Paper Menu >>
Journal Menu >>
 Journal of Mi nerals & Materials Characteriza tion & Engineering, Vol. 10, No.8, pp.727-734, 2011 jmmce.org Printed in the USA. All rights reserved 727 Synthesis and Characterization of Hydroxyapatite Powder by Sol-Gel Method for Biomedical Application Khelendra Agrawal*, Gurbhinder Singh, Devendra Puri, Satya Prakash Metallurgical and Materials Engineering Department, Indian Institute of Technology Roorkee- 247667, India *Corresponding Author: khelendraagrawal@gmail.com ABSTRACT Hydroxyapatite (HA) is effectively used as a bioimplant material because it closely resembles bone apatite and exhibits good biocompatibility. This paper describe synthesis technique of HA powder by sol-gel method. The product was sintered twice at two different temperatures 400°C to 750°C to improve its crystallinity. The final powder sintered at two temperatures was characterized by X-ray analysis, Scanning electron microscopy (SEM) and Fourier Transform Infrared Spectroscopy (FT-IR) to reveal its phase content, morphology and types of bond present within it. Thermal analysis (TG–DTA) was carried out to investigate the thermal stability of the powder. Keywords: Hydroxyapatite, bioimplant, sol-gel, X-ray diffraction, FT-IR, TG-DTA 1. INTRODUCTION Hydroxyapatite (Ca10(PO4)6(OH)2, HA) is an important inorganic biomaterial which has attracted the attention of researchers related to biomaterials field in recent years. Due to its chemical and structural similarity with the mineral phase of bone and teeth, HA is widely used for hard tissues repair. As a result, this inorganic phosphate has been studied extensively for medical applications in the form of powders, composites or even coatings [1–11]. It is also observed that dense sintered HA has many bone replacement applications and is used for repairing bone defects in dental and orthopedic sites, immediate tooth replacement, augmentation of alveolar ridges, pulp capping material and maxillo facial reconstruction, etc [12]. For substituting or repairing the bone, the designed material must has the ability to create a bond with the host living bone [13]. Hence, it is always desirable to include a high degree of crystallinity and chemical stability among the desirable properties of an ideal hydroxyapatite [14, 15]. Furthermore, HA has also been studied for other non-medical  728 Khelendra Agrawal, Gurbhinder Singh, Devendra Puri, Satya Prakash Vol.10, No.8 applications, for example, as packing media for column chromatography, gas sensors, catalysts, etc. [2, 16]. However, poor mechanical properties, e.g. low strength and toughness, restrict monolithic HA applications to those that require little or no load-bearing parts [17]. Due to its diverse applications, the materials properties accordingly need to be tailored for real world application. Hence researchers have tried to customize its properties such as bioactivity, mechanical strength, solubility and sinterability by controlling its composition, morphology and particle size [9, 10]. The chemical, structural and morphological properties of synthetic HA can be modulated by varying the method and the conditions of synthesis. Classical methods for HA powder synthesis include direct precipitation, hydrothermal techniques, hydrolysis of other calcium phosphates, as well as solid-state reactions [18, 19] and mechano-chemical methods [20, 21]. One of the most widely used methods is wet precipitation, where chemical reactions take place between calcium and phosphorus ions under a controlled pH and temperature of the solution. The precipitated powder is typically calcined at 400-600°C or even at higher temperature in order to obtain a stoichiometric, apatitic structure. In some cases, a well- crystallized HA phase was only developed while approaching a sintering temperature of 1200°C. However, fast precipitation during phosphate solution titration (to calcium solution) leads to chemical in homogeneity in the final product. Slow titration and diluted solutions must be used to improve chemical homogeneity and stoichiometry of the resulting HA. Careful control of the solution condition is critical in the wet precipitation. Otherwise, a decrease of solution pH below about 9 could lead to the formation of Ca-deficient HA structure [17]. Most of the wet methods are time-consuming because the formation of HA phase and the rinsing of unnecessary anions all take time. A method involving non-aqueous systems to synthesize HA has also been reported [22], in which a viscous solution was first obtained by hydrolysis and oxidation of a mixed acetone solution of calcium nitrate (Ca(NO3)2.4H2O) and phenydichlorophosphine (C6H5PCl2). After the viscous solution was dried and calcined at above 700°C, HA Powder was obtained. However, the method is usually not so simple due to the need of hydrolysis and oxidation steps. Sol–gel technique have attracted much attention recently [5, 6, 9-11] due to its well-known inherent advantages to generate glass, glass–ceramic and ceramics powders. These include homogeneous molecular mixing, low processing temperature, the ability to generate sized particles, the tremendous flexibility to generate nanocrystalline powders, bulk amorphous monolithic solids and thin films [23]. The sol–gel process is easily applicable to surface coating and it allows the preparation of high-quality HA thin films on metal substrates [7-10]. Thus, the sol–gel process can be usefully utilized to synthesize both HA powders and HA films under significantly mild conditions. The versatility of the sol-gel method opens a great opportunity to form thin film coatings in a rather simple process, an alternative to thermal spraying which is currently widely used for biomedical applications [24, 25]. Accordingly, the objective of the present work is to synthesis of HA powder from sol-gel method and carried out its characterization. 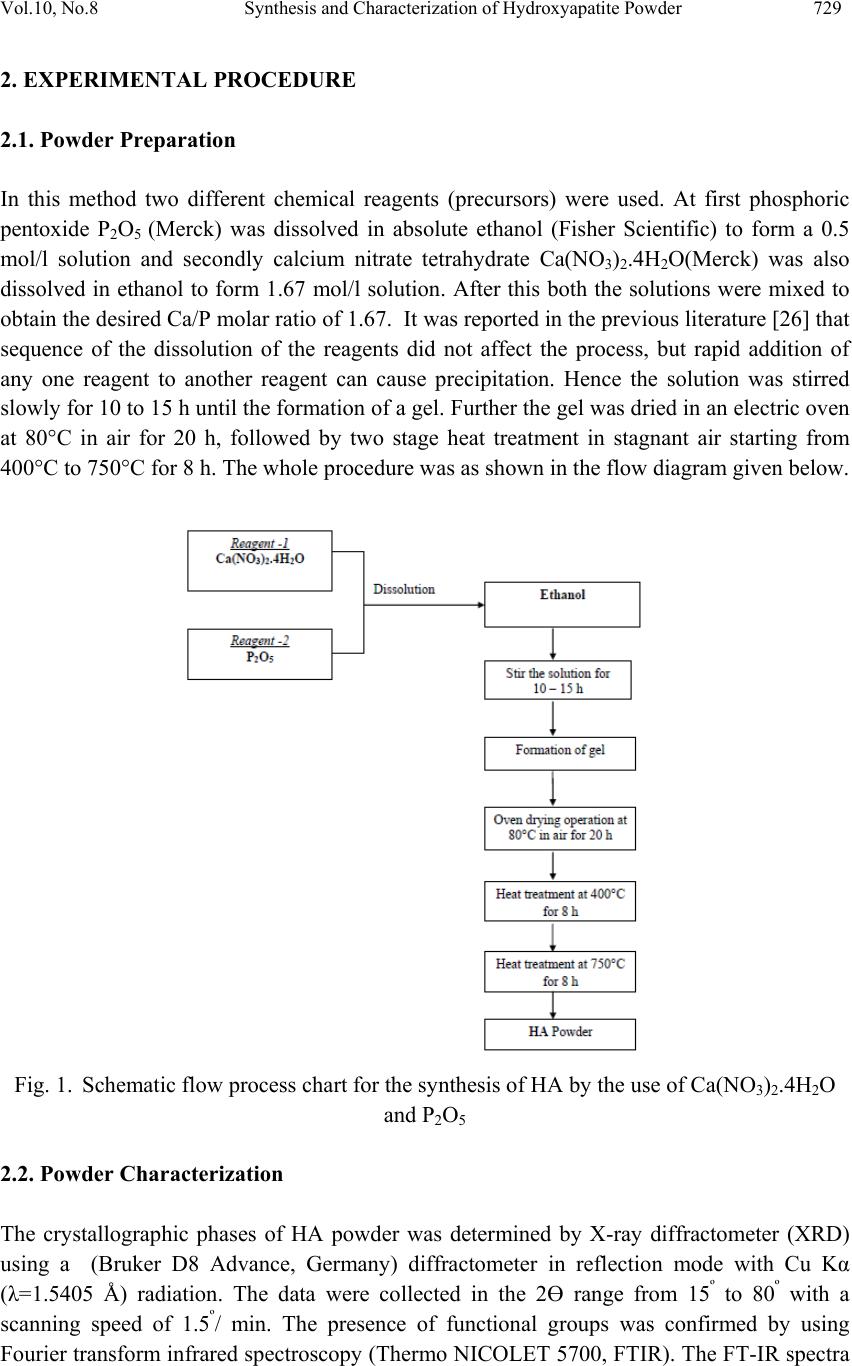 Vol.10, No.8 Synthesis and Characterization of Hydroxyapatite Powder 729 2. EXPERIMENTAL PROCEDURE 2.1. Powder Preparation In this method two different chemical reagents (precursors) were used. At first phosphoric pentoxide P2O5 (Merck) was dissolved in absolute ethanol (Fisher Scientific) to form a 0.5 mol/l solution and secondly calcium nitrate tetrahydrate Ca(NO3)2.4H2O(Merck) was also dissolved in ethanol to form 1.67 mol/l solution. After this both the solutions were mixed to obtain the desired Ca/P molar ratio of 1.67. It was reported in the previous literature [26] that sequence of the dissolution of the reagents did not affect the process, but rapid addition of any one reagent to another reagent can cause precipitation. Hence the solution was stirred slowly for 10 to 15 h until the formation of a gel. Further the gel was dried in an electric oven at 80°C in air for 20 h, followed by two stage heat treatment in stagnant air starting from 400°C to 750°C for 8 h. The whole procedure was as shown in the flow diagram given below. Fig. 1. Schematic flow process chart for the synthesis of HA by the use of Ca(NO3)2.4H2O and P2O5 2.2. Powder Characterization The crystallographic phases of HA powder was determined by X-ray diffractometer (XRD) using a (Bruker D8 Advance, Germany) diffractometer in reflection mode with Cu Kα (λ=1.5405 Å) radiation. The data were collected in the 2Ө range from 15º to 80º with a scanning speed of 1.5º/ min. The presence of functional groups was confirmed by using Fourier transform infrared spectroscopy (Thermo NICOLET 5700, FTIR). The FT-IR spectra 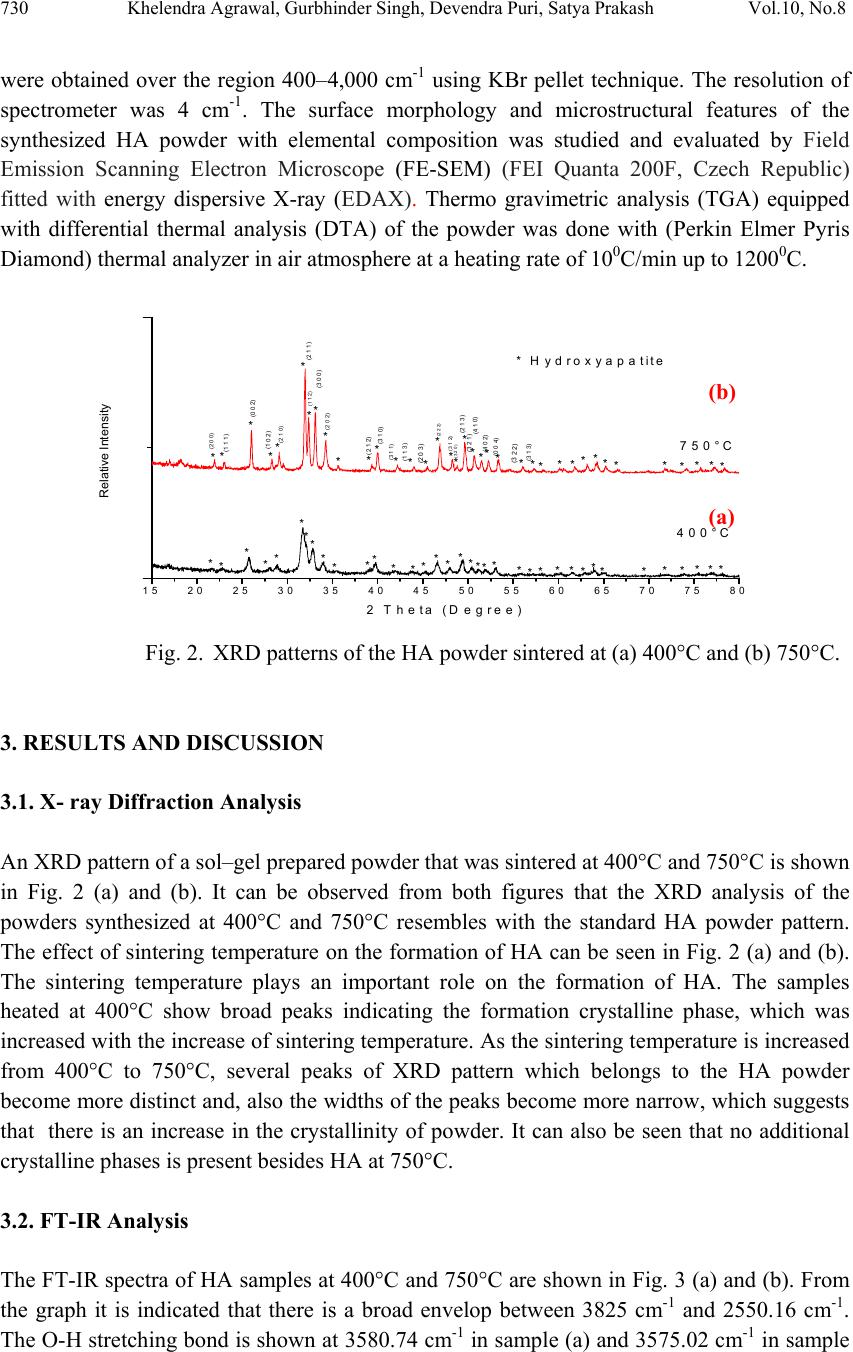 730 Khelendra Agrawal, Gurbhinder Singh, Devendra Puri, Satya Prakash Vol.10, No.8 were obtained over the region 400–4,000 cm-1 using KBr pellet technique. The resolution of spectrometer was 4 cm-1. The surface morphology and microstructural features of the synthesized HA powder with elemental composition was studied and evaluated by Field Emission Scanning Electron Microscope (FE-SEM) (FEI Quanta 200F, Czech Republic) fitted with energy dispersive X-ray (EDAX). Thermo gravimetric analysis (TGA) equipped with differential thermal analysis (DTA) of the powder was done with (Perkin Elmer Pyris Diamond) thermal analyzer in air atmosphere at a heating rate of 100C/min up to 12000C. 15 20 25 30 35 40 45 50 55 60 65 70 75 80 Relative Intensity 2 Theta (D egree) 400°C 750°C *** ********* ** **** ** ******* * *** * * * * * ** * *** * * * * ***** ** * * *********** ****** * Hydroxyapatite (2 0 0) (1 1 1) (0 0 2) (1 0 2) (2 1 0) (2 1 1) (1 1 2) (3 0 0) (2 0 2) (2 1 2) (3 1 0) (3 1 1) (1 1 3) (2 0 3) (2 2 2) * (3 1 2) (3 2 0) (2 1 3) (3 2 1) (4 1 0) (4 0 2) (0 0 4) (3 2 2) (3 1 3) Fig. 2. XRD patterns of the HA powder sintered at (a) 400°C and (b) 750°C. 3. RESULTS AND DISCUSSION 3.1. X- ray Diffraction Analysis An XRD pattern of a sol–gel prepared powder that was sintered at 400°C and 750°C is shown in Fig. 2 (a) and (b). It can be observed from both figures that the XRD analysis of the powders synthesized at 400°C and 750°C resembles with the standard HA powder pattern. The effect of sintering temperature on the formation of HA can be seen in Fig. 2 (a) and (b). The sintering temperature plays an important role on the formation of HA. The samples heated at 400°C show broad peaks indicating the formation crystalline phase, which was increased with the increase of sintering temperature. As the sintering temperature is increased from 400°C to 750°C, several peaks of XRD pattern which belongs to the HA powder become more distinct and, also the widths of the peaks become more narrow, which suggests that there is an increase in the crystallinity of powder. It can also be seen that no additional crystalline phases is present besides HA at 750°C. 3.2. FT-IR Analysis The FT-IR spectra of HA samples at 400°C and 750°C are shown in Fig. 3 (a) and (b). From the graph it is indicated that there is a broad envelop between 3825 cm-1 and 2550.16 cm-1. The O-H stretching bond is shown at 3580.74 cm-1 in sample (a) and 3575.02 cm-1 in sample (a) (b) 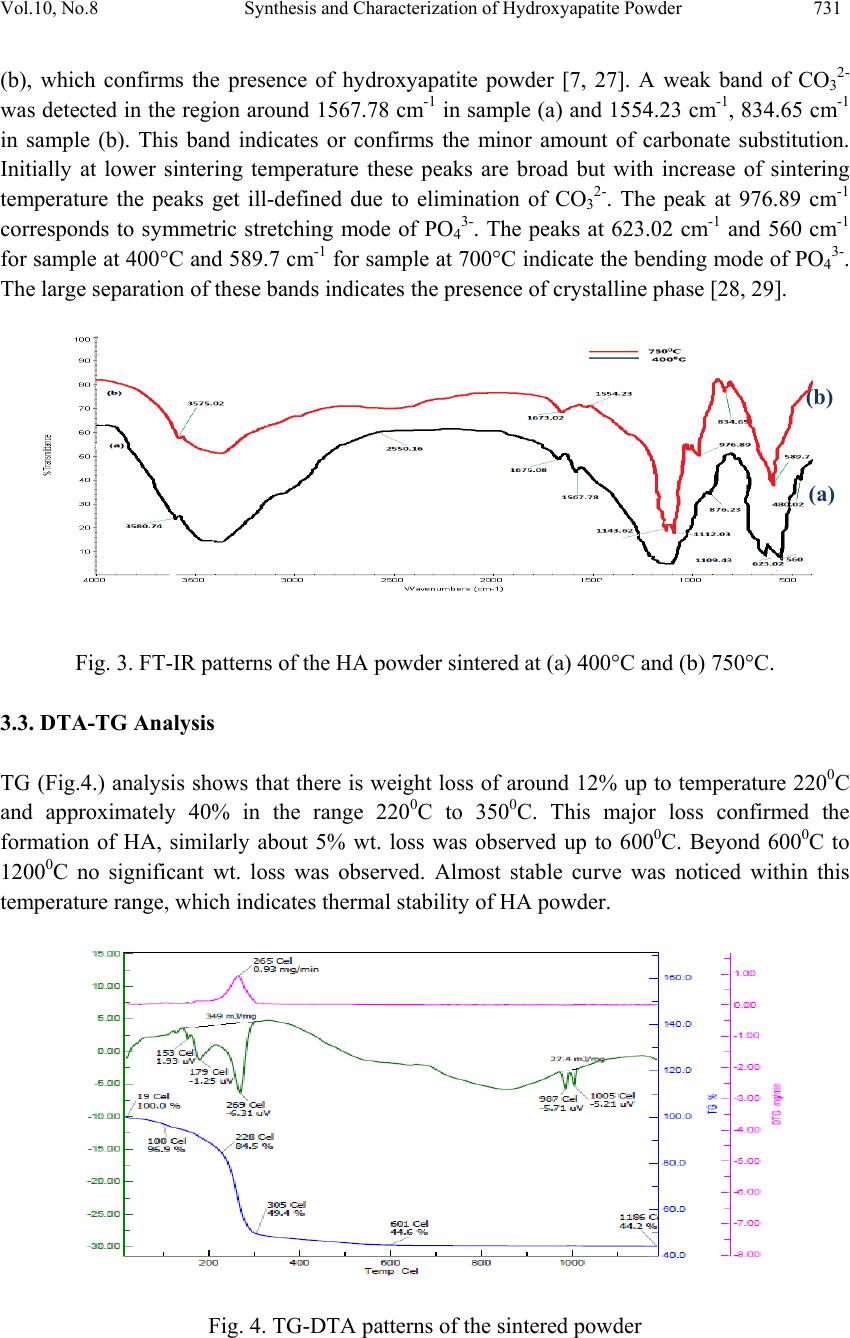 Vol.10, No.8 Synthesis and Characterization of Hydroxyapatite Powder 731 (b), which confirms the presence of hydroxyapatite powder [7, 27]. A weak band of CO32- was detected in the region around 1567.78 cm-1 in sample (a) and 1554.23 cm-1, 834.65 cm-1 in sample (b). This band indicates or confirms the minor amount of carbonate substitution. Initially at lower sintering temperature these peaks are broad but with increase of sintering temperature the peaks get ill-defined due to elimination of CO32-. The peak at 976.89 cm-1 corresponds to symmetric stretching mode of PO43-. The peaks at 623.02 cm-1 and 560 cm-1 for sample at 400°C and 589.7 cm-1 for sample at 700°C indicate the bending mode of PO43-. The large separation of these bands indicates the presence of crystalline phase [28, 29]. Fig. 3. FT-IR patterns of the HA powder sintered at (a) 400°C and (b) 750°C. 3.3. DTA-TG Analysis TG (Fig.4.) analysis shows that there is weight loss of around 12% up to temperature 2200C and approximately 40% in the range 2200C to 3500C. This major loss confirmed the formation of HA, similarly about 5% wt. loss was observed up to 6000C. Beyond 6000C to 12000C no significant wt. loss was observed. Almost stable curve was noticed within this temperature range, which indicates thermal stability of HA powder. Fig. 4. TG-DTA patterns of the sintered powder (a) (b)  732 Khelendra Agrawal, Gurbhinder Singh, Devendra Puri, Satya Prakash Vol.10, No.8 In DTA curve initially there are series of small curves occur which is followed by a broad curve between approx. (2150C to 3300C). This is occurring because evaporation of water in calcium nitrate tetrahydrate Ca(NO3)2.4H2O happens. Similarly the other endothermic peaks in the curve related to the removal or addition of other groups during the synthesis of HA powder. However in the starting at 2000C a sharp exotherm indicates the crystallization of HA. 3.4. SEM- Analysis Fig. 5. SEM micrographs of synthesized HA powder sintered at 7500C temperature for 8 hr at (a) 100X (b) 500X The above Figure 5 shows the SEM images of the synthesized powder obtained after heat- treatment at 7500C for 8 h in stagnant air. The powder appears to be of crushed angular shape when observed at 100X in Figure 5(a). Figure 5(b) which was taken at higher magnification reveals single particle of HA is made of agglomeration of nano sized grains. These grains may be agglomerated due to the formation of the gel during the synthesis process. 4. CONCLUSION This study presents an alternative method to form pure, stable, good crystalline nanosized HA powder at low temperature maximum (7500C) as compared to other existing methods where temperature of treatment is more than 8000C to achieve all the above characteristics. Also it was observed that the crystallinity of the synthesis powder can be improved further by increasing the sintering temperature. The nanosized HA powder produced can be highly useful as a bone replacement material. (a) (b)  Vol.10, No.8 Synthesis and Characterization of Hydroxyapatite Powder 733 REFERENCES [1] W. Weng, G. Shen, G. Han, 2000, “Low temperature preparation of hydroxyapatite coatings on titanium alloy by a sol-gel route”, Materials Science Letters, Vol. 19, pp. 2187- 2188. [2] K. Cheng, W. Weng, G. Han, P. Du, G. Shen, J. Yang, J.M.F. Ferreira, 2003, “The effect of triethanolamine on the formation of sol–gel derived fluoroapatite/hydroxyapatite solid solution”, Journal of Materials Chemistry and Physics, Vol. 78, pp. 767-771. [3] R.E. Riman, W.L. Suchanek, K. Byraopa, C-W. Chen, P. Shuk, C.S. Oakes, 2002, “Solution synthesis of hydroxyapatite designer particulates”, Solid State Ionics, Vol. 151, pp. 393- 402. [4] L-Y. Huang, K-W. Xu, J. Lu, 2000, “A study of the process and kinetics of electrochemical deposition and the hydrothermal synthesis of hydroxyapatite coatings”, Journal of Materials Science: Materials in Medicine, Vol. 11, pp. 667-673. [5] W. Weng, S. Zhang, K. Cheng, H. Qu, P. Du, G. Shen, J. Yuan, G. Han, 2003, “Sol– gel preparation of bioactive apatite films”, Surface and Coatings Technology, Vol. 167, pp. 292-296. [6] W. Weng, G. Han, P. Du, G. Shen, 2002, “The effect of citric acid addition on the formation of sol–gel derived hydroxyapatite”, Materials Chemistry and Physics, Vol. 74, pp. 92-97. [7] D. Choi, K. Marra, P.N. Kumta, 2004, “Chemical synthesis of hydroxyapatite/poly (caprolactone) composite”, Materials Research Bulletin, Vol. 39, pp. 417-432. [8] W. Weng, J.L. Baptista, 1998, “Alkoxide route for preparing hydroxyapatite and its coatings”, Biomaterials, Vol. 19, pp. 125-131. [9] K. Cheng, W. Weng, G. Han, P. Du, G. Shen, J. Yang, J.M.F. Ferreira, 2003, “Sol–gel derived fluoridated hydroxyapatite films”, Materials Research Bulletin, Vol. 38, pp. 89-97. [10] K. Cheng, G. Shen, W. Weng, G. Han, J.M.F. Ferreira, J. Yang, 2001, “Synthesis of hydroxyapatite /fluoroapatite solid solution by a sol-gel method”, Materials Letters, Vol. 51, pp. 37- 41. [11] W. Weng, G. Han, P. Du, G. Shen, J. Yang, 2002, “The effect of citric acid addition on sol–gel preparation of apatite films”, Materials Chemistry and Physics, Vol. 77, pp. 578-582. [12] V. Shiny, P. Ramesh, M.C. Sunny, H.K. Varma, 2000, “Extrusion of hydroxyapatite to clinically significant shapes”, Materials Letters, Vol. 46, pp. 142-146. [13] T. Kokubo, H.M. Kim, M. Kawashita, 2003, “Novel bioactive materials with different mechanical properties”, Biomaterials, Vol. 24, pp. 2161-2175. [14] L.J. Jha, S.M. Best, J.C. Knoles, I. Rehman, J.D. Santos, W. Bonfield, 1997, “Preparation and characterization of fluoride-substituted apatites”, Journal of Materials Science: Materials in Medicine, Vol. 8, pp. 185-191.  734 Khelendra Agrawal, Gurbhinder Singh, Devendra Puri, Satya Prakash Vol.10, No.8 [15] Y.C. Tsui, C. Doyle, T.W. Clyne, 1998, “Plasma sprayed hydroxyapatite coatings on titanium substrates Part 1: Mechanical properties and residual stress levels”, Biomaterials, Vol. 19, pp. 2015-2029. [16] W. Weng, J.L. Baptista, 1997, “A new synthesis of hydroxyapatite”, Journal of the European Ceramic Society, Vol. 17, pp. 1151-1156. [17] Dean-Mo Liu, T. Troczynski, Wenjea J. Tseng, 2001, “Water-based sol-gel synthesis of hydroxyapatite: process development”, Biomaterials, Vol. 22, pp. 1721-1730. [18] W. Suchanek, M. Yoshimura, 1998, “Processing and properties of hydroxyapatite- based biomaterials for use as hard tissue replacement implants”, Journal of Materials Research, Vol. 13, pp. 94-117. [19] J.C. Elliott, 1994, “Structure and Chemistry of the Apatites and Other Calcium Orthophosphates”, Elsevier Science, The Netherlands [20] M. Toriyama, A. Ravaglioli, A. Krajewski, G. Gelotti, A. Piancastelli, 1997, “Synthesis of hydroxyapatite-based powders by mechano-chemical method and their sintering”, Journal of the European Ceramic Society, Vol. 16, pp. 429-436. [21] M. Otsuka, Y. Matsuda, J. Hsu, J. Fox, W. Higuchi, 1994, “Mechanochemical synthesis of bioactive material: Effect of environmental conditions on the phase transformation of calcium phosphates during grinding”, Bio-Medical Materials and Engineering, Vol. 4, pp. 357-362. [22] U. Partenfelder, A. Engel, C. Russel, 1993, “A pyrolytic route for the formation of hydroxyapatite/fluoroapatite solid solutions”, Journal of Materials Science: Materials in Medicine, Vol. 4, pp. 292-295. [23] C.J. Brinker, G.W. Scherer, 1990, “Sol–Gel Science”, Academic Press, Boston, pp. 787. [24] Haddow DB, James PF, Van Noort R, 1998, “Sol-gel derived calcium phosphate coatings for biomedical applications”, Journal of Sol-Gel Science and Technology, Vol. 13, pp. 261-265. [25] Chai CS, Ben-Nissan B, Pyke S, Evans L, 1995, “Sol-gel derived hydroxyapatite coatings for biomedical applications”, Materials and Manufacturing Processes, Vol. 10, pp. 205-216. [26] Il-Seok kim, Prashant N.Kumata, “Sol-gel synthesis and characterization of nanostructured hydroxyapatite powder”, Materials science and engineering B, Vol. 111, pp. 232-236, 2004. [27] W. Weng, J.L. Baptista, 1998, “Sol-gel derived porous hydroxyapatite coatings”, Journal of materials science: materials in medicine”, Vol. 9, pp. 159-163. [28] LeGeros, R.Z.,“Calcium Phosphate in Oral Biology and Medicine”, Karger: Basel, 1991. [29] Tas, A.C, 2000, “Synthesis of biomimetic Ca hydroxyapatite powders at 370C in synthetic body fluids”, Biomaterials, Vol. 21, pp. 1429. |