organic content was greater in the saprist peat, which can be attributed to the higher degree of decompo- sition [35], longer exposure to weathering (including me- chanical activities such burrowing by worms, soil move- ment and coverage), and deeper zone of formation [20]. Consequently the ash or inorganic content was greater in the fibric peat (since the one test gives both the organic and inorganic fractions and their sum is the total mate- rial). The bulk density (wet and dry) of the saprist peat was higher as this peat was dominated by more of the smaller particle size fractions (as shown in Table 2). This is also related to the greater water retaining capacity that the saprist peat exhibited as the smaller particles would con- tribute to a greater overall surface area. The particle size distributions of the air-dried homo- genized peat soils (in Table 3) are showing that only 13% of the saprist peat had particles > 2000 μm whereas 34% of the fibric peat had particles > 2000 μm. Fractions > 2000 µm in the fibrist peat were woody undecomposed materials. Fractions < 2000 μm and >850 µm were mostly fibre of unidentifiable decomposing materials. 3.2. Metal Content The average metal concentrations in each peat sample are presented in Table 4. Also detected at concentrations below 1 mg/kg were As, Co, and Pb. Although the pres- ence of these metals could be due to both natural and anthropogenic sources, the results show that these peat soils have a natural affinity for the detected metals with calcium and iron being the predominant metals. 3.3. Surface Morphology The micrographs of the twelve peat fractions ranged in resolution from 150 times to 2200 times but the at 1000 times it was easiest to identify pores without damaging the structure so these micrographs are presented and the fractions ≤ 425 µm gave the best images of the pores and Copyright © 2012 SciRes. JMMCE 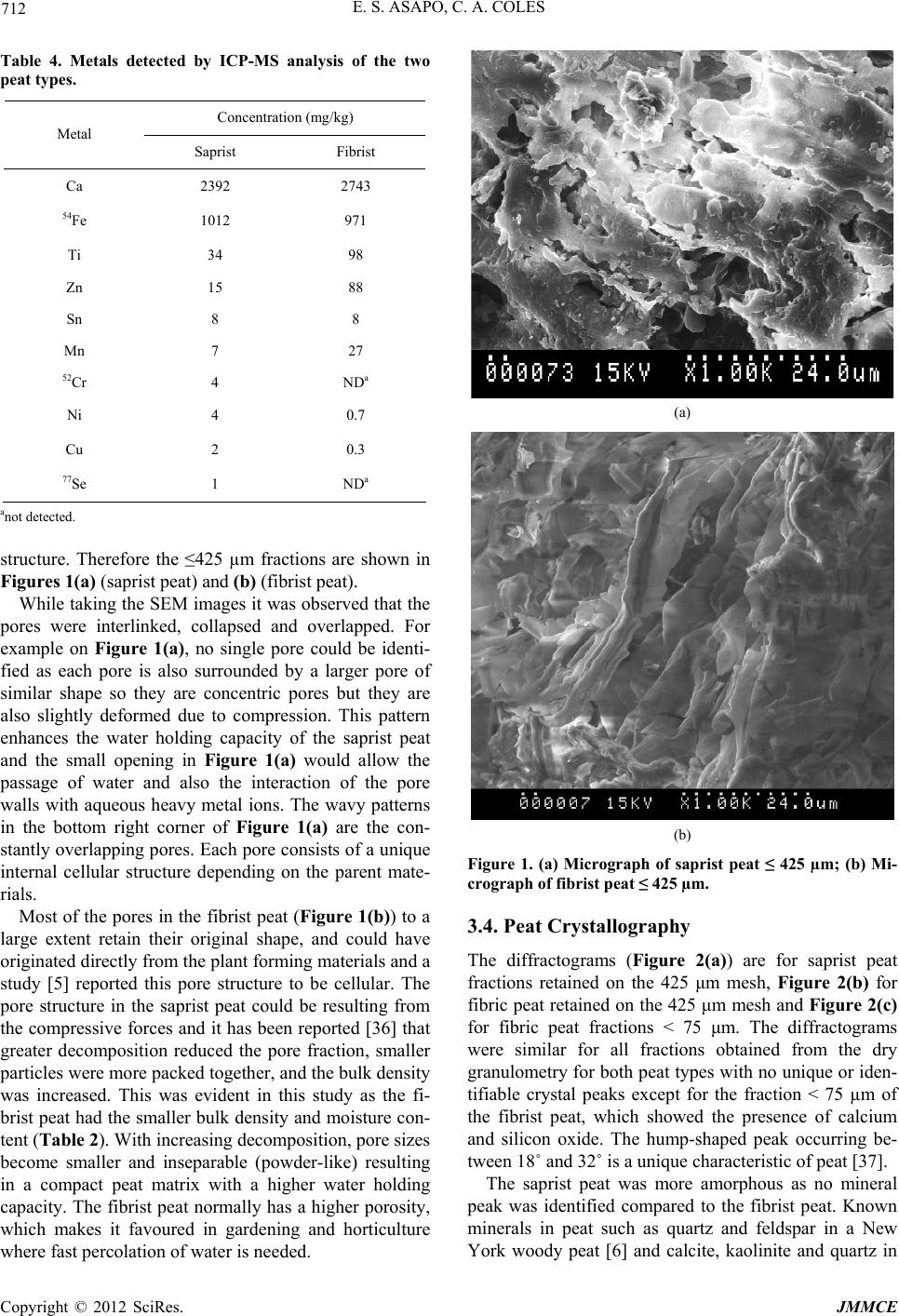 E. S. ASAPO, C. A. COLES 712 Table 4. Metals detected by ICP-MS analysis of the two peat types. Concentration (mg/kg) Metal Saprist Fibrist Ca 2392 2743 54Fe 1012 971 Ti 34 98 Zn 15 88 Sn 8 8 Mn 7 27 52Cr 4 NDa Ni 4 0.7 Cu 2 0.3 77Se 1 NDa anot detected. structure. Therefore the ≤425 µm fractions are shown in Figures 1(a) (saprist peat) and (b) (fibrist peat). While taking the SEM images it was observed that the pores were interlinked, collapsed and overlapped. For example on Figure 1(a), no single pore could be identi- fied as each pore is also surrounded by a larger pore of similar shape so they are concentric pores but they are also slightly deformed due to compression. This pattern enhances the water holding capacity of the saprist peat and the small opening in Figure 1(a) would allow the passage of water and also the interaction of the pore walls with aqueous heavy metal ions. The wavy patterns in the bottom right corner of Figure 1(a) are the con- stantly overlapping pores. Each pore consists of a unique internal cellular structure depending on the parent mate- rials. Most of the pores in the fibrist peat (Figure 1(b)) to a large extent retain their original shape, and could have originated directly from the plant forming materials and a study [5] reported this pore structure to be cellular. The pore structure in the saprist peat could be resulting from the compressive forces and it has been reported [36] that greater decomposition reduced the pore fraction, smaller particles were more packed together, and the bulk density was increased. This was evident in this study as the fi- brist peat had the smaller bulk density and moisture con- tent (Table 2). With increasing decomposition, pore sizes become smaller and inseparable (powder-like) resulting in a compact peat matrix with a higher water holding capacity. The fibrist peat normally has a higher porosity, which makes it favoured in gardening and horticulture where fast percolation of water is needed. (a) (b) Figure 1. (a) Micrograph of saprist peat ≤ 425 µm; (b) Mi- crograph of fibrist peat ≤ 425 µm. 3.4. Peat Crystallography The diffractograms (Figure 2(a)) are for saprist peat fractions retained on the 425 μm mesh, Figure 2(b) for fibric peat retained on the 425 μm mesh and Figure 2(c) for fibric peat fractions < 75 μm. The diffractograms were similar for all fractions obtained from the dry granulometry for both peat types with no unique or iden- tifiable crystal peaks except for the fraction < 75 µm of the fibrist peat, which showed the presence of calcium and silicon oxide. The hump-shaped peak occurring be- tween 18˚ and 32˚ is a unique characteristic of peat [37]. The saprist peat was more amorphous as no mineral peak was identified compared to the fibrist peat. Known minerals in peat such as quartz and feldspar in a New York woody peat [6] and calcite, kaolinite and quartz in Copyright © 2012 SciRes. JMMCE 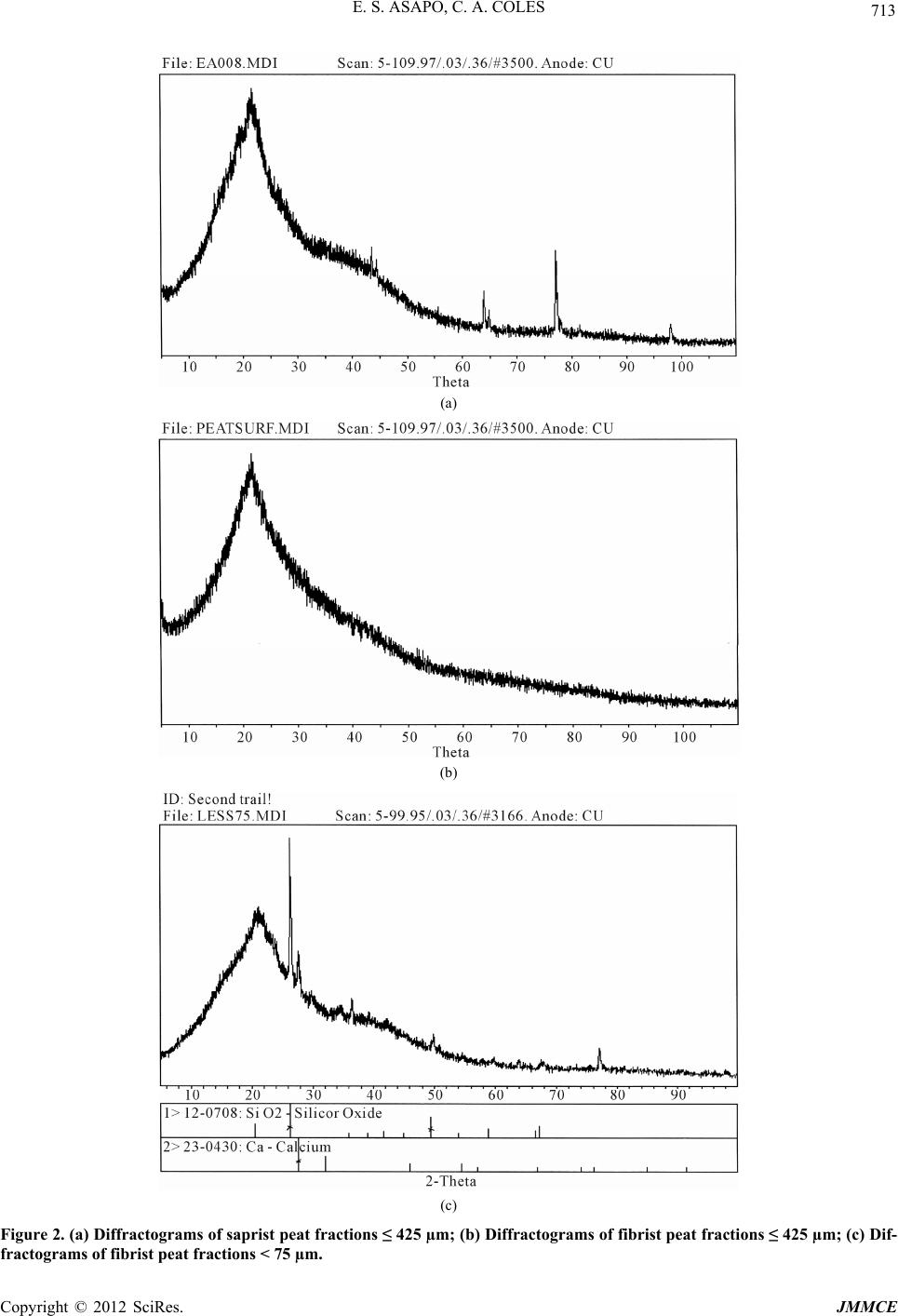 E. S. ASAPO, C. A. COLES Copyright © 2012 SciRes. JMMCE 713 (a) (b) (c) Figure 2. (a) Diffractograms of saprist peat fractions ≤ 425 µm; (b) Diffractograms of fibrist peat fractions ≤ 425 µm; (c) Dif- fractograms of fibrist peat fractions < 75 µm. 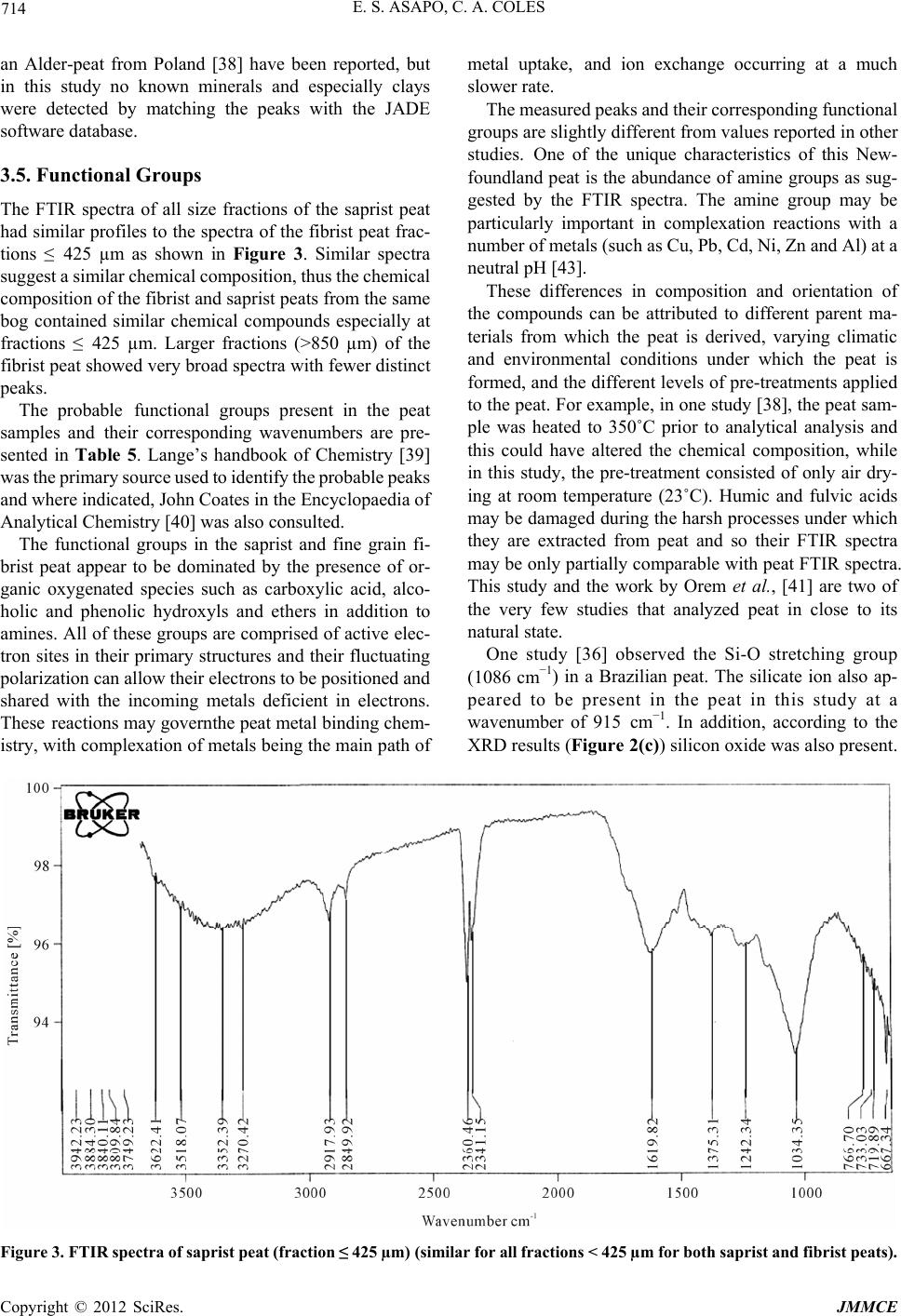 E. S. ASAPO, C. A. COLES 714 an Alder-peat from Poland [38] have been reported, but in this study no known minerals and especially clays were detected by matching the peaks with the JADE software database. 3.5. Functional Groups The FTIR spectra of all size fractions of the saprist peat had similar profiles to the spectra of the fibrist peat frac- tions ≤ 425 µm as shown in Figure 3. Similar spectra suggest a similar chemical composition, thus the chemical composition of the fibrist and saprist peats from the same bog contained similar chemical compounds especially at fractions ≤ 425 µm. Larger fractions (>850 µm) of the fibrist peat showed very broad spectra with fewer distinct peaks. The probable functional groups present in the peat samples and their corresponding wavenumbers are pre- sented in Table 5. Lange’s handbook of Chemistry [39] was the primary source used to identify the probable peaks and where indicated, John Coates in the Encyclopaedia of Analytical Chemistry [40] was also consulted. The functional groups in the saprist and fine grain fi- brist peat appear to be dominated by the presence of or- ganic oxygenated species such as carboxylic acid, alco- holic and phenolic hydroxyls and ethers in addition to amines. All of these groups are comprised of active elec- tron sites in their primary structures and their fluctuating polarization can allow their electrons to be positioned and shared with the incoming metals deficient in electrons. These reactions may governthe peat metal binding chem- istry, with complexation of metals being the main path of metal uptake, and ion exchange occurring at a much slower rate. The measured peaks and their corresponding functional groups are slightly different from values reported in other studies. One of the unique characteristics of this New- foundland peat is the abundance of amine groups as sug- gested by the FTIR spectra. The amine group may be particularly important in complexation reactions with a number of metals (such as Cu, Pb, Cd, Ni, Zn and Al) at a neutral pH [43]. These differences in composition and orientation of the compounds can be attributed to different parent ma- terials from which the peat is derived, varying climatic and environmental conditions under which the peat is formed, and the different levels of pre-treatments applied to the peat. For example, in one study [38], the peat sam- ple was heated to 350˚C prior to analytical analysis and this could have altered the chemical composition, while in this study, the pre-treatment consisted of only air dry- ing at room temperature (23˚C). Humic and fulvic acids may be damaged during the harsh processes under which they are extracted from peat and so their FTIR spectra may be only partially comparable with peat FTIR spectra. This study and the work by Orem et al., [41] are two of the very few studies that analyzed peat in close to its natural state. One study [36] observed the Si-O stretching group (1086 cm−1) in a Brazilian peat. The silicate ion also ap- peared to be present in the peat in this study at a wavenumber of 915 cm−1. In addition, according to the XRD results (Figure 2(c)) silicon oxide was also present. Figure 3. FTIR spectra of saprist peat (fraction ≤ 425 µm) (similar for all fractions < 425 µm for both saprist and fibrist peats). Copyright © 2012 SciRes. JMMCE  E. S. ASAPO, C. A. COLES 715 Table 5. Probable functional groups from FTIR spectra. Wavenumber (cm−1) Probable Functional Group Assigned Band, cm−1 Comparable Studies 3518 Primary amines (aliphatic) 3550 - 3300 (m)a Secondary amines 3550 - 3400 (w) 3352 Normal polymeric OH stretch1 [21] 3270 Ammonium ion 3300 - 3030 (s)b 2918 Carboxylic acids -CO2H, OH stretching 3000 - 2500 [41] 2850 Carboxylic acids -CO2H, OH stretching 3000 - 2500 [41] Methylene (CH2) C-H asymmetric/symmetric stretch1 [21] 2360 Tertiary amines R1R2R3NH+ 2700 - 2250 2341 Aliphatic CN 1620 Primary amines (aliphatic) 1650 - 1560 (m)a C=C conjugated with aromatic ring 1640 - 1610 (m) [41] α, β unsaturated carbonyl compounds 1640 - 1590 (m) 1412 Ammonium ion 1430 - 1390 (s)b Vinyl C-H in-plane bend1 1375 =C(CH3)2 Alkane residues attached to C 1380 (m) [41] Nitro C-NO2 aromatic 1380 - 1320 (s)c 1242 Aromatic ethers, aryl –O stretch (Φ-O-H)1 [42] 1150 Tertiary alcohol C-O stretch1 [21] 1034 Hydroxyl O-H primary aliphatic alcohols 1085 - 1030d [41,42] -O-CH3 ethers (w-m) c 1030 Peroxides -O-O- 1150 - 10301e (m-s) Alkyl [41,42] 915 Silicate ion1 845 Nitro C-NO2 aromatic 865 - 835c 825 Peroxides -O-O- 900 - 830 (w)e 767 -CH2- Rocking vibration 720 Saturated CH2c 720 [42] 667 Hydroxyl O-H primary aliphatic alcohols 700 - 600d 1John Coates in Encyclopedia of Analytical Chemistry; aprimary amine bands at 3550 - 3300 and 1650 - 1560; bam- monium ion bands at 3300 - 3030 and 1430 - 1390; cnitro C-NO2 aromatic bands at 1380 - 1320 and 865 - 835; dpri- mary aliphatic alcohols bands at 1085 - 1030 and 700 - 600; eperoxide bands at 1150 - 1030 and 900 - 830. Analysis of the 13C NMR spectra as shown in Figure 4 (similar for all other fractions for both peat types), for the fraction ≤ 425 µm supported the functional groups iden- tified by the FTIR spectra as shown in Table 6. 4. Conclusions Characterization of a saprist peat was undertaken as a prelude to its evaluation as an adsorbent for removing metals from wastewaters. The CEC (70 meq/100g) was higher than that of the poorly humified peat (45 meq/100g) making the highly humified peat a better cation exchanger. Although the metal content of the fibrist peat was greater this could have been due to its closer contact to the upper layer of the bog and exposure to windblown transport. The ash content of the fibrist peat was almost twice that of saprist peat, which might be due to the different zones of formation and accumulation of inorganic materials such s silicon oxide at the upper layer of the bog. a Copyright © 2012 SciRes. JMMCE 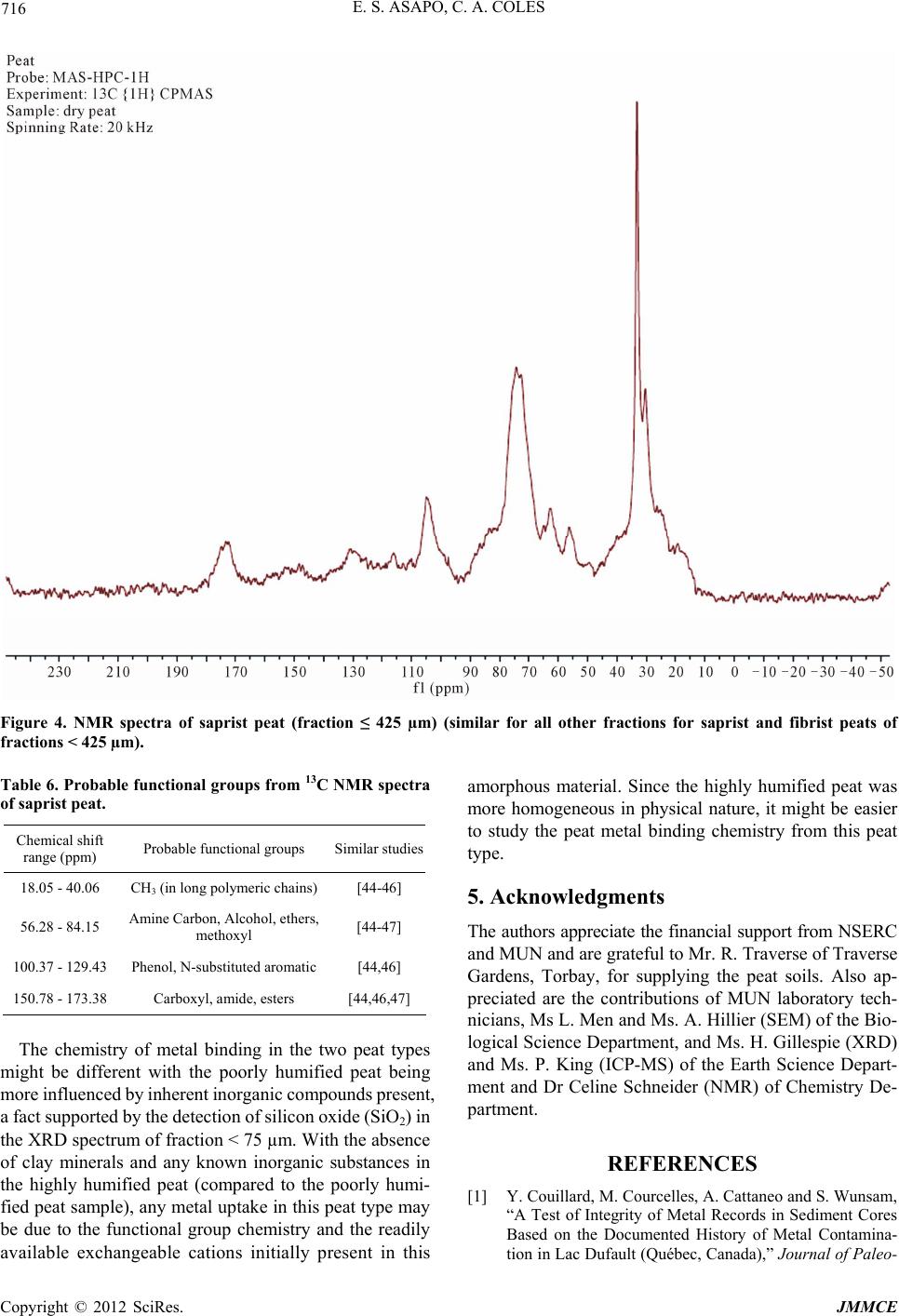 E. S. ASAPO, C. A. COLES 716 Figure 4. NMR spectra of saprist peat (fraction ≤ 425 µm) (similar for all other fractions for saprist and fibrist peats of fractions < 425 µm). Table 6. Probable functional groups from 13C NMR spectra of saprist peat. Chemical shift range (ppm) Probable functional groups Similar studies 18.05 - 40.06 CH3 (in long polymeric chains) [44-46] 56.28 - 84.15 Amine Carbon, Alcohol, ethers, methoxyl [44-47] 100.37 - 129.43 Phenol, N-substituted aromatic [44,46] 150.78 - 173.38 Carboxyl, amide, esters [44,46,47] The chemistry of metal binding in the two peat types might be different with the poorly humified peat being more influenced by inherent inorganic compounds present, a fact supported by the detection of silicon oxide (SiO2) in the XRD spectrum of fraction < 75 µm. With the absence of clay minerals and any known inorganic substances in the highly humified peat (compared to the poorly humi- fied peat sample), any metal uptake in this peat type may be due to the functional group chemistry and the readily available exchangeable cations initially present in this amorphous material. Since the highly humified peat was more homogeneous in physical nature, it might be easier to study the peat metal binding chemistry from this peat type. 5. Acknowledgments The authors appreciate the financial support from NSERC and MUN and are grateful to Mr. R. Traverse of Traverse Gardens, Torbay, for supplying the peat soils. Also ap- preciated are the contributions of MUN laboratory tech- nicians, Ms L. Men and Ms. A. Hillier (SEM) of the Bio- logical Science Department, and Ms. H. Gillespie (XRD) and Ms. P. King (ICP-MS) of the Earth Science Depart- ment and Dr Celine Schneider (NMR) of Chemistry De- partment. REFERENCES [1] Y. Couillard, M. Courcelles, A. Cattaneo and S. Wunsam, “A Test of Integrity of Metal Records in Sediment Cores Based on the Documented History of Metal Contamina- tion in Lac Dufault (Québec, Canada),” Journal of Paleo- Copyright © 2012 SciRes. JMMCE  E. S. ASAPO, C. A. COLES 717 limnology, Vol. 32, No. 2, 2004, pp. 149-162. doi:10.1023/B:JOPL.0000029429.13621.68 [2] R. Parker and C. Dumaresq, “Effluent Characterization, Water Quality Monitoring and Sediment Monitoring in the Metal Mining EEM Program,” Water Quality Re- search Journal of Canada, Vol. 37, No. 1, 2002, pp. 219- 228. [3] G. Annadurai, R.-S. Juang and D.-J. Lee, “Adsorption of Heavy Metals from Water Using Banana and Orange Peels,” Water Science and Technology, Vol. 37, No. 1, 2002, pp. 185-190. [4] M. A. Zarraa, “Study on the Removal of Chromium (VI) from Wastes Solutions by Adsorption onto Sawdust in Vessels,” Advertising Science and Technology, Vol. 12, No. 2, 1995, pp. 129-138. [5] B. Coupal and J.-M. Lalancette, “The Treatment of Waste- waters with Peat Moss,” Water Research, Vol. 10, No. 12, 1976, pp. 1071-1076. doi:10.1016/0043-1354(76)90038-5 [6] P. R. Bloom and M. B. McBride, “Metal Ion Binding and Exchange with Hydrogen Ions in Acid-Washed Peat,” Soil Science Society of America Journal, Vol. 43, No. 4, 1979, pp. 687-692. doi:10.2136/sssaj1979.03615995004300040012x [7] G. Blanachard, M. Muanaye and G. Martin, “Removal of Heavy Metals from Water by Means of Natural Zeolites,” Water Research, Vol. 18, No. 12, 1984, pp. 1501-1507. doi:10.2136/sssaj1979.03615995004300040012x [8] S. E. Bailey, T. J. Olin, R. M. Bricka and D. D. Adrian, “A Review of Potentially Low-Cost Sorbents for Heavy Metals,” Water Research, Vol. 33, No. 11, 1999, pp. 2469-2479. doi:10.1016/S0043-1354(98)00475-8 [9] C. B. Dissanayake and S. V. R. Weerasooriya, “Peat as a Metal-Trapping Material in Purification of Industrial Ef- fluents,” International Journal of Environmental Studies, Vol. 17, No. 3, 1981, pp. 233-238. doi:10.1080/00207238108709912 [10] S. Babel and T. A. Kurniawan, “Low-Cost Adsorbents for Heavy Metals Uptake from Contaminated Water: A Re- view,” Journal of Hazardous Materials, Vol. 97, No. 1-3, 2003, pp. 119-243. [11] US Market Information, “Personal Communication,” 2008. [12] USGS 2006a, Mineral Commodity Summaries (accessed 10 November 2008). http://minerals.usgs.gov/minerals/pubs/mcs/ [13] USGS 2006b, Mineral Commodity Summaries (accessed 10 November 2008). http://miinerals.usgs.gov/minerals/pubs/mcs/ [14] USGS 2006c, Mineral Commodity Summaries (accessed 10 November 2008). http://minerals.usgs.gov/minerals/pubs/mcs/ [15] J. I. Pérez, E. Hontoria, M. Zamorano and M. A. Gόmez, “Wastewater Treatment Using Fibrist and Saprist Peat: A Comparative Study,” Journal of Environmental Science and Health A, Vol. 40, No. 5, 2005, pp. 1021-1032. [16] I. Twardowska, J. Kyziol, T. Goldrath and Y. Avnimelech, “Adsorption of Zinc onto Peat from Peatlands of Poland and Israel,” Journal of Geochemical Exploration, Vol. 66, No. 1-2, 1999, pp. 387-405. [17] A. Martinez-Cortizas, X. Ponteveda-Pombai, E. Garcia- Rodeja, J. C. Nóvoa-Muñoz and W. Shotyk, “Mercury in a Spanish Peat Bog: Archive of Climate Change and At- mospheric Metal Deposition,” Science, Vol. 284, No. 5416, 1999, pp. 939-942. doi:10.1126/science.284.5416.939 [18] E. Bohlin, M. Hämäläinen and T. Sunden, “Botanical and Chemical Characterization of Peat Using Multivariate Methods,” Soil Science, Vol. 147, No. 4, 1989, pp. 252- 263. doi:10.1097/00010694-198904000-00004 [19] B. Nordén, E. Bohlin, M. Nilsson, Å. Albano and C. Röckner, “Characterization of Particle Size Fractions of Peat: An Integrated Biological, Chemical, and Spectro- scopic Approach,” Soil Science, Vol. 153, No. 5, 1992, pp. 382-396. doi:10.1097/00010694-199205000-00006 [20] P. J. Spedding, “Peat,” Fuel, Vol. 67, No. 7, 1988, pp. 883-900. doi:10.1016/0016-2361(88)90087-7 [21] J. Niemeyer, Y. Chen and J.-M. Bollag, “Characterization of Humic Acids, Composts, and Peat by Diffuse Reflec- tance Fourier-Transform Infrared Spectroscopy,” Soil Sci- ence Society of America Journal, Vol. 56, No. 1, 1992, pp. 135-140. doi:10.2136/ sssaj 199 2.03615 995005 6000100 21x [22] A. Baran, “Characterization of Carex Peat Using Extinc- tion Values of Humic Acids,” Bioresource Technology, Vol. 85, No. 1, 2002, pp. 99-101. doi:10.1016/S0960-8524(02)00072-X [23] H. Li, L. E. Parent, A. Karam and C. Tremblay, “Potential of Sphagnum Peat for Improving Soil Organic Matter, Water Holding Capacity, Bulk Density and Potato Yield in a Sandy Soil,” Plant and Soil, Vol. 265, No. 1-2, 2004, pp. 355-365. [24] D. Gondar, R. Lopez, S. Fiol, J. M. Antelo and F. Arce, “Characterization and Acid-Base Properties of Fulvic and Humic Acids Isolated from Two Horizons of an Ombro- trophic Peat Bog,” Geoderma, Vol. 126, No. 3-4, 2005, pp. 367-374. [25] S. S. Fong and M. Mohamed, “Chemical Characterization of Humic Substances Occurring in the Peats of Sarawak, Malaysia,” Organic Geochemistry, Vol. 38, No. 6, 2007, pp. 967-976. doi:10.1016/j.orggeochem.2006.12.010 [26] P. Burba, A.-M. Beer and J. Lukanov, “Metal Distribu- tion and Binding in Balneological Peats and Their Aque- ous Extracts,” Fresenius’ Journal of Analytical Chemistry, Vol. 37, No. 4, 2001, pp. 419-425. doi:10.1007/s002160100810 [27] Y. S. Ho, D. A. J. Wase and C. F. Forster, “Batch Nickel Removal from Aqueous Solution by Sphagnum Moss Peat,” Water Research, Vol. 29, No. 5, 1995, pp. 1327- 1332. doi:10.1016/0043-1354(94)00236-Z [28] R. H. Crist, J. R. Martin, J. Chonko and D. R. Crist, “Up- take of Metals on Peat Moss: An ion Exchange Process,” Environmental Science & Technology, Vol. 30, No. 8, 1996, pp. 2456-2461. doi:10.1021/es950569d [29] L. Ringqvist, A. Holmgren and I. Őborn, “Poorly Humi- fied Peat as an Adsorbent for Metals in Wastewater,” Water Research, Vol. 36, No. 9, 2002, pp. 2394-2404. doi:10.1016/S0043-1354(01)00430-4 [30] R. H. Kadlec and G. A. Keoleian, “Metal Ion Exchange Copyright © 2012 SciRes. JMMCE  E. S. ASAPO, C. A. COLES Copyright © 2012 SciRes. JMMCE 718 on Peat in Peat and Water,” Elsevier Applied Science Publishers Ltd., Amsterdam, 1986, pp. 61-93. [31] I. Kuziemska and B. Quant, “Peat as a Sorbent for Heavy Metal Removal from Water and Wastewater,” Proceed- ings of Green, the International Symposium on Geotech- nics Related to Environment 2, Wolverhampton, 28 June- 1 July 1998, pp. 308-312. [32] F. Pollet, C. M. Lane, F. Gover and J. H. McKillop, “Peat Resources of Newfoundland,” Mineral Resources Report, No. 2, 1968, pp. 4-5. [33] ASTM, “Annual Books of ASTM Standards, Section 4: Construction,” ASTM, West Conshohocken, 2006. [34] B. H. Sheldrick, Ed., “Analytical Methods Manual,” Ag- riculture and Agri-Food Canada, Ottawa, 1984. [35] T. J. Malterer, E. S. Verry and J. Erjavec, “Fiber Content and Degree of Decomposition in Peats: Review of Na- tional Methods,” Soil Science Society of America Journal, Vol. 56, No. 4, 1992, pp. 1200-1211. doi:10.2136/sssaj1992.03615995005600040033x [36] S. Bozkhurt, M. Lucisano, L. Moreno and I. Neretnieks, “Peat as a Potential Analogue for the Long-Term Evolu- tion in Landfills,” Earth-Science Reviews, Vol. 53, No. 1-2, 2001, pp. 95-147. doi:10.2136/sssaj1992.03615995005600040033x [37] L. P. C. Romăo, J. R. Lead, J. C. Rocha, L. C. de Oliveira, A. H. Rosa, A. G. R. Mendonça and A. de S. Ribeiro, “Structure and Properties of Brazilian Peat: Analysis by Spectroscopy and Microscopy,” Journal of the Brazilian Chemical Society, Vol. 18, No. 4, 2007, pp. 714-720. doi:10.1590/S0103-50532007000400008 [38] I. Twardowska and J. Kyziol, “Binding and Chemical Fractionation of Heavy Metals in Typical Peat Matter,” Fresenius Journal of Analytical Chemistry, Vol. 354, No. 5-6, 1996, pp. 580-586. [39] N. A. Lange and J. G. Speight, “Lange’s Handbook of Chemistry,” 16th Edition, McGraw-Hill Inc., Boston, 2005. [40] J. Coates, “Interpretation of Infrared Spectra: A Practical Approach,” In: R. A. Meyers, Ed., Encyclopedia of Ana- lytical Chemistry, John Wiley & Sons, Hoboken, 2000, pp. 10815-10837. [41] W. H. Orem, S. G. Neuzil, H. E. Lerch and C. B. Cecil, “Experimental Early-Stage Coalification of a Peat Sample and a Peatified Wood Sample from Indonesia,” Organic Geochemistry, Vol. 24, No. 2, 1996, pp. 111-125. doi:10.1016/0146-6380(96)00012-5 [42] R. R. E. Artz, S. J. Chapman, A. H. J. Robertson, J. M. Potts, F. Laggoun-Défarge, S. Gogo, L. Comot, J. R. Disnar and A. J. Francez, “FTIR Spectroscopy Can Be Used as a Screening Tool for Organic Matter Quality Regenerating Cutover Peatlands,” Soil Biology & Biochemistry, Vol. 40, No. 2, 2008, pp. 515-527. doi:10.1016/j.soilbio.2007.09.019 [43] S. E. Cabaniss, “Quantitative Structure-Property Rela- tionships for Predicting Metal Binding by Organic Li- gands,” Environmental Science & Technology, Vol. 42, No. 14, 2008, pp. 5210-5216. doi:10.1021/es7022219 [44] C. M. Preston, D. E. Axelson, M. Lévesque, S. P. Mathur, H. Dinel and R. L. Dudley, “Carbon-13 NMR and Chemi- cal Characterization of Particle-Size Separates of Peats Differing in Degree of Decomposition,” Organic Geo- chemistry, Vol. 14, No. 4, 1989, pp. 393-403. doi:10.1016/0146-6380(89)90005-3 [45] J. A. Baldrock, J. M. Oades, A. G. Waters, X. Peng, A. M. Vassallo and M. A. Wilson, “Aspects of the Chemical Structure of Soil Organic Materials as Revealed by Soli- State 13C NMR Spectroscopy,” Biogeochemistry, Vol. 16, No. 1, 1992, pp. 1-42. [46] J.-D. Mao, W.-G. Hu, K. Schmidt-Rohr, G. Davies, E. A. Ghannour and B. Xing, “Quantitative Characterization of Humic Substances by Solid-State Carbon-13 NMR,” Soil Science Society of America Journal, Vol. 64, No. 3, 2000, pp. 873-884. doi:10.2136/sssaj2000.643873x [47] G. Almendros, H. Knicker and J. Gonzalez-Vila, “Rear- rangement of Carbon and Nitrogen forms in Peat after Progressive Thermal Oxidation as Determined by Solid State 13C and 15N-NMR Spectroscopy,” Organic Geo- chemistry, Vol. 34, No. 11, 2003, pp. 1559-1568. doi:10.1016/S0146-6380(03)00152-9
|