 Vol.2, No.6, 535-543 (2010) Natural Science http://dx.doi.org/10.4236/ns.2010.26067 Copyright © 2010 SciRes. OPEN ACCESS Phylogeny of γ-proteobacteria inferred from comparisons of 3’ end 16S rRNA gene and 5’ end 16S-23S ITS nucleotide sequences Sabarimatou Yakoubou1,2, Jean-Charles Côté1* 1Agriculture and Agri-Food Canada, Research Centre, Gouin Blvd, St-Jean-sur-Richelieu, Québec, Canada; *Corresponding Author: Jean-Charles.Cote@agr.gc.ca 2Département des Sciences Biologiques, Université du Québec à Montréal, CP 8888, Succ. “Centre-Ville” Montréal, Québec, Canada Received 27 January 2010; revised 25 March 2010; accepted 12 April 2010. ABSTRACT The phylogeny of γ-proteobacteria was inferred from nucleotide sequence comparisons of a short 232 nucleotide sequence marker. A total of 64 γ-proteobacterial strains from 13 Orders, 22 families, 40 genera and 59 species were analyz- ed. The short 232 nucleotide sequence marker used here was a combination of a 157 nucleot- ide sequence at the 3’ end of the 16S rRNA gene and a 75 nucleotide sequence at the 5’ end of the 16S-23S Internal Transcribed Spacer (ITS) sequence. Comparative analyses of the 3’ end of the 16S rRNA gene nucleotide sequence sho- wed that the last 157 bp were conserved among strains from same species and less conserved in more distantly related species. This 157 bp sequence was selected as the first part in the construction of our nucleotide sequence marker. A bootstrapped neighbor-joining tree based on the alignment of this 157 bp was constructed. This 157 bp could distinguish γ-proteobacterial species from different genera from same family. Closely related species could not be distingu- ished. Next, an alignment of the 16S-23S ITS nucleotide sequences of alleles from same bac- terial strain was performed. The first 75 bp at the 5’ end of the 16S-23S ITS was highly conserved at the intra-strain level. It was selected as the second part in the construction of our nucleotide sequence marker. Finally, a bootstrapped neigh- bor-joining tree based on the alignment of this 232 bp sequence was constructed. Based on the topology of the neighbour-joining tree, four maj- or Groups, Group I to IV, were revealed with sev- eral sub-groups and clusters. Our results, bas- ed on the 232 bp sequence were, in general, in agreement with the phylogeny of γ-proteobact- eria based on the 16S rRNA gene. The use of this 232 bp sequence as a phylogenetic marker presents several advantages over the use of the entire 16S rRNA gene or the generation of exten- sive phenotypic and genotypic data in phyloge- netic analyses. First, this marker is not allele-de- pendant. Second, this 232 bp marker contains 157 bp from the 3’ end of the 16S rRNA gene and 75 bp from the 5’ end of the 16S-23S ITS. The 157 bp allows discrimination among distantly related species. Owing to its higher rate of nucl- eotide substitutions, the 75 bp adds discrimina- ting power among closely related species from same genus and closely related genera from same family. Because of its higher percentage of nucleotide sequence divergence than the 16S rRNA gene, the 232 bp marker can better discri- minate among closely related γ-proteobacterial species. Third, the method is simple, rapid, suit- ed to large screening programs and easily acce- ssible to most laboratories. Fourth, this marker can also reveal γ-proteobacterial species which may appear misassigned and for which additi- onal characterization appear warranted. Keywords: γ-Proteobacteria; 16S rRNA; 16S-23S ITS; Phylogeny 1. INTRODUCTION The phylum proteobacteria or ‘‘purple bacteria and their relatives’’ encompasses bacteria with a wide variety of phenotype and physiological attributes and habitats [1,2]. Proteobacteria have been classified based on the homo- logy of 16S ribosomal RNA or by hybridization of ribo- somal DNA with 16S and 23S ribosomal RNA [3-6]. They have been subdivided in five major classes: α-, β-, γ-, δ- and ε- [7-9]. Most γ-proteobacteria are Gram-negative. This class  S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 536 comprises 14 Orders and more than 200 genera. The γ-proteobacteria exhibit a wide range of metabolic diver- sity. Most are chemo-organotrophs, some are phototro- phs or chemolitotrophs [1,2,10,11]. This class includes several medically and scientifically important bacteria. Some genera are human (Klebsiella, Shigella, Salmon- ella, Yersina, Vibrio), animal (Pasteurella) or plant path- ogens (Pseudomonas, Xanthomonas , Xylella). Others are obligate endosymbionts (Buchnera, Soda lis, Wiggleswo- rthia and Coxiella) [10-12]. Because of their biological importance, γ-proteobacteria are extensively studied. The 16S ribosomal RNA (rRNA) gene has been est- ablished as the macromolecule of choice for phylogen- etic analyses [5,13]. The current phylogeny of γ-proteo- bacteria is based on the homology of 16S rDNA nucleo- tide sequences [3-6,11,14]. The 16S-23S internal transcribed spacer (ITS) region is more variable than the 16S rRNA gene. It has been used, among others, in the study of specific γ-proteoba- cterial diversity at the species level, including Escheri- chia, Ha emophilus , Xanthomonas, Klebsiella and Pseud- omonas [15-19]. Additional approaches, based on different genes, have been used for the study of γ-proteobacterial phylogeny [12,20-25]. Very recently, Gao et al., [26] have used a combination of phylogenomic and comparative genomic approaches to reconstruct the phylogeny of γ-proteob- acteria. In an earlier work on the bacterial Gram-positive Ba- cillus genus and related genera [27], a short 220 bp nu- cleotide sequence “marker” was used to reconstruct their phylogeny. This 220 bp marker was a combination of a 150 bp sequence at the 3’ end of the 16S rRNA gene and a 70 bp sequence at the 5’ end of the 16S-23S ITS se- quence. Owing to its higher rate of nucleotide substitu- tion, the 70 bp sequence at the 5’ end of the 16S-23S ITS sequence added a greater discriminatory power among closely related species than 16S rRNA gene nucleotide sequences alone. They showed that the phylogeny in- ferred from the 220 bp marker was in agreement with then current classifications based on phenetic and mo- lecular data. The marker also indentified species which appeared misassigned. It also created new clusters sug- gesting the creation of new taxa levels. In a very recent study, we [28] have tested whether or not this marker could reconstruct the phylogeny of the bacterial Gram- positive Order of the Bacilla les. In the current study, we further assess the usefulness of a similar marker among 13 of the 14 γ-proteobacterial Orders. The last 157 bp at the 3’ end of the 16S rRNA gene was combined with the first 75 bp at the 5’ end of the 16S-23S Internal Transcribed Spacer (ITS) to yield a single 232 bp DNA marker. This marker was used to reconstruct the phylogeny of γ-proteobacteria. A total of 64 γ-proteobacteria from 13 Orders, 22 families, 40 gen- era and 59 species was analyzed. 2. MATERIALS AND METHODS 2.1. Bacterial Species and Strains A total of 64 γ-proteobacterial species and strains were analyzed. They were selected on the basis that their com- plete genome sequences were freely available in Gen- Bank, at the National Center for Biotechnology Informa- tion (NCBI) completed microbial genomes database (http://www.ncbi.nlm.nih.gov/genomes/MICROBES/Co mplete.html). They encompassed 13 Orders, 22 families, 40 genera and 59 species. These 13 γ-proteobacterial Or- ders included six Aeromonadales families, one Cardiob- acteriales family, two Chromatiales families, one Enter- obacteriales family, two Legionellales families, one Me- thylococcales family, two Oceanospirillales families, one Pasteurellales family, two Pseudomonadales fami- lies, two Thiotrichales families, one Vibrionales family and one Xanthomonada les family. All bacterial species and strains and the GenBank accession number for their fully sequenced genome are listed in Table 1. 2.2. Sequences Analysis The 16S rRNA gene nucleotide sequences were retriev- ed from GenBank (Table 1) for the 64 γ-proteobacteria species and strains under study. First, all 64 sequences were aligned using ClustalW [29] (data not shown). Next, the 3’ end of the 16S rRNA gene nucleotide sequ- ences of alleles from same bacterial strain, of alleles from different strains from same species, and of alleles from different species from same genus (data not shown) were aligned using ClustalW [29]. The length of the nucleotide sequence most conserved was determined at 157 bp. Likewise, the 16S-23S Internal Transcribed Spacer (ITS) nucleotide sequences of alleles from same bacterial strain were also aligned using ClustalW. The length of the nucleotide sequence most conserved was determined at 75 bp. These two most conserved nucleotide sequences, the 157 bp at the 3’ end of 16S, and the 75 bp at the 5’ end of 16S-23S ITS were combined into a single 232 bp seq- uence for each bacterial species and strain under study. This 232 bp sequence will be used here as a phylogen- etic marker for the γ-proteobacteria under study. 2.3. Phylogenetic trees Two neighbor-joining trees were constructed [30], a first one based on the alignment of the last 157 bp at the 3’ end of the 16S rRNA gene described above, a second  S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 537 537 one based on the alignment of the 232 bp sequence also described above. Both trees were bootstrapped using 1,000 random samples of sites from the alignment, all using CLUSTAL W [29] at the DNA Data Bank of Jap- an (DDBJ) (http://clustalw.ddbj .nig.ac.jp/top-e.html ), with the Kimura’s parameter method [31]. The neighbor- joining tree was drawn using TreeView (version 1.6.6) [32,33]. Table 1. γ-proteobacteria strains used in this study. Orders, Families, Genera, Species Strain/Source GenBank Accession no. Orders, Families, Genera, Species Strain/Source GenBank Accession no. Aeromonadales Aeromonodaceae Aeromonas hydrophyla Aeromonas salmonida ATCC 7966 A449 NC_008570.1 NC_009348.1 Shigella flexineri Sodalis glossinidus Wigglesworthia glossinidia Yersinia enterocolitica Yersinia pestis Yersinia pseudotuberculosis 2457T morsitans 8081 CO92 IP31758 NC_004741.1 NC_007712.1 NC_004344.2 NC_008800.1 NC_003143.1 NC_009708.1 Legionellales Coxiellaceae Coxiella burneti Coxiella burnetii Legionellaceae Legionella pneumophila Legionella pneumophila Dugway 5j108-111 RSA 493 Lens Corby NC_009727.1 NC_002971.3 NC_006369.1 NC_009494.1 Methylococcales Methylococcaceae Methylococcus capsulatus Bath NC_002977.6 Alteromonadales Alteromonadaceae Marinobacter aquaeolei Saccharophagus degradans Colwelliaceae Colwellia psychrerythrea Idiomarinaceae Idiomarina ihoihiensis Pseudoalteromonadaceae Pseudoalteromonas atlantica Pseudoalteromonas haloplanktis Shewanellaceae Shewanella amazonensis Shewanella denitrificans Shewanella frigidimarina Shewanella oneidensis VT8 2-40 34H L2TR T6c TAC125 SB2B OS217 NCIMB 400 MR-1 NC_008740.1 NC_007912.1 NC_003910.7 NC_006512.1 NC_008228.1 NC_007481.1 NC_008700.1 NC_007954.1 NC_008345.1 NC_004347.1 Cardiobacteriales Cardiobacteriaceae Dichelobacter nodosus VCS1703A NC_009446.1 Pseudomonadales Moraxellaceae Acinetobacter baumannii Acinetobacter sp. Psychrobacter arcticus Psychrobacter cryohalolentis Pseudomonadaceae Pseudomonas fluorescens Pseudomonas syringea ATCC 17978 ADP1 273-4 K5 Pf5 DC3000 NC_009085.1 NC_005966.1 NC_007204.1 NC_007969.1 NC_004129.6 NC_004578.1 Chromatiales Chromatiaceae Nitrosococcus oceani Ectothiorhodospiraceae Alkalilimnicola ehrlichei Halorhodospira halophila ATCC 19707 MLHE-1 SL1 NC_007484.1 NC_008453.1 NC_008789.1 Thiotrichales Francisellaceae Francisella philomiragia Francisella tularensis Piscirickettsiaceae Thiomicrospira crunogena philomiragia horlatica XCL-2 NC_010336.1 NC_007880.1 NC_007520.2 Vibrionales Vibrionaceae Photobacterium profundum Vibrio cholerae Vibrio parahaemolyticus Vibrio vulnificus SS9 N16961 RIMD 2210633 CMCP6 NC_006370.1 NC_002505.1 NC_004603.1 NC_004459.2 Enterobacteriales Enterobacteriaceae Buchnera aphidicola Citrobacter koseri Enterobacter sakazakii Enterobacter sp Escherichia coli Escherichia coli Klebsiella pneumoniae Photorhabdus luminescens Salmonella enterica Salmonella enterica Salmonella enterica Shigella boydii Shigella dysenteriae APS ATCC BAA-895 ATCC BAA-894 638 CFT073 O157:H7 Sakai 342 NTUH-K2044 TT01 Ty2 arizonae Sb227 Sd197 NC_002528.1 NC_009792.1 NC_009778.1 NC_009436.1 NC_004431.1 NC_002695.1 NC_011283.1 NC_012731.1 NC_005126.1 NC_004631.1 NC_010067.1 NC_007613.1 NC_007606.1 Xanthomonadales Xanthomonadaceae Xanthomonas axonopodis Xanthomonas campestris Xylella fastidiosa 306 8004 9a5c NC_003919.1 NC_007086.1 NC_002488.3  S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 538 3. RESULTS AND DISCUSSION In a previous study [27], on the bacterial genus Bacillus and closely-related genera, we reported the development of a short DNA marker that could be used to reconstruct their phylogeny. This marker was a combination of the last 150 bp at the 3’ end of the 16S rRNA gene and the first 70 bp at the 5’ end of the 16S-23S rRNA internal transcribed spacer (ITS) into a single 220 bp “marker”. It could cluster Bacillus species and species from closely related genera into taxa akin to genera and could also distinguish closely related species. The 3’ end of the 16S rRNA gene contained three regions that were known to be highly conserved among bacteria [34]. The 5’ end of the 16S-23S rRNA ITS was conserved among alleles from same strains [27]. In the current study on γ-proteobacteria, we further assessed the usefulness of this marker. The sizes of the 3’ end of the 16S rRNA gene and the 5’ end of the 16S-23S rRNA ITS retained here for the construction of our phylogenetic marker for γ-proteobacteria are slightly different at 157 and 75 bp, respectively, for a total marker size of 232 bp. These sizes were selected as fol- lows: first, an alignment of the 16S rRNA gene nucleo- tide sequence of alleles from same strain showed that these sequences were highly conserved. The intra-strain alleles shared 99% nucleotide sequence identities. Al- leles from species from same genus, however, covered a wider spectrum of nucleotide sequence identities. Where- as alleles from Pseudomonas (Ps.) fluorescens Pf-5 and from Ps. syringae pv. tomato strain DC3000 share 98% nucleotide sequence identities, alleles from Vibrio (V.) cholerae and from V. parahaemolyticus RIMD 2210633 share 92% nucleotide sequence identities. Comparative analyses of the 3’ end of the 16S rRNA gene nucleotide sequence showed that the last 157 bp were in many cases highly conserved among strains from same species. This is exemplified by Salmonella (Sal.) enterica arizonae and Sal. enterica Ty2 which share 99% nucleotide se- quence identities over the last 157 bp at the 3’ end of the 16S rRNA gene. Species from same genus share lower nucleotide sequence identities. This is exemplified by Shewanella (She) amazonensis and She. denetrificans which share 94% nucleotide sequence identities over the last 157 bp at the 3’ end of the 16S rRNA gene. A bootstrapped neighbor-joining tree based on the align- ment of this 157 bp located at the 3’ end of the 16S rRNA gene was constructed (Figure 1). Although, in most cases, this 157 bp could distinguish species from different genera from same family, in some cases, close- ly related species from different genera from same fam- ily appeared undistinguishable. This is the case for Mann- heimia succiniciproducens and both Actinobacillus (Act), species, Act. pleuropneumonia and Act. succinogenes. This is also true for Xylella (Xy.) fastidiosa and both Xanthomonas ( X.) species, X. axonopodis pv. citri str. 306 and X. campestris pv. campestris str.8004. And this is true for the human and animal pathogenic Entero- bacteriaceae: the Yersinia, Shigella, Kleb siella, Escheri- chia, Enterobacter and Citrobacter species. In some cases, this 157 bp could distinguish species from same genus as exemplified by the Psychrobacter, Pseudomo- nas, Shewan ella and Vibrio species. In other cases, closely related species from same genus could not be distinguished as exemplified by the Francisella , Aero- monas, Actinobacillus, Haemophilus, Acinetobacter and Xanthomonas species. In all cases, this 157 bp could distinguish species from different families, with one exception: She. amazonensis and Photobacterium (Ph.) profundum, members of the Shewanellacea and Vibrionacea family, respectively. Both appear undistinguishable. Clearly, this 157 bp seq uence cannot distinguish closely-related species. An ad- ditional DNA sequence appears necessary to better disti- nguish closely-related species. Next, an alignment of the 16S-23S ITS nucleotide se- quences of alleles from same bacterial strain was carried with a subset of the bacteria under study: Xanthomonas campestris pv. campestris str. 8004, Ps. syringae pv. tomato str. DC3000, Act. succinogenes 130Z, E. coli K12, V. parahaemolyticus RIMD 2210633 and Shigella flexneri 2a str. 301 (Figure 2). The total number of al- leles vary from two to ten for X. campestris pv. campes- tris str. 8004 and V. parahaemolyticus RIMD 2210633, respectively. The allelic sequences were highly ho- mologous for some species and highly hererologous for others. Xanthomonas campestris pv. campestris str. 8004 and Ps. syringae pv. tomato str. DC3000 carry two and five identical alleles, respectively. Actinoba cillus suc- cinogenes 130Z, E. coli K12, V. parahaemolyticus RIMD 2210633 and Sh igella flexneri 2a str. 301 carry five, eight, ten and six alleles respectively, with varying level of heterogeneity, where highly homologous alleles are grouped together and can be distinguised from dif- ferent alleles in same strain (Figure 2). Alleles carry from zero to four tRNA genes. An alignment of the nu- cleotide sequences among alleles at the intra-strain level required the introduction of several gaps. The first 75 bp at the 5’ end of the 16S-23S ITS, however, was highly conserved at the intra-strain level. It was retained here for the construction of our phylogenetic marker. The two conserved nucleotide sequences identified above, the 157 bp at the 3’ end of 16S rRNA gene and the 75 bp at the 5’ end of 16S-23S ITS, were combined into a single 232 bp sequence. This will be used here as a phyloge- netic marker for the γ-proteobacteria under study.  S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 539 539 Buchnera aphidicola Thiomicrospira crunogena Pseudoalteromonas atlantica Pseudoalteromonas haloplanktis Francisella philomiragia Francisella tularensis 98 52 eromonas salmonicida eromonas hydrophilaMannheimia succiniciproducens ctinobacillus pleuropneumonia ctinobacillus succinogenes 65 65 Haemophilus ducrey Haemophilus influenzae Pasteurella multocida 57 59 81 Shewanella amazonensis Photobacterium profundum 85 Methylococcus capsulatu lcanivorax borkumensis Psychrobacter cryohalolentis Psychrobacter arcticu 94 Idiomarina ioihiensis Chromohalobacter salexigens Coxiella burnetii RSA493 Coxiella burnetii Dugway 5J108-111 100 cinetobacter baumanni cinetobacter sp ADP1 98 Dichelobacter nodosu ylella fastidiosa anthomonas axonopodis anthomonas campestris 88 88 65 60 Nitrosococcus oceani lkalilimnicola ehrliche Halorhodospira halophila Marinobacter aquaeole Legionella pneumophila str.Corby Legionella pneumophila str.Lens 89 Pseudomonas fluorescen Pseudomonas syringae Saccharophagus degradans 59 64 57 Salmonella enterica Photorhabdus luminescens Wigglesworthia glossinidia Shewanella oneidensis Colwellia psychrerythraea Shewanella denitrificans Shewanella frigidimarina Vibrio vulnificus 55 Vibrio cholerae Vibrio parahaemolyticus Enterobacter sakazaki Citrobacter koser Enterobacter sp Escherichia coli. Klebsiella pneumoniae Shigella boydi Shigella dysenteriae Shigella flexneri Yersinia enterocolitica Yersinia pestis Sodalis glossinidius Yersinia pseudotuberculosis 51 0.01 Figure 1. Bootstrapped neighbor-joining tree of γ –proteobacteria species inferred from the alignment of a 157 nucleotide sequence at the 3’ end of the 16S rRNA gene. Bootstrap values higher than 50% are indicated (expressed as percentage of 1000 replication). The horizontal bar represents 1% nucleotide difference. A bootstrapped neighbor-joining tree based on the alignment of a 232 bp sequence was constructed (Figure 3). Based on the topology of the neighbor-joining tree, four major Groups, Group I to IV are revealed (Figure 3). Based on nucleotide sequence identities, sub-groups and clusters can be formed. Group I contains seven Ord- ers and nine families. Group II contains eight Orders and eleven families. Group III contains three Orders and five families. Group IV contains one Order and one family. Of the 13 Orders under study, four are present in more than Group one. The Thio trichales are present in Groups I and III. The Alteromonadales are present in Groups I, II and III. The Legionellales are present in Groups I and II. The Vibrionales are present in Groups I and III. All other nine Orders are present in a single Group. Of the 22 families under study, four are present in more than one Group. The Alteromon adaceae are present in Groups I and II. The Pseudoalteromonadaceae (Pse) are present in Groups I and III. The Vibrionaceae are pre- sent in Groups I and III. The Shewanellaceae are present in Groups I and III. All other 18 families are present in a single Group. All species from same genus are present in  S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 540 same Group with the exception of Pse. haloplanktis and Pse. atlantica present in Group I and III, respectively. Group I can be sub-divided into six sub-groups, sub- group I-1 to I-6. Sub-group I-1 contains both species of the Francisellaceae family. Sub-group I-2 contains three species, members of two families: Pseudomonadacea e and Alteromonadaceae. Sub-group I-3 comprises Pse. haloplanktis and both Legionella pneumophila strains. Although in the same sub-group, Pse. haloplanktis sh- ows 20% nucleotide sequence divergence with the two Legionella strains. Both Legionella strains are tightly grouped together and form cluster I-3-1. Sub-group I-4 contains Ph. profundum and She. amazonensis, two spe- cies from two different families, Vibrionaceae and She- wanellaceae, respectively. Although they appear very si- milar on the neighbor-joining tree, both sequences show 15% nucleotide divergence. Sub-group I-5 contains both Aeromonas species, tightly grouped together. Sub-group I-6 contains all six Pasteurellaceae species. The Mann- heimia species is tightly grouped with the two Actino- bacillus species and form cluster I-6-1. Both Haemop- hilus species form cluster I-6-2. In Group I, clusters co- mprised species from same genus or closely related spe- cies from different genera from same family. All other branches corresponded to families. Group II can be sub-divided into four sub-groups, sub-group II-1 to II-4, and one ungrouped species, Me- thylococcus capsulatus. Sub-group II-1 comprises mem- bers of four families from four Orders. Members of sub-group II-1 share up to 30% nucleotide sequence di- vergences. Closely related species can be further gro- uped together. This is the case for the two Acinetobacter species and all three Xanthomonadaceae species which form cluster II-1-1 and II-1-2, respectively. Sub-group II-2 contains Nitrosoccocus oceani and Halorhodospira halophila, member of the Chromatiaceae and Ectothior- hodospira cea family respectively. Both species show 42% nucleotide sequence divergences. Both families belong to the Chromatiales Order. Sub-group II-3 con- tains six species, from five families and three Orders. Members of sub-group II-3 share up to 24% nucleotide sequence divergences. Two closely related species, Psy- chrobacter (Psy) cryohalolentis and Psy. arcticus form cluster II-3-1. Sub-group II-4 comprises both strains of Coxiella burnetti, members of the Coxelliacea e family. In Group II, clusters comprised species from same genus or closely related species from different genera from same family. All other branches corresponded to families. Pseudomonas syringae pv. tomato str. DC3000 PSTPTO _R01 PSTPTO _R04 PSTPTO _R07 PSTPTO _R02 PSTPTO _R015 Xanthomonas campestris pv. campestris str. 8004 XC_4386 XC_4393 rrsA rrsH rrrsC rrsE 100 200 300400 500 600 700800 850 Asuc_R 0 01 7 Asuc_R 0 04 9 Asuc_R0019 Asuc_R0035 Asuc_R0054 Actinobacillus succinogenes 130Z Shigella flexneri 2astr. 301 Vibrio paraheaemolyticus RIMD 2210633 Vpr003 Vpr006 Vpr001 Vpr007 Vpr005 Vpr004 Vpr002 Vpr022 Vpr025 Vpr019 rrsA rrsB rrsC rrsE rrrsH rrrsG rrrsB rrrsD rrrsG Escherichia coli K12 substr. MG 1655 tRNAIle tRNA AlatRNAGlutRNA LystRNA Val Figure 2. Schematic representation of allelic 16S-23S rDNA Internal Transcribed Spacer of γ –prot- eobacteria.The non filed boxes represent regions of homologus nucleotide sequences between allelic ITS of the same bacteria. Filed boxes represent tRNA. The blank spaces between boxes represent non conservation regions between allelic ITS of the same bacteria. 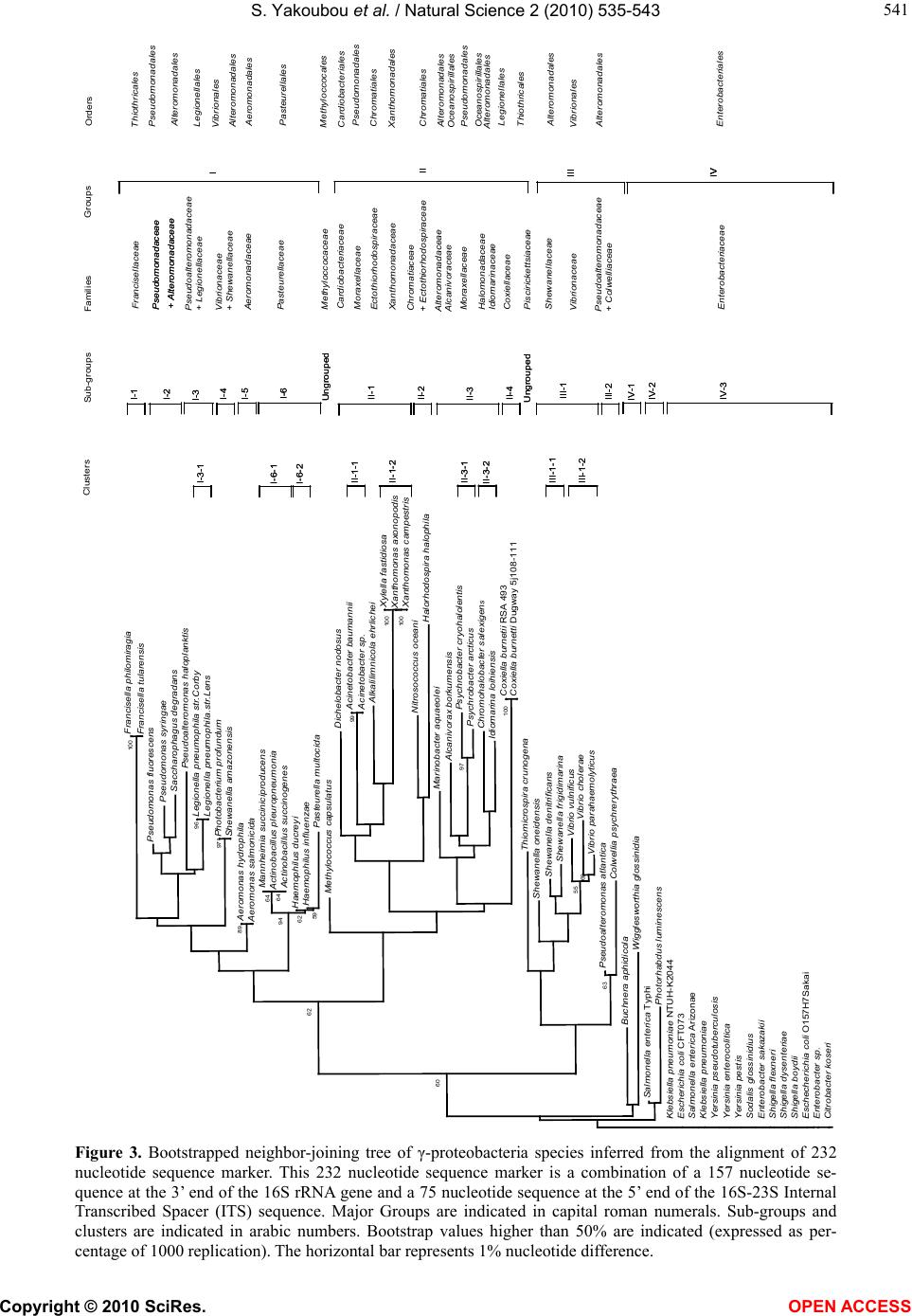 S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 541 541 Francisella philomiragia Francisella tularensis 100 Pseudomonas fluorescens Pseudomonas syringae Saccharophagusdegradans Pseudoalteromonashaloplanktis Legionella pneumophila str.Corby Legionella pneumophila.str.Lens 96 Photobacterium profundum Shewanella amazonensis 971 Aeromonashydrophila Aeromonassalmonicida 89 Mannheimia succiniciproducens Actinobacillus pleuropneumonia Actinobacillus succinogenes 64 64 Haemophilusducreyi Haemophilusinfluenzae Pasteurella multocida 59 62 94 Methylococcus capsulatus Dichelobacter nodosus Acinetobacter baumannii Acinetobacter sp. 99 Alkalilimnicolaehrlichei Xylella fastidiosa Xanthomonas axonopodis Xanthomonas campestris 100 100 Nitrosococcus oceani Halorhodospirahalophila Marinobacter aquaeolei Alcanivoraxborkumensis Psychrobactercryohalolentis Psychrobacter arcticus 97 Chromohalobacter salexigen s Idiomarina loihiensis Coxiella burnetii RSA 493 100 62 Thiomicrospira crunogena Shewanella oneidensis Shewanella denitrificans Shewanella frigidimarina Vibrio vulnificus Vibrio cholerae Vibrio parahaemolyticus 69 55 Pseudoalteromonasatlantica Colwellia psychrerythraea 63 60 Buchnera aphidicola Wigglesworthia glossinidia Salmonella enterica Typhi Photorhabdusluminescens Klebsiella pneumoniae NTUH-K2044 Escherichia coli CFT073 Salmonella enterica Arizonae Klebsiella pneumoniae Yersinia pseudotuberculosis Yersinia enterocolitica Yersinia pestis Sodalis glossinidius Enterobactersakazakii Shigella flexneri Shigella dysenteriae Shigella boydii Eschecherichia coli O157H7Sakai Enterobactersp. Citrobacter koseri Sub-groups Families Groups Clusters Orders Francisellaceae Pseudomonadaceae + Alteromonadaceae Pseudomonadaceae + Alteromonadaceae Pseudoalteromonadaceae + Legionellaceae Vibrionaceae + Shewanellaceae Aeromonadaceae Pasteurellaceae Cardiobacteriaceae Methyloccocaceae Moraxellaceae Ectothiorhodospiraceae Xanthomonadaceae Chromatiaceae + Ectothiorhodospiraceae Alteromonadaceae Alcanivoraceae Moraxellaceae Halomonadaceae Idiomarinaceae Coxiellaceae Piscirickettsiaceae Shewanellaceae Vibrionaceae Pseudoalteromonadaceae + Colwelliaceae Enterobacteriaceae Thiothricales Pseudomonadales Alteromonadales Legionellales Vibrionales Alteromonadales Aeromonadales Pasteurellales Meth yloc coc al es Cardiobacteriales Pseudomonadales Chromatiales Xanthomonadales Chromatiales Alteromonadales Oceanospirillales Pseudomonadales Oceanospirillales Alteromonadales Legionellales Alteromonadales Thiothricales Vibrionales Enterobacteriales Alteromonadales II I III IV II I IIIIII IVIV I-1 I-2 I-3 I-4 I-5 I-6 Ungrouped II-1 II-2 II-3 II-4 Ungrouped III-1 III-2 IV-2 IV-1 IV-3 I-1 I-2 I-3 I-4 I-5 I-6 Ungrouped II-1 II-2 II-3 II-4 Ungrouped III-1 III-2 IV-2 IV-1 IV-3 I-6-1 I-6-2 II-1-1 II-1-2 II-3-1 III-1-1 III-1-2 II-3-2 I-3-1 I-6-1 I-6-2 II-1-1 II-1-2 II-3-1 III-1-1 III-1-2 II-3-2 I-3-1 Coxiella burnetti Dugway5j108-111 Figure 3. Bootstrapped neighbor-joining tree of γ-proteobacteria species inferred from the alignment of 232 nucleotide sequence marker. This 232 nucleotide sequence marker is a combination of a 157 nucleotide se- quence at the 3’ end of the 16S rRNA gene and a 75 nucleotide sequence at the 5’ end of the 16S-23S Internal Transcribed Spacer (ITS) sequence. Major Groups are indicated in capital roman numerals. Sub-groups and clusters are indicated in arabic numbers. Bootstrap values higher than 50% are indicated (expressed as per- centage of 1000 replication). The horizontal bar represents 1% nucleotide difference.  S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 542 Group III can be sub-divided into two sub-groups, sub-groups III-1 and III-2, and one ungrouped species, Thiomicrospira crunogena. Sub-group III-1 contains three Shewallenaceae and three Vibrionaceae species. She. denitrificans and She. frigidimarina, and the three Vibrio species form two clusters, III-1-1 and III-1-2, respectively. Sub-group III-2 contains two genera from two families of the same Order. Both species, on two separate branches, show 23% nucleotide sequence di- vergence. In Group III, both clusters comprised species from same genus. All other branches corresponded to families. Group IV contains all the Enterobacteriaceae species under study. Three sub-groups can be revealed: sub- group IV-1 to IV-3. Sub-group IV-1 contains two insect obligate endosymbionts. Sub-group IV-2 contains Salm- onella enterica Ty2 and Photorhabtus luminescens. All other Enterobacteriaceae species are in sub-group IV-3. The latter are closely related to each others. Our results, based on the 232 bp phylogenetic marker described here are, in general, in agreement with the phylogeny of γ-proteobacteria based on the 16S rRNA gene with some exceptions. In the neighbor-joining tree, clusters comprised species from same genus or closely related species from different genera from same family. All other branches corresponded to families. As indi- cated above, of the 22 families under study, 18 are pre- sent in a single Group and four are present in more than one Group. These latter four families encompass the marine bacteria [35]. They are Vibrionaceae, in Groups I and III; and the Alteromonas-related protobacteria (Ivanova et al., 2004): Alteromon adaceae, in Groups I and II; Pseudoalteromon adaceae, in Groups I and III; and Shewanellaceae, in Groups I and III. Interestingly, within a Group, these marine bacteria are found in close proximity of one another. It reflects the varying level of heterogeneity among Alteromonas-related protobacteria. The grouping showed here is based on a 232 bp marker. The biological significance of this grouping is unknown. Clearly, however, the phylogenetic analyses of these related marine heterotrophic bacteria is a work in pro- gress [36]. 4. CONCLUSIONS In conclusion, the use of this 232 bp marker presents several advantages over the use of the entire 16S rRNA gene or the generation of extensive phenotypic and gen- otypic data in phylogenetic analyses. First, this marker is not allele-dependant. The 3’ end of the 16S rRNA gene is highly conserved at the intra-strain level. We have shown here that although the16S-23S ITS allelic se- quences can be very heterogeneous within a strain, the first 75 bp, however, are conserved among alleles from same strain in γ-proteobacteria. Clearly, any allele would generate the same results. Second, this 232 bp marker contains 157 bp from the 3’ end of the 16S rRNA gene and 75 bp from the 5’ end of the 16S-23S ITS. The 157 bp is highly conserved among closely related species. Owing to its higher rate of nucleotide substitutions, the 75 bp adds discriminating power among closely related species from same genus and closely related genera from same family. Because of its higher percentage of nucleo- tide sequence divergence than the 16S rRNA gene, the 232 bp marker can better discriminate among closely related γ-proteobacteria species. Third, the method is simple, rapid, suited to large screening programmes and easily accessible to most laboratories. More importantly, however, this 232 bp marker can group γ-proteobacteria families and genera in accordance with established phy- logenies, with the exceptions indicated above. It can also reveal γ-proteobacteria species which may appear mis- assigned and for which additional characterization app- ear warranted. REFERENCES [1] Stackebrandt, E., Murray, R.G.E. and Trüper, H.G. (1988) Proteobacteria classis nov., a name for the phylogenetic taxon that includes the “purple bacteria and their rela- tives”. International Journal of Systematic Bacteriology, 38(3), 321-325. [2] Gupta, R.S. (2000) The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiology Reviews, 24(4), 367-402. [3] Fox, G.E., Stackebrandt, E., Hespell R.B., Gibson J., Maniloff, J., Dyer, T.A., Wolfe, R.S., Balch, W.E., Tanner, R.S., Magrum, L.J., Zablen, L.B., Blakemore, R., Gupta, R., Bonen, L., Lewis, B.J., Stahl, D.A., Luehrsen, K.R., Chen, K.N. and Woese, C.R. (1980) The phylogeny of prokaryotes. Science, 209(4455), 457-463. [4] Woese, C.R., Stackebrandt, E., Macke, T.J. and Fox, G. E. (1985a) A phylogenetic definition of the major eubacterial taxa. Systematic and Applied Microbiology, 6, 143-151. [5] Woese, C.R. (1987) Bacterial Evolution. FEMS Mic- robiology Reviews, 51(2), 221-271. [6] De Ley, J. (1992) The proteobacteria: Ribosomal RNA cistron similarities and bacterial taxonomy. In: by Balows, A., Trüper, H.G., Dworkin, M., Harder, W., and Schleifer, K.H., Eds., The Prokaryotes, 2nd Edition, Springer-Verlag, Berlin, 2111-2140. [7] Woese, C.R., Stackebrandt, E., Weisburg, W.G., Paster, B. J., Madigan, M.T., Fowler, V.J., Hahn, C.M., Blanz, P., Gupta, R., Nealson, K.H. and Fox, G.E. (1984a) The phylogeny of purple bacteria: The alpha subdivision. Systematic and Applied Microbiology, 5, 315-326. [8] Woese, C.R., Weisburg, W.G., Paster, B.J., Hahn, C.M., Tanner, R.S., Krieg, N.R., Koops, H.-P., Harms, H. and Stackebrandt, E. (1984b) The phylogeny of purple bacte- ria: The beta subdivision. Systematic and Applied Micro-  S. Yakoubou et al. / Natural Science 2 (2010) 535-543 Copyright © 2010 SciRes. OPEN ACCESS 543 543 biology, 5, 327-336. [9] Woese, C.R., Weisburg, W.G., Hahn, C.M., Paster, B.J., Zablen, L.B., Lewis, B.J., Macke, T.J., Ludwig, W. and Stackebrandt, E. (1985b) The phylogeny of purple bacte- ria: The gamma subdivision. Systematic and Applied Mi- crobiology, 6, 25-33. [10] Brenner, D.J., Krieg, N.R., Staley, J.T. and Garrity, G.M., (Eds.) (2005) Bergey’s manual of systematic bacteriology, 2nd Edition, Vol. 2 (The P roteobacteria), part C (The Al- pha-, Beta-, Delta-, and Epsilonproteobacteria), Springer, New York. [11] Kersters, K., Devos, P., Gillis, M., Swings, J., Vandamme, P. and Stackebrandt, E. (2006) Introduction to the Proteo- bacteria. In: Dworkin, M., Falkow, S., Rosenberg, E. Schleifer, K.H. and Stackebrandt, E., Eds., The Pro- karyotes: A Handbook on the Biology of Bacteria. Springer, New York, 3-37. [12] Belda, E., Moya, A. and Silva, F.J. (2005) Genome rear- rangement distances and gene order phylogeny in γ-pro- teobacteria. Molecular Biology and Evolution, 22(6), 1456- 1467. [13] Woese, C.R., Kandler, O. and Wheelis, M.L. (1990) To- wards a natural system of organisms: Proposal for the domains archaea, bacteria, and eucarya. Proceedings of the National Academy of Sciences, USA, 87(12), 4576- 4579. [14] Ludwig, W. and Klenk, H.P. (2005) Overview: A phy- logenetic backbone and taxonomic framework for pro- karyotic systematics. In: Brenner, D.J. Krieg, N.R. Staley J.T. and Garrity, G.M., Eds., Bergey’s Manual of System- atic Bacteriology, Springer-Verlag, Berlin, 49-65. [15] Anton, A.I., Martinez-Murcia, A.J. and Rodriguez-Val- era, F. (1998) Sequence diversity in the 16S-23S inter- genic spacer region (ISR) of the rRNA operons in repre- sentatives of the Escherichia coli ECOR collection. Jou- rnal of Molecular Evolution, 47(1), 62-72. [16] Giannino, V., Rappazzo, G., Scuto, A., Di Marco, O., Privitera, A., Santagati, M. and Stefani, S. (2001) rrn op- erons in Haemophilus parainfluenzae and mosaicism of conserved and species-specific sequences in the 16S-23S rDNA long spacer. Research in Microbiology, 152(5), 461-468. [17] Goncalves, E.R. and Rosato, Y.B. (2002) Phylogenetic analysis of Xanthomonas species based upon 16S-23S rDNA intergenic spacer sequences. International Journal of Systematic and Evolutionary Microbiology, 52(Pt 2), 355-361. [18] Wang, M., Cao, B., Yu, Q., Liu, L., Gao, Q., Wang, L. and Feng, L. (2008) Analysis of the 16S-23S rRNA gene internal transcribed spacer region in Klebsiella species. Journal of Clinical Microbiology, 46(11), 3555-3563. [19] Tambong, J.T., Xu, R. and Bromfield, E.S.P. (2009) In- tercistronic heterogeneity of the 16S-23S rRNA spacer region among Pseudomonas strains isolated from subter- ranean seeds of hog peanut (Amphicarpa bracteata). Mi- crobiology, 155(Pt 8), 2630-2640. [20] Kunisawa, T. (2001) Gene arrangements and phylogeny in the class Proteobacteria. Journal of Theoretical Biol- ogy, 213(1), 9-19. [21] Lerat, E., Daubin, V. and Moran, N.A. (2003) From gene trees to organismal phylogeny in prokaryotes: The case of the γ-proteobacteria. Public Library of Science Biol- ogy, 1(1), e19. [22] Brown, J.R. and Volker, C. (2004) Phylogeny of γ –prot- eobacteria: Resolution of one branch of the universal tree? Bioessays, 26(5), 463-468. [23] Ciccarelli, F.D., Doerks, T., von Mering, C., Creevey, C. J., Snel, B. and Bork, P. (2006) Toward automatic recon- struction of a highly resolved tree of life. Science, 311 (5765), 1283-1287. [24] Mrazek, J., Spormann, A.M. and Karlin, S. (2006) Ge- nomic comparisons among γ-proteobacteria. Environm- ental Microbiology, 8(2), 273-288. [25] Lee, H.Y. and Côté, J.-C. (2006) Phylogenetic analysis of γ-proteobacteria inferred from nucleotide sequence com- parisons of the house-keeping genes adk, aroE and gdh: comparisons with phylogeny inferred from 16S rRNA gene sequences. Journal of General and Applied Micro- biology, 52(3), 147-158. [26] Gao, B., Mohan, R. and Gupta, R.S. (2009) Phyloge- nomics and protein signatures elucidating the evolution- ary relationships among the Gammaproteobacteria. In- ternational Journal of Systematic and Evolutionary Mi- crobiology, 59(2), 234-247. [27] Xu, D. and Côté, J.-C. (2003) Phylogenetic relationships between Bacillus species and related genera inferred from comparison of 3’ end 16S rDNA and 5’ end 16S-23S ITS nucleotide sequences. International Journal of Systematic and Evolutionary Microbiology, 53(Pt 3), 695-704. [28] Yakoubou, S., Xu, D. and Côté, J.-C. Phylogeny of Baci- llales inferred from 3’ 16S rDNA and 5’ 16S-23S ITS nucleotide sequences. Nature Science, in Press. [29] Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) Clustal W: Improving the sensitivity of progressive mul- tiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research, 22(22), 4673-4680. [30] Saitou, N. and Nei, M. (1987) The neighbor-joining me- thod: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4(4), 406-425. [31] Kimura, M. (1983) The neutral theory of molecular evo- lution. Cambridge University Press, Cambridge. [32] Page, R.D.M. (1996) TreeView: An application to display phylogenetic trees on personal computers. Computer Ap- plication in the Biosciences, 12(4), 357-358. [33] Page, R.D.M. (2000) TreeView—tree drawing software for apple macintosh and windows. http://taxonomy.zoo- logy.gla.ac.uk/rod/treeview.html [34] Gürtler, V. and Stanisich V.A. (1996) New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology, 142(Pt 1), 3-16. [35] Suen, G., Goldman, B.S. and Welch, R.D. (2007) Pred- icting prokaryotic ecological niches using genome se- quence analysis. Public Library of Science Biology ONE, 2(8), e743. [36] Ivanova, E.P., Flavier, S. and Christen, R. (2004) Phylo- genetic relationships among marine Alteromonas-like pr- oteobacteria: Emended description of the family Altero- monadaceae and proposal of Pseudoalteromonadaceae fam. nov., Colwelliaceae fam. nov., Shewanellaceae fam. nov., Moritellaceae fam. nov., Ferrimonadaceae fam. nov., Idiomarinaceae fam. nov. and Psychromona-daceae fam. nov. International Journal of Systematic and Evolu- tionary Microbiology, 54(Pt 5), 1773-1788.
|