Journal of Cancer Therapy
Vol.4 No.1(2013), Article ID:28139,11 pages DOI:10.4236/jct.2013.41035
PLGA-Polymer Encapsulating Tumor Antigen and CpG DNA Administered into the Tumor Microenvironment Elicits a Systemic Antigen-Specific IFN-γ Response and Enhances Survival
![]()
1Department of Biomedical Engineering, Rutgers University, Piscataway, USA; 2The Cancer Institute of New Jersey and Departments of Surgery, Robert Wood Johnson Medical School, University of Medicine and Dentistry of New Jersey, Piscataway, USA.
Email: *lattimec@umdnj.edu, *yarmush@rci.rutgers.edu
Received November 14th, 2012; revised December 16th, 2012; accepted December 24th, 2012
Keywords: Cytokine; Immune Response; Immunomodulation; Macrophage; Microencapsulation; Vaccine
ABSTRACT
Critical to the generation of an effective therapeutic antitumor immune response is the elicitation of effective antigen presentation coupled with overcoming tumor-immune escape mechanisms. Towards this end, we aimed to understand the therapeutic effectiveness of a polymer based vaccine approach at enhancing the anti-tumor responses in a tumorbearing mouse model. While we and others have previously demonstrated the effectiveness of PLGA based systems in delivering antigen etc., studies scarcely focus on understanding the immunological mechanisms of polymer based therapies in tumor bearing treatment models. Considering tumors modulate the immune system and consequently the efficacy of therapies, understanding treatment mechanisms in the presence of tumor will help lead to more efficacious treatment options. We demonstrate here that a poly(lactic-co-glycolic acid) (PLGA) based delivery system encapsulating tumor antigen (OVA) and the TLR9 agonist CpG motif DNA administered into the tumor microenvironment initiates an effective type 1 mediated (IFN-γ producing) anti-tumor response in a syngeneic murine model of T cell lymphoma (E.G7-OVA). Although E.G7-OVA tumors spontaneously generate antigen specific CTLs in draining lymph nodes (LN), tumors progress rapidly. Modulation of the tumor microenvironment via local PLGA based therapy led to the generation of a systemic antigen specific Th1 response, absent in the non-polymer delivery method, subsequently associated with reduced tumor growth and prolongation of survival. These studies provide further insight into the use of a PLGA-based therapeutic approach at modulating the tumor microenvironment and highlight the need for analyzing the treatment effects in a tumor bearing model.
1. Introduction
Studies from our and other laboratories clearly demonstrate that the tumor microenvironment represents a complex interface between the immune system and the tumor, leading to poor antigen presentation, elevated levels of IL-10 or increased Treg activity [1-5]. Identifying immune escape mechanisms and developing targeted strategies to overcome these mechanisms has thus been a focus of recent studies [6-8]. Fundamentally, work from our lab has focused on approaches to modulate the tumor microenvironment to enhance effective systemic immunity. The studies here demonstrate that intratumoral immunization using a polymer complex is effective at generating a systemic immune response and enhancing survival.
Poly(lactic-co-glycolic acid) (PLGA) is an FDA approved resorbable polymer widely utilized as a delivery vehicle that offers numerous advantages over other delivery methods of therapeutic agents. As we and others have shown, PLGA enhances antigen presentation, increases cross presentation, ensures co-delivery of antigen and adjuvant to antigen presenting cells, and protects from proteolytic degradation of the encapsulated moieties [9-13]. Studies presented in this report build on our prior demonstration that polymer encapsulating CpG motif oligonucleotides effectively activates antigen-presenting dendritic cell populations in vitro [13]. In this prior work we have demonstrated enhanced expression of CD80, CD86, MHCII and secretion of IL-12 associated with the stimulation of Th1 T cell responses. Although we and others have shown the effectiveness of PLGA based systems to modulate an immune response, there is a lack of attention on elucidating the mechanistic and antitumor therapeutic responses to these PLGA treatments in tumor bearing models [14-16]. Considering the perturbations introduced by the tumor microenvironment, understanding the responses to treatment regimens within tumor models will help guide the design and potential efficacy of these therapeutic options.
In efforts to accomplish this and understand the immunological responses to a polymer therapy in a tumor model, tumor associated antigen ovalbumin (OVA) and the immune adjuvant CpG motif DNA were encapsulated in PLGA and administered intratumorally to E.G7-OVA bearing mice. We find that intratumoral treatment using the combined moieties stimulates the development of a tumor-specific interferon (IFN)-γ producing response associated with a reduction in tumor growth and prolongation of survival. In examining the anti-tumor CTL and IFN-producing cell responses, we show that despite a significant CTL response in the local lymph nodes of tumor bearing, non-treated mice, tumor progresses. We demonstrate that treatment with the PLGA based therapy results in the generation of a new Th1 response and results in decreased tumor growth. Taken together these results provide further understanding of the polymer based delivery system and emphasize the significance of analyzing immunological responses in tumor bearing models.
2. Materials and Methods
2.1. Tumor Model
The tumor cell line E.G7-OVA was obtained from ATCC and maintained in TCM media (RPMI 1640 based media supplemented with 10% FBS, 2 mM L-Glutamine, essential AA, non-essential AA, 110 mg/L Na-Pyruvate, 50 IU/ml penicillin/streptomycin) at 37˚C and 5% CO2. Cells were split two days prior to implantation to ensure consistent growth. E.G7-OVA cells were derived from the C57BL/6 (H-2b) mouse lymphoma cell line EL4. EL4 cells were transfected by electroporation with plasmid pAc-neo-OVA containing a complete copy of chicken ovalbumin (OVA) mRNA. E.G7-OVA cells constitutively synthesize and secrete OVA [17].
2.2. PLGA Based Delivery System
Poly(lactic-co-glycolic acid) (PLGA) based vesicles were generated using the water/oil/water double emulsion method [13,18], with the following modification. In brief, 0.1g of PLGA (Sigma-Aldrich, MW 7 - 17 kDa, 50:50 ratio) was dissolved in 0.4 ml of chloroform (SigmaAldrich). 50 mg/ml of OVA (ovalbumin, Sigma-Aldrich) and/or 5 mg/ml of CpG DNA (ODN-1668, phosphorothiated TCCATGACGTTCCTGATGCT, IDT) were added to the PLGA mixture at 0.05 ml per 0.4 ml of chloroform/PLGA. A microtip sonicator (Branson Ultrasonics) created the primary emulsion at 60% magnitude for 5 sec pulses and repeated for 4 cycles. This primary emulsion was combined with a 2 ml solution of 7% PVA (Sigma-Aldrich, MW 31 - 50 kDa, 87% - 89% hydrolyzed) and sonicated for another round. Secondary emulsion was added drop-wise to an 8 ml bath of a 7% PVA solution under constant stirring overnight at 4˚C. Final product was collected by centrifuging at 16,000 xg for 2 hours, washed with dH2O, resuspended in 2% sucrose and lyophilized.
To determine loading efficiency, FITC labeled BSA (Sigma-Aldrich) was dissolved to 50 mg/ml and Cy5 labeled CpG DNA (IDT) was dissolved to 5 mg/ml and incorporated into the polymer fabrication as before. Lyophilized polymer was weighed and lysed with 3 M NaOH. Supernatant was collected and scanned on a fluorescent plate reader at 495 nm and 655 nm, corresponding to the labeled BSA and CpG DNA respectively. Standard curves were generated and loading concentrations calculated.
Scanning electron microscopy (SEM) was performed on washed and dried PLGA vesicle samples. Samples were first sputter coated with gold for 2 min at 30 - 40 mA. Imaging was performed at the indicated magnifications with a 20 kV electron beam.
2.3. Murine Model and Therapy
Four to 6-week old female C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME) and maintained in a HEPA-filtered cage system for at least 1 week before use. For the prophylaxis studies, mice were immunized via subcutaneous (s.c.) injections into the lower left ventral abdomen with 5 mg of polymer in 100 µl of PBS, followed by another 5 mg injection of the same polymer after 2 weeks. Saline solution was used as a sham control. After an additional one week, mice were challenged with 250 × 103 EG7-OVA cells s.c. in the lower left ventral abdomen (day 0). For the treatment model, 250 × 103 E.G7-OVA cells were implanted s.c. into the lower left ventral abdomen. On days 3, 5, and 7, intratumor (i.t.) injections of 5mg of the respective polymer were administered. Mice were monitored and the tumor sizes were measured every 2 days with metric calipers by measuring the largest two diameters. Mice were sacrificed when the longest diameter of the tumor reached 1.5 cm. Kaplan Meier survival plots were generated using MedCalc commercial software. Mice were housed in AALAC approved facilities and all murine procedures were conducted in strict accordance with the UMDNJ-Robert Wood Johnson Medical School IACUC approved protocols and guidelines.
2.4. 51Cr Release Assay
Cytotoxic T Lymphocyte (CTL) assays were performed as previously reported with the following modifications [5]. Stimulators were splenocytes derived from naïve female C57BL/6 mice. Spleens were disassociated, incubated with ammonium chloride buffer (ACK, 0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM EDTA) for 5 min to lyse red blood cells, washed with TCM then filtered through a 70 µm mesh (BD Falcon). Cells were resuspended in TCM media, supplemented with 2-mercaptoethanol (2-ME), at 4 × 106 cells/ml and cultured with or without 300 µg/ml of OVA overnight. Effector cells were harvested and processed from the inguinal lymph nodes and spleens of day 16 treated mice. 3 × 106 stimulator splenocytes were then added to 7 × 106 effector cells in 2 ml total volume. On day 4, 50 µl of supernatant was harvested from each condition and analyzed for IFN-γ via a sandwich ELISA. On day 5, effector cells were harvested and added to target cells. 2 × 106 E.G7- OVA target cells were incubated with 100uCi of 51Cr for 1 hour at 37˚C and 5% CO2. 51Cr-labeled target cells (5 × 103) and effector cells were combined at known effector:target (E:T) ratios in 200 µl of TCM media. After 4hrs in 37˚C and 5% CO2, 100 µl of supernatant was collected and measured for 51Cr with a gamma counter (Packard Bioscience). Percentage of specific lysis was calculated from the formula (experimental release-spontaneous release) × 100/(maximal release in 1%SDS- 0.5%NaOH-spontaneous release).
2.5. IFN-γ Responses
Elisa assays for elicited IFN-γ were performed as we have described [5]. Briefly, purified rat anti-mouse IFN-γ (PharMingen) was diluted in coating buffer (NaHCO3) to 2 µg/ml and incubated overnight at 4˚C on 96 well flat bottom plates (Nunc). Plates were blocked with PBS/ 10%FBS for 2 hours at room temperature. Standards and sample were added at 100 µl per well and incubated overnight at 4˚C. After washing, biotin-conjugated antimouse IFN-γ (PharMingen) were dissolved to 1 µg/ml and added at room temperature for 45 min. Wells were washed and avidin-peroxidase diluted to 2.5 µg/ml in PBS/Tween/0.1%FBS for 30 minutes at room temperature was added. Enzyme activity was determined using O-phenylenediamine dihydrochloride reagent (OPD, Sigma) dissolved to 1mg/ml citrate buffer (pH 4.5) with 3%H2O2. Reaction was stopped with 3M HCl and color read at 492 nm.
2.6. Statistical Analysis
Results were expressed as the experimental averages ± SE. Statistical comparison between experimental averages was done using a two sample equal variance Student’s t-test at the p < 0.05 level.
3. Results
3.1. Polymer Delivery System
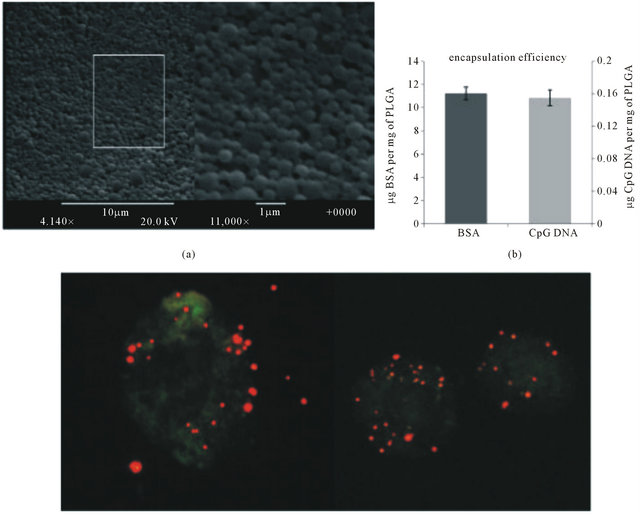
With the goal of developing a vaccine platform which would allow incorporation of both varied antigenic material and immune-regulating agents, we have adopted the FDA-approved, resorbable copolymer PLGA. Based on the water/oil/water emulsion technique [13,16], PLGA was utilized to fabricate submicron diameter vesicles. SEM images (Figure 1(a)) visualize the polymer vesicles when fabricated as described, having an average diameter of 500 nm [13]. With a starting concentration of 50 mg/ml, final encapsulation of the tumor associated antigen OVA was measured to be 11 µg of OVA per mg of polymer vesicle and CpG DNA encapsulation with a starting concentration of 5 mg/ml yielded a final 150 pg of CpG DNA per mg of vesicle (Figure 1(b)). To verify the ability of the polymers to be phagocytosed by antigen presenting cells, we delivered PLGA particles containing rhodamine labeled dextran to bone marrow derived DCs in vitro as previously demonstrated [13] (Figure 1(c)). As seen in Figure 1(c), these DCs readily take up the polymer vesicles.
3.2. Prophylactic Immunization Protects against Tumor Formation
To demonstrate the capacity of the polymer complex to effectively generate tumor-specific immunity, C57BL/6 mice received 2 s.c. injections of the polymer complex 2 weeks apart and where challenged with tumor 7 days later. PLGA encapsulating both tumor antigen and CpG was used. Unmethylated CpG motif DNA is a TLR9 agonist that stimulates B cell and antigen presenting cell functions [19]. We added antigen to the CpG DNA based on our prior studies demonstrating the efficacy of intratumoral immunization using antigen-encoding poxvirus to overcome tumor microenvironment associated immune escape and elicit a systemic response [8]. Combined CpG DNA and tumor antigen have previously been shown to stimulate IFN-γ responses manifesting antitumor activity [20,21]. Mice were pretreated with the polymer vaccine then challenged with tumor. In two independent experiments, we found significant protection against tumor formation after administration of the polymer (Fig-
 (c)
(c)
Figure 1. Polymer characterization. (a) SEM images at 4140× and 11,000× magnification provide visualization of the PLGA vesicles. Scale bars indicate 10 µm and 1 µm for the 4140× and 11,000× magnification, respectively; (b) To quantify the amount of protein and CpG DNA, fluorescently labeled BSA and CpG DNA were encapsulated within the PLGA vesicles. Standard curves were generated and the concentration of the vesicles were calculated to determine the loading efficiency; µg of BSA or CpG DNA per mg of PLGA vesicle; (c) Bone marrow derived dendritic cells were cultured for 48hrs with PLGA particles containing TMR dextran. Cells were stained with FITC labeled CD11c.
ure 2(a)). At the time when control mice needed to be sacrificed, day 17, all mice treated with antigen and adjuvant had either no palpable tumors or below 10 mm2 tumor cross-sections while sham had most tumors above 100 mm2 cross-sections (p < 0.01, Figure 2(b)). We saw no statistically significant difference between mice treated with PLGA-OVA or PLGA-OVA+ CpG DNA, as the anti-tumor response from either regimen was very effective at delaying tumor onset (Figure 2). Vaccination prolonged survival of mice in each treatment condition compared to sham (p < 0.02, Figure 2(c)). Several tumor free mice as a result of vaccination were seen, demonstrated in the Kaplan-Meier survival curves (Figure 2(c)).
3.3. Intratumoral PLGA Based Immunization of Tumor-Bearing Mice Significantly Delays Tumor Growth
To determine the therapeutic effects of the PLGA-CpG+ antigen complex, tumor bearing mice were administered the PLGA based therapy intratumorally starting 3 days post tumor implantation (3 injections 2 days apart) and the anti-tumor responses measured. A significant delay in tumor growth was seen after administration of polymer treatment (Figure 3(a)). While empty PLGA vesicles had no effect on tumor growth compared to saline treatment (not reported), we observed that all other polymer treatment conditions delayed tumor growth in compari-
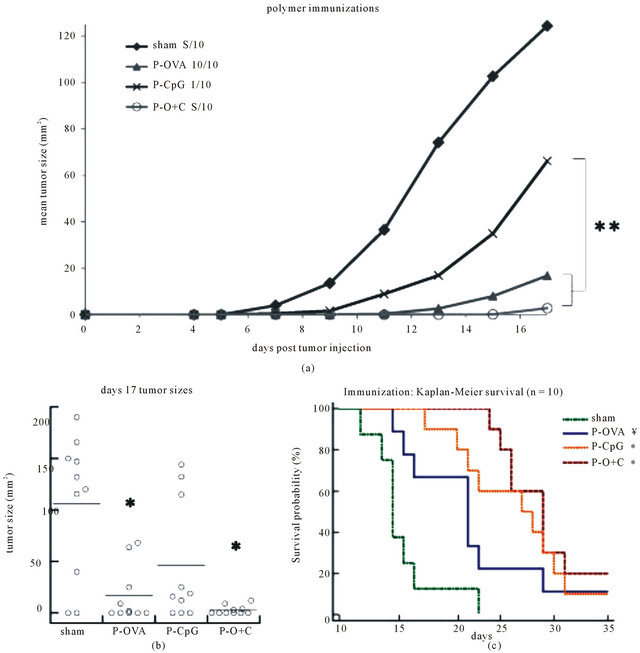
Figure 2. Prophylactic treatment with respective polymer vesicles. (a) 5 mg of indicated polymer (PLGA with OVA (▲ P-OVA), PLGA with CpG (× P-CpG), PLGA with OVA+CpG (○ P-O+C)) or PBS (♦ Sham) was injected on days −14 and −7, followed by E.G7-OVA tumor challenge on day 0. Tumor size was monitored and reported as the cross sectional area using largest two diameters. Two independent experiments with 5 mice each (10 total) were conducted. Mean tumor size measurements derived from mice bearing tumors is shown. (b) Day 17 tumor size distribution where each point represents one mouse in each respective condition. (c) Kaplan-Meier survival curves demonstrate the probability of survival in response to polymer treatment. Significance reported as p < 0.01 (*), p < 0.05 (**), or p = 0.02 (¥) in comparison to sham unless otherwise noted.
son to sham (15 mice from 3 independent experiments, p < 0.01, Figure 3(b)). Although no significant difference between PLGA-OVA and PLGA-CpG DNA treatment was observed, PLGA with both OVA and CpG DNA demonstrated an optimal delay in tumor growth (p < 0.02, Figure 3(a)). By day 17, all PLGA-OVA+CpG DNA treated mice had tumors below 50 mm2, while all other polymer conditions had numerous if not most tumors above 50 mm2 (p < 0.05, Figure 3(b)). Additionally, PLGA-OVA+CpG DNA provided the optimal survival
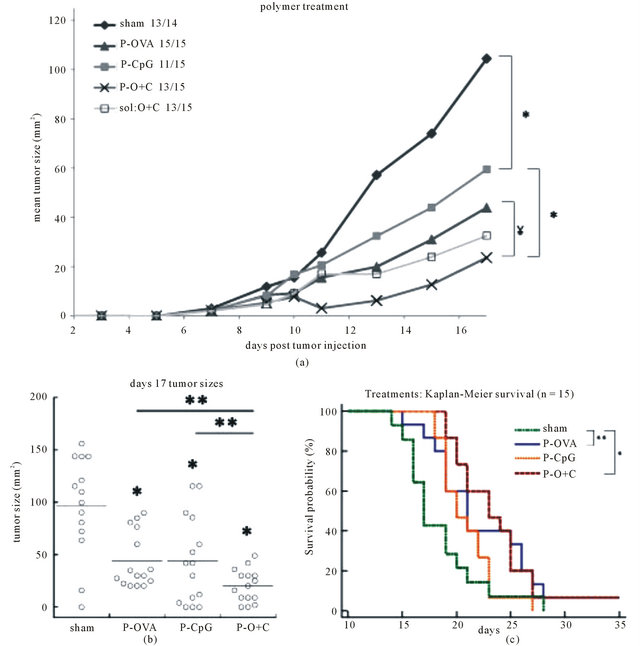
Figure 3. Tumor treatment model with polymer vesicles. (a) E.G7-OVA cells were implanted on day 0 and treatments were administered i.t. on days 3, 5 and 7 with the indicated conditions (PBS (♦ sham), PLGA with CpG (■ P-CpG), PLGA with OVA (▲ P-OVA), PLGA with OVA and CpG DNA (× P-O+C), OVA and CpG in solution/PBS (□ sol:O+C)). Tumor size reported as the cross sectional area using largest two diameters. Three independent experiments with 5 mice each (15 total) were conducted. Mean tumor size derived from mice bearing tumors is shown. (b) Day 17 tumor size distribution where each point represents one mouse in each respective condition. (c) Kaplan-Meier survival curves demonstrate the probability of survival in response to polymer treatment. Significance reported as p < 0.01 (*), p < 0.05 (**), or p = 0.02 (¥) in comparison to sham unless otherwise noted.
probability, with 2/15 mice not forming tumors (p < 0.01, Figure 3(c)). It is of note that the soluble delivery of OVA and CpG had similar effects on tumor growth as with the polymer delivery. However, the immunological responses differed as the PLGA based system provided enhanced IFN-γ production while soluble delivery did not.
3.4. E.G7-OVA Spontaneously Elicits Local but Not Systemic CTL Response
Cytotoxic T cell activity is oft hypothesized as the primary mechanism underlying antitumor responses [8,12, 14,22]. To assess the putative role of CTL in anti-tumor activity and the response of CTL to therapy in our system, both local (LN) and systemic (splenic) tumor-specific CTL activity was evaluated in tumor bearing mice. Figure 4 shows a representative result of one of three such experiments where 51Cr release assays were used to demonstrate the systemic (splenic) and local (LN) antigen specific CTL response of tumor bearing mice following therapy. We observed that tumor growth alone led to a significant local lymph node, but not systemic, CTL response reminiscent of what we observed in the MB49 system [8]. THowever, he polymer treatments neither enhanced the local (LN) CTL activity nor induced systemic CTL activity. Since polymer treatments were still able to retard tumor growth, these experiments suggest that CTL activity alone does not account for tumor regression and support the existence of dysfunctional or inadequate CTL in vivo activity. Despite significant CTL activity, tumors progressed in size as seen in Figure 3(a).
3.5. Polymer Delivery System Elevates Th1 Response
To determine the effects of polymer treatment on the generation of tumor-specific Th1 responses in tumor bearing mice, we examined the local (LN) and systemic (splenic) lymphocyte compartments for antigen-specific IFN-γ production. We found that saline (sham) treated mice expressed a low baseline level of IFN-γ in both the spleen and lymph nodes (Figure 5). Consistent with our previous findings where polymer delivery to dendritic cells increased expression of CD80, CD86, MHCII and production of IL-12 [13], mice treated with OVA and CpG DNA via PLGA polymer generated a significant tumor-specific IFN-γ (Th1) response both systemically and locally as compared to sham (p < 0.05, Figure 5). Interestingly, we found that when mice were treated with OVA and CpG DNA in soluble form, there was no induction of a type 1 response, as no significant change in IFN-γ levels was observed (Figure 5).
4. Discussion
The studies presented here demonstrate the ability of a polymer delivery system to induce a Th1 producing tumor antigen specific anti-tumor immune response in a tumor bearing mouse model. Although CTLs, considered primary effectors at inducing tumor regression, were present in tumor draining lymph nodes, tumor growth was still robust in non-treated mice. We demonstrated that PLGA based delivery of antigen and adjuvant was able to overcome this immune deficiency and generate local (LN) and systemic (splenic) IFN-γ mediated responses, slowing tumor growth. However, although both soluble and polymer delivery methods were efficient at reducing tumor growth, the cumulative data warrants further investigations into the mechanistic understanding of polymer based delivery systems in tumor bearing models.
Studies from our and other laboratories have demonstrated the necessity of analyzing therapeutic strategies in the context of tumor models by highlighting a variety of overlapping mechanisms contributing to tumor-immune escape. It has been well documented that tumors can curb the immune response by producing and/or inducing factors such as IL-10 [5,23,24], TGF-beta [25], VEGF [26], as well as activating Tregs [27]. Our prior studies in the murine bladder cancer model MB49 showed that tumorassociated IL-10 can render antigen presenting cells deficient and inhibit immune stimulation [5]. Consistent with
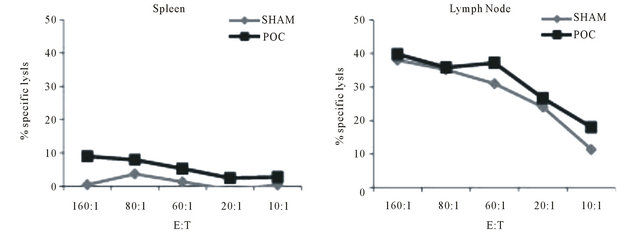
Figure 4. Representative systemic and local CTL responses against E.G7-OVA cells. OVA-restimulated spleens and lymph nodes from respective conditions were cultured with 51Cr labeled target cells, E.G7-OVA, at the indicated Effector:Target (E:T) ratios. Systemic (spleen) specific lysis was below detection after OVA restimulation. CTL response was elevated in the tumor draining lymph nodes, however no difference is seen between the different treatment conditions. Data representative of three independent experiments run in triplicate.
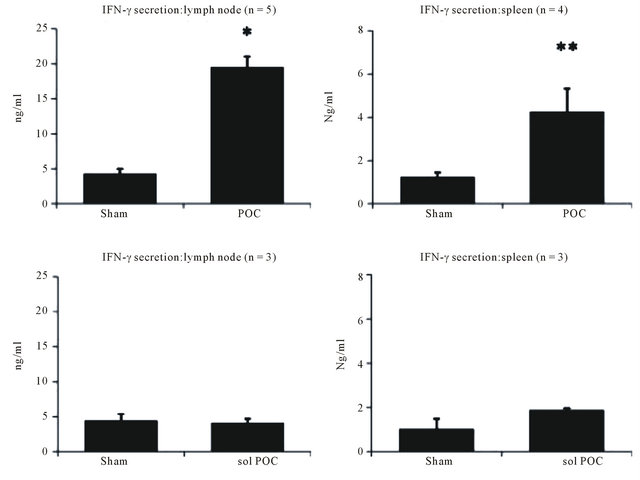
Figure 5. IFN-γ levels from restimulated spleens and lymph nodes. Systemic and local levels of IFN-γ are elevated as a result of the polymer treatment containing both OVA and CpG DNA. In vivo delivery of OVA and CpG DNA dissolved in PBS had no effect on IFN-γ expression levels. Significance reported as p < 0.01 (*) or p < 0.05 (**) in comparison to sham. Data represents the average of n = 3 − 5 independent experiments run in triplicate.
our findings here, we found in the MB49 model that local but not systemic CTLs were generated by the tumors [8]. This lack of systemic immunity could be overcome by immunizing into the tumor microenvironment but not systemically using a recombinant viral vaccine producing antigen and GMCSF. Using the E.G7-OVA model here we found a similar tumor-specific CTL population locally but not systemically, also deficient at retarding tumor progression. While the delivery of antigen and adjuvant is well known and documented to induce key immune responses, such as in the current model to retard tumor growth, understanding the mechanisms involved with these responses in a tumor bearing model provide significant insight into therapeutic strategies. To thus broaden the therapeutic immunization techniques and more importantly understand their behavior and effects on tumor bearing models, we utilized a PLGA based polymeric delivery system to administer antigen and adjuvant to tumor bearing mice.
PLGA is FDA approved, easily adaptable to different systems and cost effective. It has high bio-compatibility with minimal toxicity; the original monomers, lactic acid and glycolic acid, are byproducts of various metabolic pathways [28]. In addition, these vesicles are phagocytosed in vivo by macrophages and antigen presenting cells (APCs), facilitating the development of adaptive immunity [18]. We have recently shown that PLGA based vaccines provide a platform to increase the activation of dendritic cells [13]. We demonstrated that delivery of antigen and adjuvant via PLGA vesicles significantly enhanced the activated phenotype of antigen presenting cells in vitro. Cell surface expression of CD80, CD86, MHCII and secretion of IL-12 were upregulated as a result of treatment. Concurrently, we and others have seen that PLGA provides a more efficient method for dendritic cell stimulation as compared to soluble delivery; greater immunological responses are seen via PLGA delivery as compared to equivalent amounts delivered in solution [13,29-31].
Despite this growing evidence advocating polymer based immune therapies, studies analyzing the effects of PLGA on tumor bearing models are often limited to tumor growth analysis with little emphasis on immunologic mechanism or are limited to immunization, not treatment, regimens [14-16,22,32]. Given that tumors modulate the immune system, as highlighted previously, understanding the in vivo effects of these polymers in tumor models is critical in designing more effective therapeutic protocols. For example, studies have demonstrated that in vivo immunizations of naive mice with PLGA encapsulating OVA and CpG stimulate antigen specific CTLs [22]. As demonstrated in our study however, despite significant tumor specific CTL activity in the tumor microenvironment from non-treated mice, they are insufficient at preventing tumor growth. This observation may arguably negate the requisite for therapies that are designed with a focus on exclusively enhancing CTL activity. To therefore demonstrate and understand the efficacy of this polymer system in inducing an antigen specific anti-tumor immune response, the polymer was adopted for use in the E.G7-OVA tumor bearing mouse model.
While intratumoral injections of the PLGA vesicles failed to stimulate systemic CTL activity, the PLGA complex significantly stimulated a local (LN) and systemic (splenic) antigen specific IFN-γ mediated Th1 response. The soluble delivery of the same components failed to generate a significant antigen-specific IFN-γ producing response both locally and systemically. These results further demonstrate both the advantage of a polymer based delivery system and the difference in response that is generated. Studies from other non-tumor bearing models support these findings and show efficacy for a PLGA based antigen delivery system at enhancing humoral immunity, inducing elevated antibody levels and generating Th1 responses to specific antigens [33-37]. In particular, elevated IFN-γ is commonly attributed to a strong CD4+ mediate Th1 response [38], and has been shown to act directly on tumor cells to inhibit both proliferation and angiogenesis and to facilitate apoptosis [39-41]. The current data is thus in support of the hypothesis that local PLGA delivery to a tumor bearing host generates a strong IFN-γ mediated Th1 response that may support combating tumor growth.
It is of interest to note that delivery of OVA and CpG in both soluble form or PLGA vesicles induced similar tumor regression, while their respective Th1 responses differed. OVA is a known and well characterized foreign antigen, utilized for its high immunogenicity. It is therefore not uncommon to find that different routes of immunization will induce similar anti-tumor effects against a strong antigen. Determining the anti-tumor mechanisms created by each delivery method require further experimentation, but the processing of the antigen and adjuvant and the impact on antigen presenting cells is known to vary when delivered via polymer vesicles or other delivery methods [42,43]. This can be hypothesized to explain the enhanced IFN-γ response as a result of PLGA delivery. With elevated IFN-γ being a hallmark of CD4+ mediate anti-tumor activity, increasing a Th1 response may be critical for mounting successful anti-tumor responses in models where the antigen is not foreign or immunogenic. Taken together, these results further demonstrate the need to verify immunological analysis of in vitro and non-tumor bearing models in tumor systems. This sets an impetus for understanding mechanistically how polymer delivery systems modulate the immune response in this and other tumor models.
In conclusion, the results demonstrate the importance of analyzing the immune response of proposed therapies in tumor bearing models. While the PLGA based delivery system provides an effective means to deliver antigen and adjuvant into the tumor microenvironment and stimulate a CTL response, a CTL response alone is insufficient at reducing tumor growth. As shown, tumor cells are able to elicit local antigen specific CTL responses in mice without vaccination or stimulation, but are insufficient at perturbing tumor growth. The data suggest tumor cells fail to prime for a functional type 1 (IFN-γ) response in normal mice. Treatment with PLGA provides a means of escaping immune tolerance, and although additional experimentation would be required to verify, the data suggests a role for a tumor-specific IFN-γ mediated response. With a broader understanding of the immune responses in tumor bearing hosts and the specific therapeutic targets which serve to modulate those responses, we can better assess and subsequently design improved therapeutic vaccine delivery strategies.
5. Acknowledgements
This research was supported by Shared Resources of The Cancer Institute of New Jersey (P30CA072720). This work was supported by grants from the NCI R01CA- 42908, Rutgers/UMDNJ NIH Biotechnology Training Program NIHT32GM008339, and the New Jersey Commission on Cancer Research Predoctoral Fellowship
REFERENCES
- G. Raimondi, M. S. Turner, A. W. Thomson and P. A. Morel, “Naturally Occurring Regulatory T Cells: Recent Insights in Health and Disease,” Critical Reviews in Immunology, Vol. 27, No. 1, 2007, pp. 61-95. doi:10.1615/CritRevImmunol.v27.i1.50
- P. Neeson and Y. Paterson, “Effects of the Tumor Microenvironment on the Efficacy of Tumor Immunotherapy,” Immunological Investigations, Vol. 35, 2006, pp. 359-394. doi:10.1080/08820130600755009
- J. R. Wilczynski, M. Radwan and J. Kalinka, “The Characterization and Role of Regulatory T Cells in Immune Reactions,” Frontiers in Bioscience, Vol. 13, No. 6, 2008, pp. 2266-2274. doi:10.2741/2840
- A. F. Ochsenbein, “Principles of Tumor Immunosurveillance and Implications for Immunotherapy,” Cancer Gene Therapy, Vol. 9, No. 12, 2002, pp. 1043-1055. doi:10.1038/sj.cgt.7700540
- A. S. Yang and E. C. Lattime, “Tumor-Induced Interleukin 10 Suppresses the Ability of Splenic Dendritic Cells to Stimulate CD4 and CD8 T-Cell Responses,” Cancer Research, Vol. 63, No. 9, 2003, pp. 2150-2157.
- M. O. Lasaro and H. C. Ertl, “Targeting Inhibitory Pathways in Cancer Immunotherapy,” Current Opinion in Immunology, Vol. 22, No. 3, 2010, pp. 385-390. doi:10.1016/j.coi.2010.04.005
- T. L. Whiteside, “Inhibiting the Inhibitors: Evaluating Agents Targeting Cancer Immunosuppression,” Expert Opinion on Biological Therapy, Vol. 10, No. 7, 2010, pp. 1019-1035. doi:10.1517/14712598.2010.482207
- A. S. Yang, C. E. Monken and E. C. Lattime, “Intratumoral Vaccination with Vaccinia-Expressed Tumor Antigen and Granulocyte Macrophage Colony-Stimulating Factor Overcomes Immunological Ignorance to Tumor Antigen,” Cancer Research, Vol. 63, No. 20, 2003, pp. 6956-6961.
- R. Audran, K. Peter, J. Dannull, et al., “Encapsulation of Peptides in Biodegradable Microspheres Prolongs Their MHC Class-I Presentation by Dendritic Cells and Macrophages in Vitro,” Vaccine, Vol. 21, No. 11-12, 2003, 1250-1255. doi:10.1016/S0264-410X(02)00521-2
- H. Shen, A. L. Ackerman, V. Cody, et al., “Enhanced and Prolonged Cross-Presentation Following Endosomal Escape of Exogenous Antigens Encapsulated in Biodegradable Nanoparticles,” Immunology, Vol. 117, No. 1, 2006, pp. 78-88. doi:10.1111/j.1365-2567.2005.02268.x
- S. Hamdy, A. Haddadi, R. W. Hung and A. Lavasanifar, “Targeting Dendritic Cells with Nano-Particulate PLGA Cancer Vaccine Formulations,” Advanced Drug Delivery Reviews, Vol. 63, No. 10-11, 2011, pp. 943-955. doi:10.1016/j.addr.2011.05.021
- Y. Waeckerle-Men, E. U. Allmen, B. Gander, et al., “Encapsulation of Proteins and Peptides into Biodegradable Poly(D,L-lactide-co-glycolide) Microspheres Prolongs and Enhances Antigen Presentation by Human Dendritic Cells,” Vaccine, Vol. 24, No. 11, 2006, pp. 1847-1857. doi:10.1016/j.vaccine.2005.10.032
- K. Nikitczuk, E. Lattime, R. Schloss and M. Yarmush, “Analysis of Dendritic Cell Stimulation Utilizing a MultiFaceted Nanopolymer Deliver System and the Immune Modulator 1-Methyl Tryptophan,” Nano LIFE, Vol. 1, No. 3, 2010, pp. 1-12. doi:10.1142/S1793984410000171
- A. Heit, F. Schmitz, T. Haas, et al., “Antigen Co-Encapsulated with Adjuvants Efficiently Drive Protective T Cell Immunity,” European Journal of Immunology, Vol. 37, No. 8, 2007, pp. 2063-2074. doi:10.1002/eji.200737169
- M. Diwan, P. Elamanchili, H. Lane, et al., “Biodegradable Nanoparticle Mediated Antigen Delivery to Human Cord Blood Derived Dendritic Cells for Induction of Primary T Cell Responses,” Journal of Drug Targeting, Vol. 11, No. 8-10, 2003, pp. 495-507. doi:10.1080/10611860410001670026
- P. Elamanchili, C. M. Lutsiak, S. Hamdy, et al., “Pathogen-Mimicking Nanoparticles for Vaccine Delivery to Dendritic Cells,” Journal of Immunotherapy, Vol. 30, No. 4, 2007, pp. 378-395. doi:10.1097/CJI.0b013e31802cf3e3
- M. W. Moore, F. R. Carbone and M. J. Bevan, “Introduction of Soluble Protein into the Class I Pathway of Antigen Processing and Presentation,” Cell, Vol. 54, No. 6, 1988, pp. 777-785. doi:10.1016/S0092-8674(88)91043-4
- K. D. Newman, P. Elamanchili, G. S. Kwon and J. Samuel, “Uptake of Poly(D,L-lactic-co-glycolic acid) Microspheres by Antigen-Presenting Cells in Vivo,” Journal of Biomedical Materials Research, Vol. 60, No. 3, 2002, pp. 480-486. doi:10.1002/jbm.10019
- A. M. Krieg, “Therapeutic Potential of Toll-Like Receptor 9 Activation,” Nature Reviews. Drug Discovery, Vol. 5, No. 6, 2006, pp. 471-484. doi:10.1038/nrd2059
- A. M. Krieg, “CpG Motifs in Bacterial DNA and Their Immune Effects,” Annual Review of Immunology, Vol. 20, 2002, pp. 709-760. doi:10.1146/annurev.immunol.20.100301.064842
- A. M. Krieg, “Development of TLR9 Agonists for Cancer Therapy,” The Journal of Clinical Investigation, Vol. 117, No. 5, 2007, pp. 1184-1194. doi:10.1172/JCI31414
- M. Mueller, E. Schlosser, B. Gander and M. Groettrup, “Tumor Eradication by Immunotherapy with Biodegradable PLGA Microspheres—An Alternative to Incomplete Freund ’s adjuvant,” International Journal of Cancer, Vol. 129, No. 2, 2011, pp. 407-416. doi:10.1002/ijc.25914
- T. Sato, P. McCue, K. Masuoka, et al. “Interleukin 10 Production by Human Melanoma,” Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, Vol. 2, No. 8, 1996, pp. 1383-1390.
- B. K. Halak, H. C. Maguire Jr. and E. C. Lattime, “Tumor-Induced Interleukin-10 Inhibits Type 1 Immune Responses Directed at a Tumor Antigen as Well as a NonTumor Antigen Present at the Tumor Site,” Cancer Research, Vol. 59, No. 4, 1999, pp. 911-917.
- T. F. Gajewski, Y. Meng and H. Harlin, “Immune Suppression in the Tumor Microenvironment,” Journal of Immunotherapy, Vol. 29, 2006, pp. 233-240. doi:10.1097/01.cji.0000199193.29048.56
- D. I. Gabrilovich, H. L. Chen, K. R. Girgis et al., “Production of Vascular Endothelial Growth Factor by Human Tumors Inhibits the Functional Maturation of Dendritic Cells,” Nature Medicine, Vol. 2, No. 10, 1996, pp. 1096- 1103. doi:10.1038/nm1096-1096
- V. C. Liu, L. Y. Wong, T. Jang, et al., “Tumor Evasion of the Immune System by Converting CD4+CD25- T Cells into CD4+CD25+ T Regulatory Cells: Role of TumorDerived TGF-Beta,” Journal of Immunology, Vol. 178, No. 5, 2007, pp. 2883-2892.
- K. A. Athanasiou, G. G. Niederauer and C. M. Agrawal, “Sterilization, Toxicity, Biocompatibility and Clinical Applications of Polylactic Acid/Polyglycolic Acid Copolymers,” Biomaterials, Vol. 17, No. 2, 1996, pp. 93-102. doi:10.1016/0142-9612(96)85754-1
- J. Tel, A. J. Lambeck, L. J. Cruz, et al., “Human Plasmacytoid Dendritic Cells Phagocytose, Process, and Present Exogenous Particulate Antigen,” Journal of Immunology, Vol. 184, No. 8, 2010, pp. 4276-4283.
- W. Ma, T. Smith, V. Bogin, et al., “Enhanced Presentation of MHC Class Ia, Ib and Class II-Restricted Peptides Encapsulated in Biodegradable Nanoparticles: A Promising Strategy for Tumor Immunotherapy,” Journal of Translational Medicine, Vol. 9, 2011, p. 34. doi:10.1186/1479-5876-9-34
- P. Elamanchili, M. Diwan, M. Cao and J. Samuel, “Characterization of Poly(D,L-lactic-co-glycolic acid) Based Nanoparticulate System for Enhanced Delivery of Antigens to Dendritic Cells,” Vaccine, Vol. 22, No. 19, 2004, pp. 2406-2412. doi:10.1016/j.vaccine.2003.12.032
- Z. Zhang, S. Tongchusak, Y. Mizukami, et al., “Induction of Anti-Tumor Cytotoxic T Cell Responses through PLGA-Nanoparticle Mediated Antigen Delivery,” Biomaterials, Vol. 32, No. 14, 2011, pp. 3666-3678. doi:10.1016/j.biomaterials.2011.01.067
- D. T. O’Hagan, H. Jeffery and S. S. Davis, “Long-Term Antibody Responses in Mice Following Subcutaneous Immunization with Ovalbumin Entrapped in Biodegradable Microparticles,” Vaccine, Vol. 11, No. 9, 1993, pp. 965-969. doi:10.1016/0264-410X(93)90387-D
- Y. Men, C. Thomasin, H. P. Merkle, et al., “A Single Administration of Tetanus Toxoid in Biodegradable Microspheres Elicits T cell and Antibody Responses Similar or Superior to Those Obtained with Aluminum Hydroxide,” Vaccine, Vol. 13, No. 7, 1995, pp. 683-689. doi:10.1016/0264-410X(94)00046-P
- H. C. Ertl, I. Varga, Z. Q. Xiang, et al., “Poly(DL-lactide-co-glycolide) Microspheres as Carriers for Peptide Vaccines,” Vaccine, Vol. 14, No. 9, 1996, pp. 879-885. doi:10.1016/0264-410X(96)00005-9
- K. J. Maloy, A. M. Donachie, D. T. O’Hagan and A. M. Mowat, “Induction of Mucosal and Systemic Immune Responses by Immunization with Ovalbumin Entrapped in Poly(lactide-co-glycolide) Microparticles,” Immunology, Vol. 81, No. 4, 1994, pp. 661-667.
- A. Moore, P. McGuirk, S. Adams et al., “Immunization with a Soluble Recombinant HIV Protein Entrapped in Biodegradable Microparticles Induces HIV-Specific CD8+ Cytotoxic T Lymphocytes and CD4+ Th1 Cells,” Vaccine, Vol. 13, No. 18, 1995, pp. 1741-1749. doi:10.1016/0264-410X(95)00184-3
- R. Kennedy and E. Celis, “Multiple Roles for CD4+ T Cells in Anti-Tumor Immune Responses,” Immunological Reviews, Vol. 222, 2008, pp. 129-144. doi:10.1111/j.1600-065X.2008.00616.x
- Z. Qin, J. Schwartzkopff, F. Pradera, et al., “A Critical Requirement of Interferon Gamma-Mediated Angiostasis for Tumor Rejection by CD8+ T Cells,” Cancer Research, Vol. 63, No. 14, 2003, pp. 4095-4100.
- G. L. Beatty and Y. Paterson, “Regulation of Tumor Growth by IFN-Gamma in Cancer Immunotherapy,” Immunologic Research, Vol. 24, No. 2, 2001, pp. 201-210. doi:10.1385/IR:24:2:201
- W. H. Schmiegel, J. Caesar, H. Kalthoff, et al., “Antiproliferative Effects Exerted by Recombinant Human Tumor Necrosis Factor-Alpha (TNF-Alpha) and InterferonGamma (IFN-Gamma) on Human Pancreatic Tumor Cell Lines,” Pancreas, Vol. 3, No. 2, 1988, pp. 180-188. doi:10.1097/00006676-198804000-00012
- Y. Waeckerle-Men and M. Groettrup, “PLGA Microspheres for Improved Antigen Delivery to Dendritic Cells as Cellular Vaccines,” Advanced Drug Delivery Reviews, Vol. 57, No. 3, 2005, pp. 475-482. doi:10.1016/j.addr.2004.09.007
- J. E. Eyles, Z. C. Carpenter, H. O. Alpar and E. D. Williamson, “Immunological Aspects of Polymer Microsphere Vaccine Delivery Systems,” Journal of Drug Targeting, Vol. 11, No. 8-10, 2003, pp. 509-514. doi:10.1080/10611860410001670017
NOTES
*Corresponding authors.