Journal of Biophysical Chemistry
Vol.2 No.3(2011), Article ID:6732,12 pages DOI:10.4236/jbpc.2011.23041
Computational modeling studies on anti-HIV-1 non-nucleoside reverse transcriptase inhibition by dihydroalkoxybenzyloxopyrimidines analogues: an electrotopological atomistic approach
1Department of Applied Chemistry, SGSITS, Indore, India, *Corresponding Author: sukusap@yahoo.com, nsapre@sgsits.ac.in
2Department of Chemistry, Bherulal Patidar Post Graduate College, Mhow, Indore, India;
3Department of Computer Applications, SV College of Engineering, Indore, India.
Received 8 October 2010; revised 17 January 2011; accepted 20 February 2011.
Keywords: AIDS; HIV-1; NNRTIs; DABOs; QSAR; pIC50; Kier Hall E-State Indices; MLR; NN; k-NN
ABSTRACT
For the first time we report quantitative structure activity relationship (QSAR) studies based on Kier-Hall Electrotopological State (E-State) Indices for Dihydroalkoxybenzyloxopyrimidines (DABO) derivatives acting as NNRTIs of HIV-1. A dataset of 74 compounds was compiled from published studies and randomly subdivided into training and test sets. To understand the pharmacophoric effect, Kier-Hall Electrotopological State descriptors namely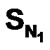 ,
, 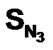 , SF, SAr, SS, SO,
, SF, SAr, SS, SO,  , SCl, SY (Y = S-alkyl and NH-alkyl), SX (X = Me) and biological activity were used as independent and dependent variable respectively. Statistical results were highly encouraging for the training set [multiple linear regression [(MLR): r2 = 0.961, F = 100.41 and q2 = 0.926, neural networks (NN): r2 = 0.966, F = 115.594, degrees of freedom = 40 and k-nearest neighbour (k-NN): r2 = 0.770, q2 = 0.757, degrees of freedom = 40]. Results of validation using a test set showed the same trend as training set (NN > MLR > kNN). The above results suggest that of various functional groups present in DABO
, SCl, SY (Y = S-alkyl and NH-alkyl), SX (X = Me) and biological activity were used as independent and dependent variable respectively. Statistical results were highly encouraging for the training set [multiple linear regression [(MLR): r2 = 0.961, F = 100.41 and q2 = 0.926, neural networks (NN): r2 = 0.966, F = 115.594, degrees of freedom = 40 and k-nearest neighbour (k-NN): r2 = 0.770, q2 = 0.757, degrees of freedom = 40]. Results of validation using a test set showed the same trend as training set (NN > MLR > kNN). The above results suggest that of various functional groups present in DABO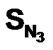 , SO, SCl, SAr and
, SO, SCl, SAr and  contribute more significantly towards activity. On the other hand
contribute more significantly towards activity. On the other hand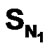 , SS, and SF do not play any role in enhancing the activity. The substitution of S-alkyl and NH-alkyl at C2 position is essential though it does not contribute much towards the activity. The substitution of methyl group at C5 position is unfavorable and exhibit negative impact on inhibitory activity. Therefore, it seems reasonable to choose E-State indices as suitable and significant descriptors for exploring the relationship between the pIC50 and the pharmacological properties of the compounds.
, SS, and SF do not play any role in enhancing the activity. The substitution of S-alkyl and NH-alkyl at C2 position is essential though it does not contribute much towards the activity. The substitution of methyl group at C5 position is unfavorable and exhibit negative impact on inhibitory activity. Therefore, it seems reasonable to choose E-State indices as suitable and significant descriptors for exploring the relationship between the pIC50 and the pharmacological properties of the compounds.
1. INTRODUCTION
Acquired Immunodeficiency Syndrome (AIDS) is a set of symptoms and infections resulting from the damage of the human immune system caused by the human immunodeficiency virus (HIV) [1]. AIDS cause very serious public health problem and economic burden. Globally, an estimated 33.4 million people are living with HIV with nearly 7500 new infections each day [2]. An anti-HIV agent may exert its activity by inhibiting a variety of steps in the life cycle of the virus [3]. Reverse transcription is a highly choreographed, multistep process in which the plus-strand RNA genome of the HIV-1is converted into a double-stranded cDNA [4]. HIV-1 reverse transcriptase (RT) is an asymmetric heterodimer composed of two related subunits, p66 (560 amino acids) and p51 (440 amino acids).p66 folds into two domains: polymerase and RNase H. The polymerase domain of p66 is divided into four subdomains fingers (amino acids 1 - 85 and 118 - 155), a palm (amino acids 86 - 117 and 156 - 237) and a thumb (amino acids 238 - 318) domain. [5] The two subunits form a stable dimer which is essential for the enzymatic activity [6]. The RT enzyme is known to be flexible, moving both the domains to accommodate inhibitors and to take on different shapes depending on the inhibitor bound to it [7]. Inhibitors of HIV-1 RT fall under two categories: the nucleoside (NRTIs) and the non-nucleoside reverse transcriptase inhibitors (NNRTIs). The NRTIs are substrate analogs that act as chain terminators, whereas the NNRTIs are a chemically diverse group of compounds that non-competitively inhibit DNA polymerization [8]. Twenty-five years ago, nucleoside analog 3’-azidothymidine (AZT) was shown to efficiently block the replication of HIV in a cell culture [9]. NNRTIs form a second class of compounds that target HIV-1 RT. [10] NNRTIs bind at an allosteric site about 10 Å away from the polymerase active site. The structural mechanism of NNRTI inhibition involves distortion of the catalytically essential triad of aspartic acid residues [11]. The non-nucleoside site, although contained within the p66 subunit, also has Glu138 from the p51 subunit located at the edge of the inhibitor pocket [12]. Over 40 amino acid substitutions have been identified to be associated with NNRTI resistance, in vitro and in vivo [13]. Almost twenty-one years ago, the NNRTIs i.e., 1-(2-2-hydroxyethoxy methyl)-6- (phenylthio) thymine (HEPT) [14,15] and Tetrahydroimidazo[4,5,1-jkj] [1,4] benzodiazepin-2(1H)-one and thione (TIBO) compounds [16,17] have been discovered. [18] Since then three more NNRTIs-nevirapine, delavirdine and efavirenz have been approved for the treatment of HIV-1 and a few more are in clinical development, including rilpivirine, etravirine and dapivirine [19]. Till date various molecular modelling studies have been performed using QSAR [20], QSPR [21], Simple docking [22] and Template docking [23] on some NNRTIs.
Pharmacophore based descriptors are of immense importance in understanding the structure activity relationships of ligand molecules. Dihydroalkoxybenzyloxopyrimidines (DABO) derivatives have been successfully used as HIV-1 reverse transcriptase inhibitors [24] and are useful precursors for deriving novel potent compounds. The first step of rational drug design often consists of performing QSAR modeling, based on congeneric structures of ligands; this procedure has been carried out by several researchers for a class of DABO derivatives [25-28]. Like other NNRTIs, DABOs also assume a “butterfly like” spatial arrangement with the methylene group or moiety serving as a bridge between the two wings of the benzene and pyrimidine rings [27].5-isopropyl-2-[(methylthiomethyl)thio]-6-benzylpyrimidin-4-(1H)-one DABO analogue was found to be very potent against HIV-1 Reverse Transcriptase (RT) [28]. Dihydro-alkyl-thio-benzyl-oxopyrimidines (S-DABOs) are common with EBUs (Emivirine, formerly MKC-442) in having a benzyl moiety at position C-6 of the pyrimidine ring while they differ from EBUs in that the alkoxy chain is linked at position C-2 instead of N-1. Sudbeck et al. have performed derivatization of DABO by modelling studies of potent 1-[(2-hydroxy ethoxy) methyl]-6-(phenylthio) thymine (HEPT) and by the substitution of benzyl moiety with phenyl thio moiety at the C-6 position of the pyrimidine ring [29]. A structure-activity relationship (SAR) profile of DABOs together with molecular modeling investigation on their putative binding mode have shown that the presence of a C-2-alkoxy (DABOs), C-2-alkylthio (S-DABOs), or C-2-alkylamino (NH-DABOs) side chain is the structural determinant for the antiviral activity of these derivatives, with the length and size of the C-2 side chain having only modulator effects on potency [30-34]. The 2,6-difluoro substitution at the C-6-benzyl moiety of S-DABOs and NH-DABOs produced favorable π- stacking interactions with the Tyr188 side chain into the non-nucleoside binding site (NNBS), leading to compounds (F2-S-DABOs and F2-NH-DABOs) active in the nanomolar range. [35] The biological testing results clearly indicated that the substitution of halogen at the C5 position of pyrimidine ring could increase the antiHIV-1 RT activity. The most active compounds showed activity in the lower micromole range with IC50 values (IC50 0.18 - 3.03 μM) comparable to nevirapine (IC50 4.12 μM). The docking showed that a new halogen bond was formed between halogen and carbonyl of TYR188 in the HIV-I RT [36].
Various statistical modeling techniques have been widely used in quantitative structure-activity relationship (QSAR) studies, such as multiple linear regression (MLR), [37] partial least squares (PLS), [37] artificial neural networks (ANN) [38]. Using such an approach, one could predict the activities of newly designed compounds before a decision is being made whether these compounds should be really synthesized and tested. Thus, for the first time a QSAR study based on KierHall Electrotopological State (E-State) [39-41] indices is performed on DABO derivatives to analyze the pharmacophoric effect on their inhibitory activities. Recently, a comparative molecular field analysis (CoMFA) study was performed in order to derive three-dimensional quantitative structure activity relationship (3D-QSAR) models of DABO analogues [28]. The rationale of the present work is to explore the structure activity relationship of E-State indices of DABO compounds and the biological activity in a quantitative manner using linear (MLR) as well as non-linear (NN and k-NN) techniques.
2. MATERIALS AND METHODS
A series of 74 compounds of DABO derivatives was taken from the literature [28] for the QSAR study. All the 74 compounds were sketched using V-life MDS software [41]. The inhibitory activity pIC50 was taken as the biological activity variable (dependent). The functional groups namely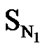 , SN3, SO, SX, SY, SAr, SS, SF, SCl, and
, SN3, SO, SX, SY, SAr, SS, SF, SCl, and 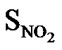 were considered as descriptor variable (independent) in the QSAR analysis. The Kier-Hall Electrotopological State (E-State) indices of such functional groups were calculated using E-Calc version 1.1 [42]. Multiple linear regression (MLR), neural network (NN) regression and k-Nearest Neighbor (k-NN) were calculated with V-Life MDS software.
were considered as descriptor variable (independent) in the QSAR analysis. The Kier-Hall Electrotopological State (E-State) indices of such functional groups were calculated using E-Calc version 1.1 [42]. Multiple linear regression (MLR), neural network (NN) regression and k-Nearest Neighbor (k-NN) were calculated with V-Life MDS software.
2.1. Kier-Hall Electrotopological State (E-State) Indices [38]
Electrotopological State (E-State) indices are widely used in QSAR modeling, including AIDS related research [21,43,44]. The large amount of variables in E-State Indices represent the structural characteristics of molecules such as information about their non-covalent interactions. It encodes electronic and topological information in a single number for each skeletal atom in the hydrogen suppressed graph of a molecule. The E-State index value (S) of an atom “I” is given by the sum of the intrinsic state value (I) and perturbations (ΔI) by the fields of all other atoms in the chemical graph, including substituent atoms. Thus, S is defined by the following set of equations:
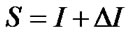
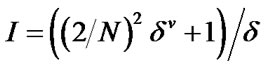
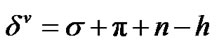
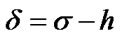
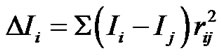
where, N = Principal quantum number (i.e., row in which the atom occurs in the periodic table)
σ = the number of sigma electron contributed by the atom.
Π = the number of pi electron contributed by the atom.
h = the number of hydrogen atoms attached to the atom n = the number of lone pair of e– on the atom r = the number of atoms in the shortest graph path connecting atom “I” and other atom “j” in the chemical graph of the whole molecule.
The resulting E-State of any atom is a numerical value depicting the accessibility of that atom to interaction across space with some reference atom (s) or group (s).
2.2. Statistical Analyses
2.2.1. Multiple Linear Regression (MLR)
Multiple linear regression (MLR) is a method used to model the relationship between two or more explanatory variables and a response variable by fitting a linear equation to the observed was employed to correlate the binding affinity and molecular descriptors. This method has been widely applied in many QSAR studies, and has proven to be a useful linear regression method to build QSAR models that may explore straightforward the properties of the chemical structure in combination with its ability of inducing a pharmacological response [45]. The advantage of MLR is its simple form and easily interpretable mathematical expression.
2.2.2. Neural Network (NN)
Intriguing approaches using machine learning methods have been studied in the field of QSAR modeling [46]. The concept of neural network was first introduced in 1943 by McCulloch and Pitts [47]. Interest in neural networks was slow until the 1980s when new computer architecture and learning algorithms began to appear. The use of neural networks (NN) in all fields has since grown substantially. In 1988, Hoskins et al. reported the first use of process control in chemistry [48]. NNs are well suited for modelling complex relationships between the variables and non-linearity in the relationships is suspected.
2.2.3. Kernel Nearest Neighbor (k-NN)
k-NN [49,50] is a simple decision scheme that requires practically no training and is asymptotically optimal, i.e., with increase in training data it converges to the optimal prediction error. For a given compound in the descriptor space, the method analyzes its k-nearest neighboring compounds from the training set and predicts the activity class that is most highly represented among these neighbors.
3. RESULTS AND DISCUSSION
Table 1 records a series of 74 DABO derivatives along with the position of substituents (X, Ar and Y), where X represents substituent such as methyl attached at C-5 position, Ar represents the aryl groups substituted with the terminal methyl group which is attached at C-6 position of the pyrimidinone ring and Y represents sulphur substituted moieties attached at C-2 position of the basic nucleus. It also records the inhibitory concentration pIC50 (where IC50 is the effective concentration of a compound required to activate 50% protection of MT-4 cell against the cytopathic effect of HIV-1) and the calculated E-State values (N1, N3, O, X, Y, Ar, S, F, Cl, and NO2) of various atoms or groups in the DABO analogues.
The correlation matrix for the correlation of E-State Indices with pIC50 for the training set of DABO derivatives is shown in Table 2. A perusal of the correlation matrix indicates that in univariate correlation 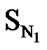 has the highest correlation potential while SX has the lowest one.
has the highest correlation potential while SX has the lowest one.
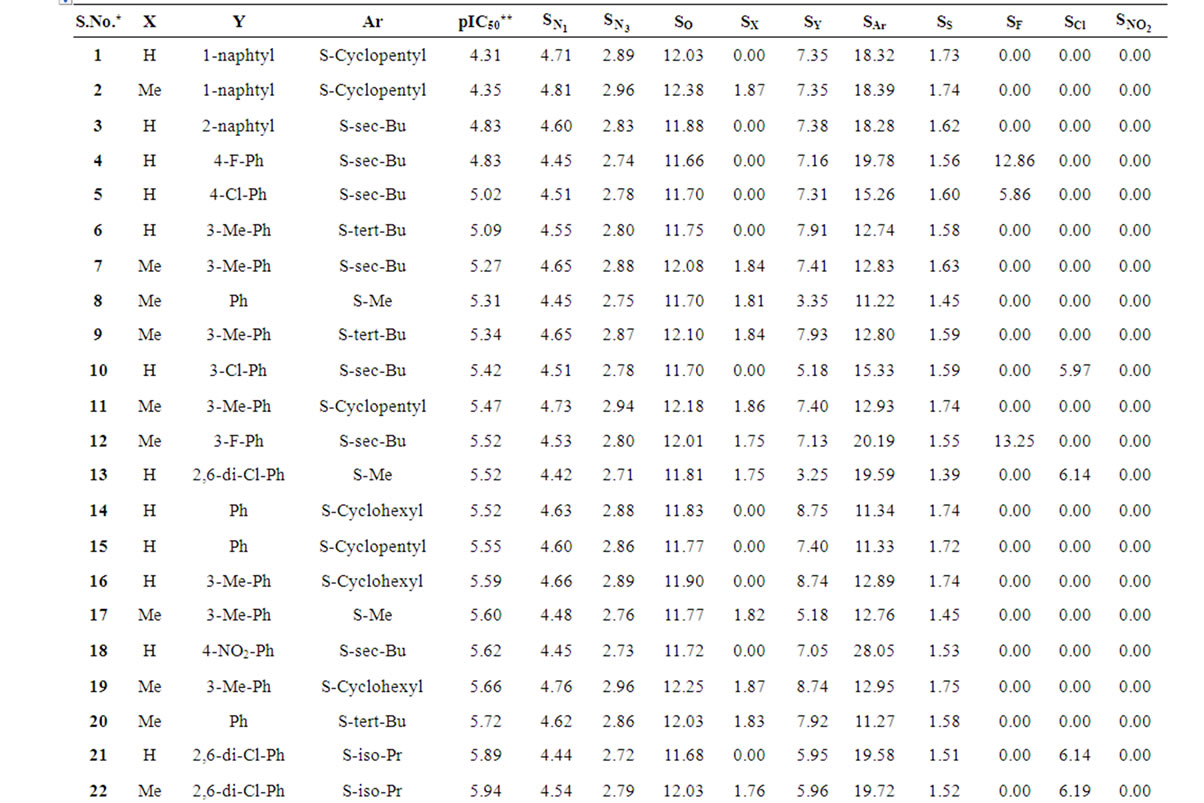
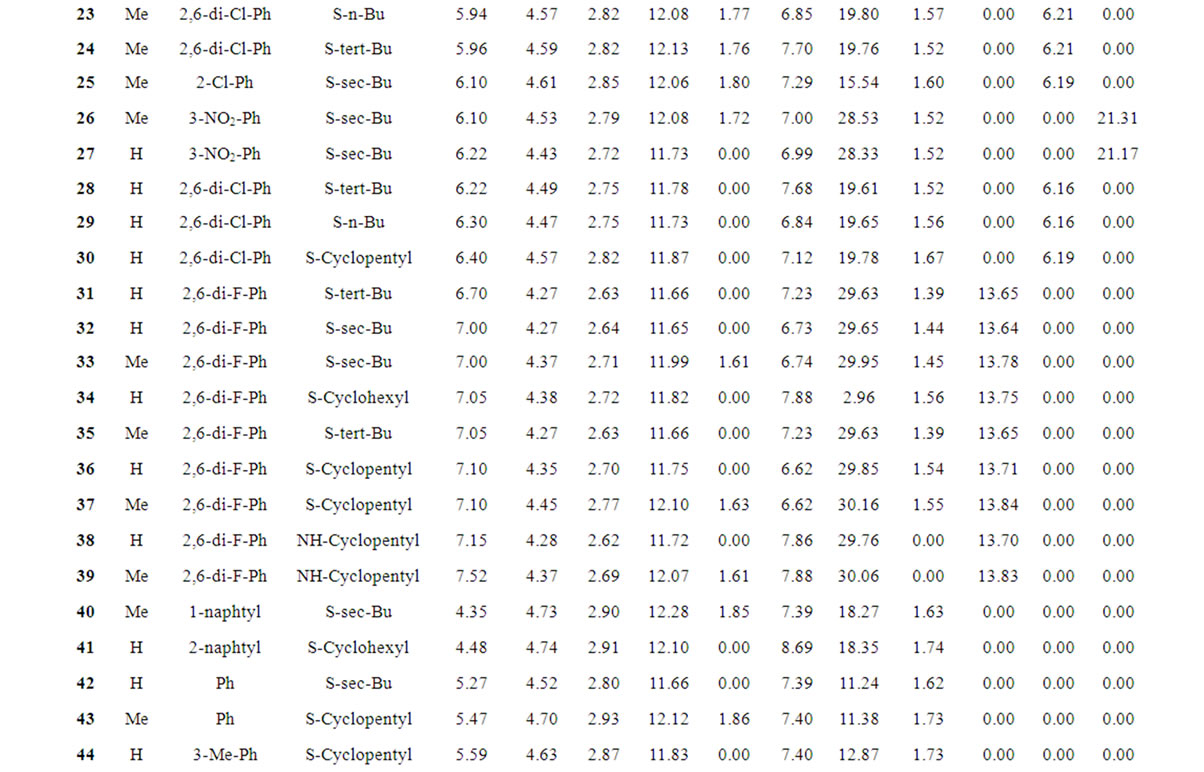
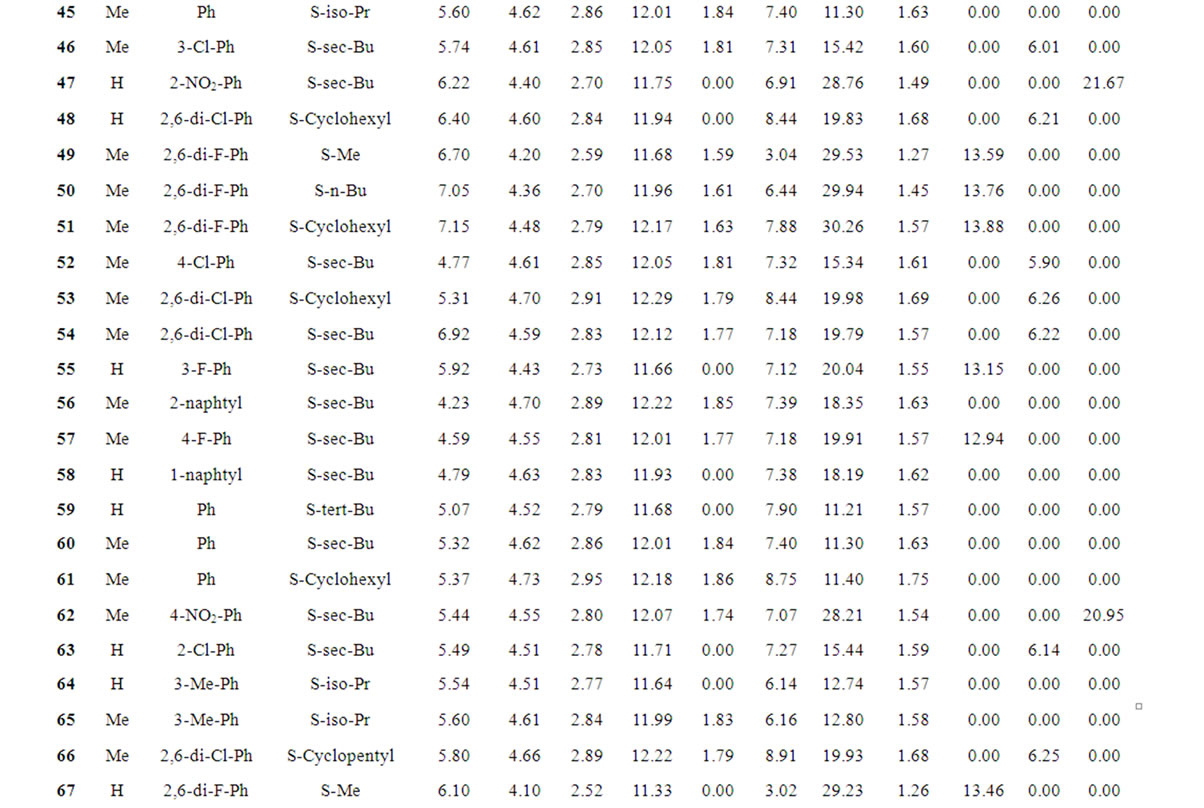
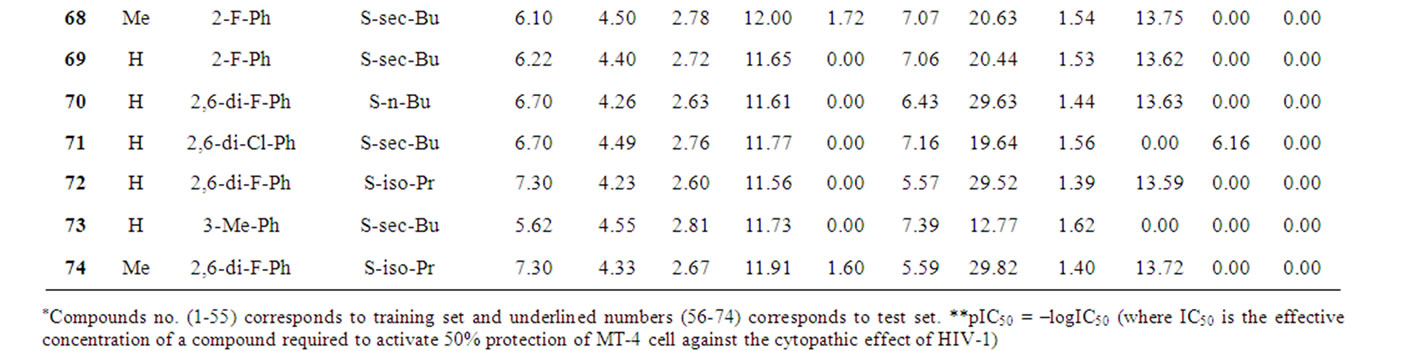
Table 1. Table represents the various substituents with the anti-HIV-1 activity (pIC50) and E-State indices of Dihydroalkoxybenzyloxopyrimidines (DABO) derivatives.
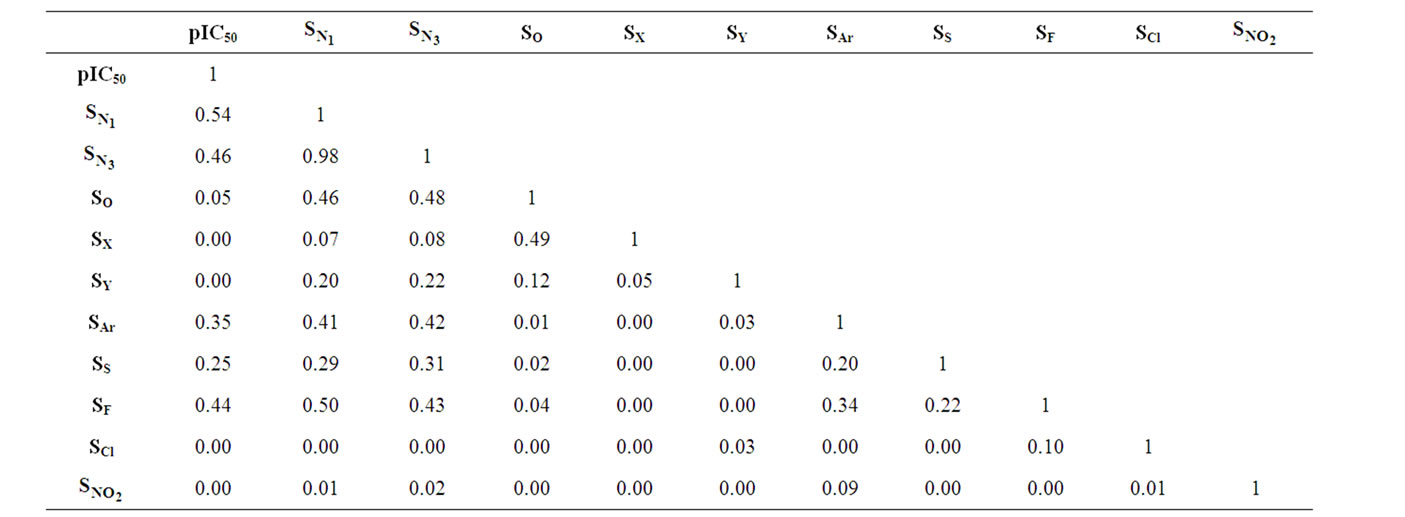
Table 2. Correlation matrix between the E-State Indices and biological activity of DABO derivatives.
The order of univariate correlations of E-State values for various substituents attached to the parent compound with inhibitory activity follow the order:
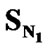 >
>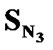 > SF > SAr > SS > SO >
> SF > SAr > SS > SO >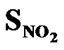 > SCl = SY > SX
> SCl = SY > SX
The QSAR models were generated using MLR, NN and k-NN techniques to study the antiviral activity of Dihydroalkoxybenzyloxopyrimidines derivatives. The dataset of 74 compounds was divided into a training set of 55 compounds for developing the MLR, NN and k-NN models and a prediction test set of 19 compounds were used to determine the external predictivity of the QSAR model. Both the training and the test sets were divided manually according to a representative range of biological activities and structural variations. Similar training and test sets were used to perform MLR, NN and k-NN methods.
3.1. Multiple Linear Regression (MLR)
The statistical analysis was performed by multiple linear regression method using back-propagation. The best model is presented by the Eq.1 for the training set of 55 compounds.
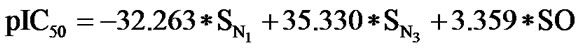
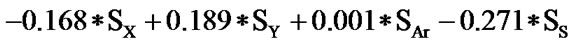
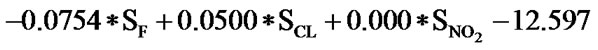 (1)
(1)
(n = 51; r2 = 0.961; F = 100.114; q2 = 0.926)
In Eq.1 a high positive coefficient for 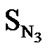 indicates that the presence of nitrogen at 3rd position in the compound enhances the activity while a high negative coefficient for
indicates that the presence of nitrogen at 3rd position in the compound enhances the activity while a high negative coefficient for 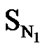 indicates the presence of nitrogen at 1st position will decrease the antiviral activity to a great extent. It has been deduced earlier that the 3-NH function enhances anti-HIV activity by donation of a hydrogen bond to the carbonyl oxygen of Lys101 in RT. [51] This result coincide with the results reported by Guang Fu Sun et al that 3-NH of DABO was essential for the inhibition of HIV RT. [52] As far as functional groups such as oxygen, nitro and sulphur are concerned, O and NO2 are the ones contributing towards enhancing the activity whilst presence of sulphur is exhibiting a retarding effect on the biological activity. Although, the coefficients for halogen groups namely F and Cl are not very significant (low coefficient) for activity, but substitution of fluorine group at benzyl group is less beneficial than chlorine moiety. This has been also observed in the work of Elena Petricci et al. [53] but their previous finding also highlighted the importance of 2, 6-difluorobenzyl substituent at position 6 for optimal activity. [54] The substitution of S-alkyl and NH-alkyl at C-2 position is essential, though it does not contribute much towards the activity. Similarly, presence of Ar exhibit negligible effect on inhibitory activity with its low positive coefficient value. On the other hand substitution of methyl group at C-5 position is unfavorable and exhibits a negative impact on the inhibitory activity. An earlier work also suggest that the DABOs are active when an H atom is present at position 5 of the pyrimidine ring of DABO derivatives [25].
indicates the presence of nitrogen at 1st position will decrease the antiviral activity to a great extent. It has been deduced earlier that the 3-NH function enhances anti-HIV activity by donation of a hydrogen bond to the carbonyl oxygen of Lys101 in RT. [51] This result coincide with the results reported by Guang Fu Sun et al that 3-NH of DABO was essential for the inhibition of HIV RT. [52] As far as functional groups such as oxygen, nitro and sulphur are concerned, O and NO2 are the ones contributing towards enhancing the activity whilst presence of sulphur is exhibiting a retarding effect on the biological activity. Although, the coefficients for halogen groups namely F and Cl are not very significant (low coefficient) for activity, but substitution of fluorine group at benzyl group is less beneficial than chlorine moiety. This has been also observed in the work of Elena Petricci et al. [53] but their previous finding also highlighted the importance of 2, 6-difluorobenzyl substituent at position 6 for optimal activity. [54] The substitution of S-alkyl and NH-alkyl at C-2 position is essential, though it does not contribute much towards the activity. Similarly, presence of Ar exhibit negligible effect on inhibitory activity with its low positive coefficient value. On the other hand substitution of methyl group at C-5 position is unfavorable and exhibits a negative impact on the inhibitory activity. An earlier work also suggest that the DABOs are active when an H atom is present at position 5 of the pyrimidine ring of DABO derivatives [25].
3.2. Neural Networks (NN)
An NN model was developed for a training set of 55 DABO derivatives. V-life MDS software has been used to perform NN analysis. A typical artificial neural network (ANN) consists of an input layer, a hidden layer and an output layer (neurons). Each neuron in any layer is fully connected with the neuron of a succeeding layer. There are neither connections between the neurons within a layer nor any direct connections between those of the input and output layers. A convergence criterion implies that the rms error value between the predicted and observed values is lower than the convergence criteria. Epoch means number or iterations in neural network runs. Pruning limit indicates the limit of variance of weights. Following parameters were set for the NN regression: max training epoch was set to 100,000 with pruning limit 0.3, two hidden neurons were set in the hidden layer. The convergence criteria were set to 0.01 and the variance cut off was 0.01 without scaling the data.
3.3. k-Nearest Neighbour (k-NN)
The k-NN model was built using V-life Molecular Design Software. The k-NN regression analysis was performed using following parameters: data were preprocessed using auto scaling option from the software, number of max neighbour = 5, number of min neighbour = 2 and variance cut-off = 0.01. The prediction ability method used was distance based weighted average.
The corresponding observed and predicted values of the pIC50 values of 55 compounds studied by MLR, NN and k-NN in this work are given in Table 3. The predictive model building abilities of all the three methods were analyzed and compared. Also, the quality of correlation is demonstrated by their respective residual values i.e. the difference between observed and calculated pIC50 values. The respective residual values thus obtained are also given in Table 3.
The models predictive power was judged based on various statistical parameters namely correlation coefficient (r2), Fischer statistical or fitness value (F) and cross validation coefficient (q2). All these statistical parameters were computed as defined in the V-Life MDS software.
A plot of observed and calculated pIC50 values of NN, MLR and k-NN for the training set of DABO derivatives is presented in Figure 1.
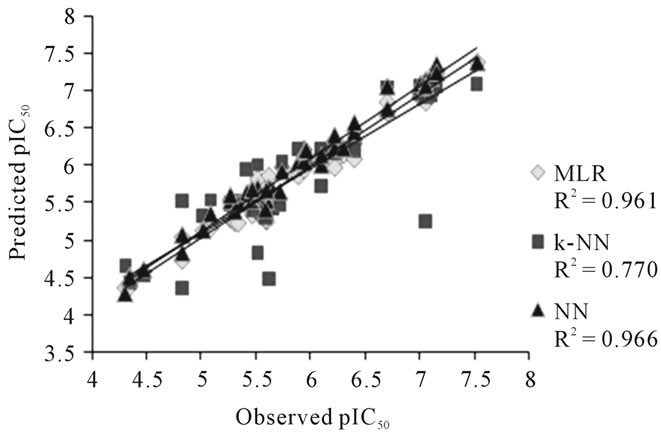
Figure 1. Graph between observed and calculated pIC50 values for training set.
3.4. Uni-column Statistics
The min and max values in both train and test set should be compared in a way that
• The maximum of the test should be less than or equal to maximum of training set.
• The minimum of the test set should be greater than or equal to minimum of the training set.
The above observation show that the test set is interpolative i.e. derived within the min-max range of the training set. The mean and standard deviation of the training and test set provides insight to the relative difference of mean and point density distribution (along mean) of the test set. In this case the mean in the training set slightly higher than the test set shows the presence of relatively more active molecules as compared to the inactive ones. Also a relatively higher standard deviation in training set indicated that training set has widely distributed activity of the molecules as compared to the test set.
Uni-column statistics for training set:

Uni-column statistics for test set:

A test set of 19 compounds are reported in Table 4 and is used to verify the validity of the QSAR model of training set. Table 4 also represents the predicted and observed values obtained by the statistical methods (MLR, NN and k-NN) showing a degree of relatedness.
A plot of observed and calculated pIC50 values of MLR, k-NN and NN for the test set of DABO derivatives is presented in Figure 2.
Methods namely MLR, NN and k-NN regression are widely used methods for building QSAR model. It is simple to interpret a regression model, in which contribution of each descriptor could be observed by the magnitude of its regression coefficient. In case of MLR, high value of squared regression coefficient r2 = 0.961 was observed. The results obtained from NN were the most significant and gave a very high value of squared regression coefficient r2 = 0.966. Though, the results of k-NN were significant with squared regression coefficient r2 = 0.770, but were inferior amongst all the three methods. The results of all the three methods could be cross validated with the squared regression coefficient between observed activity and predicted activity values of the test set compounds. For the best linear model, i.e., MLR (r2 =
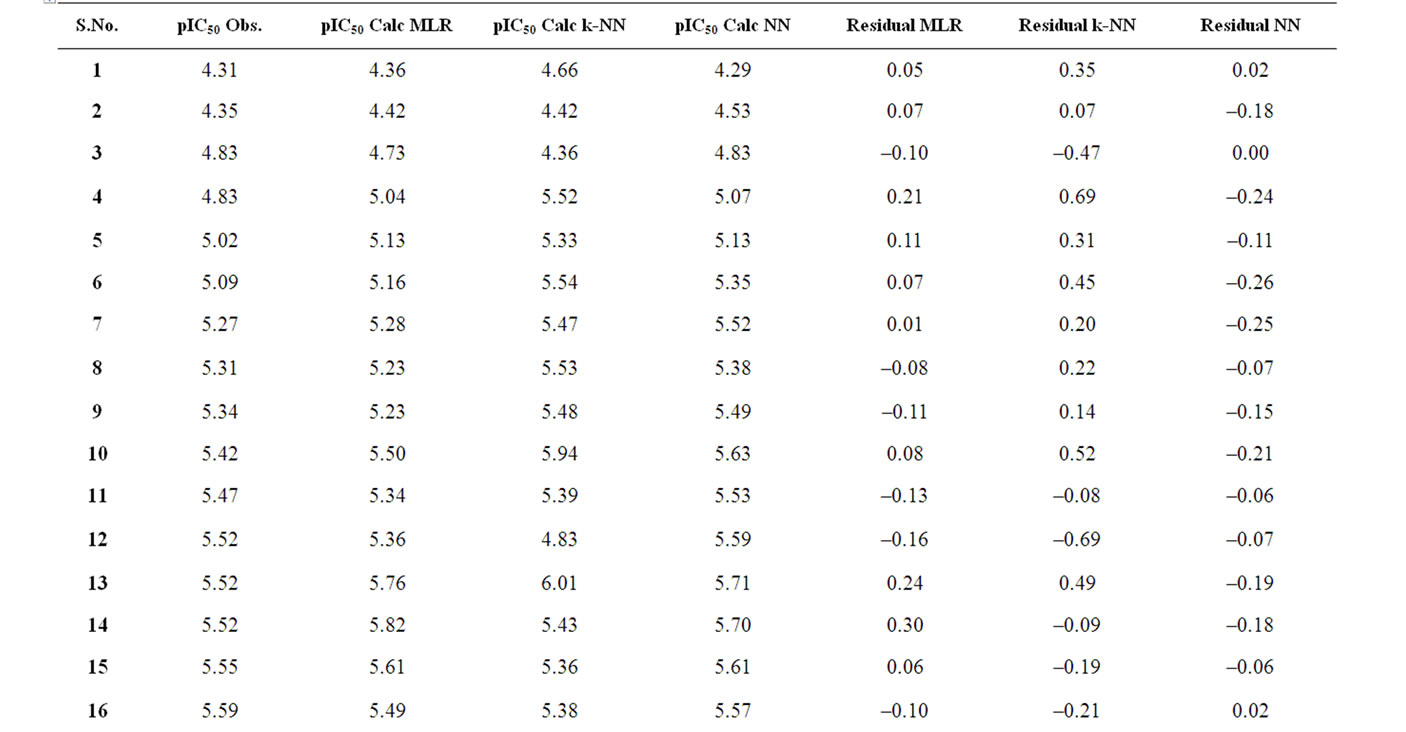
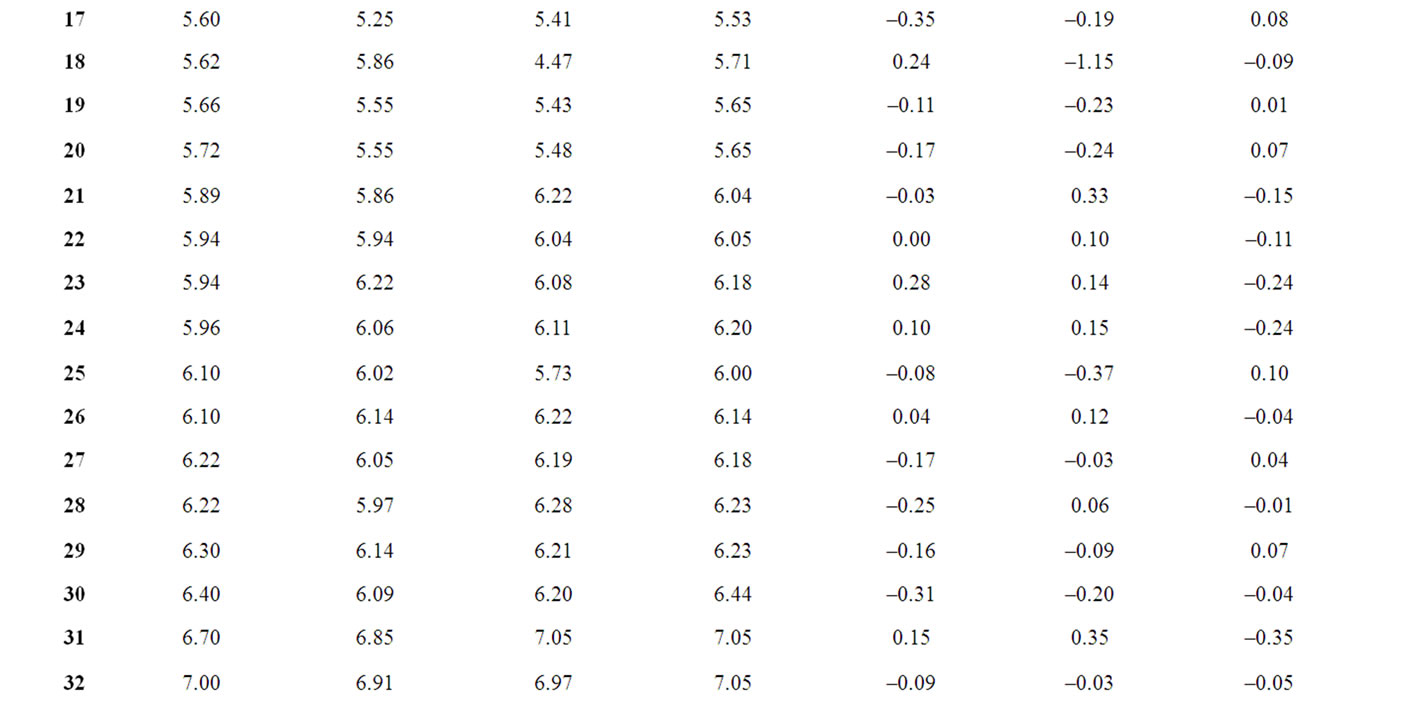
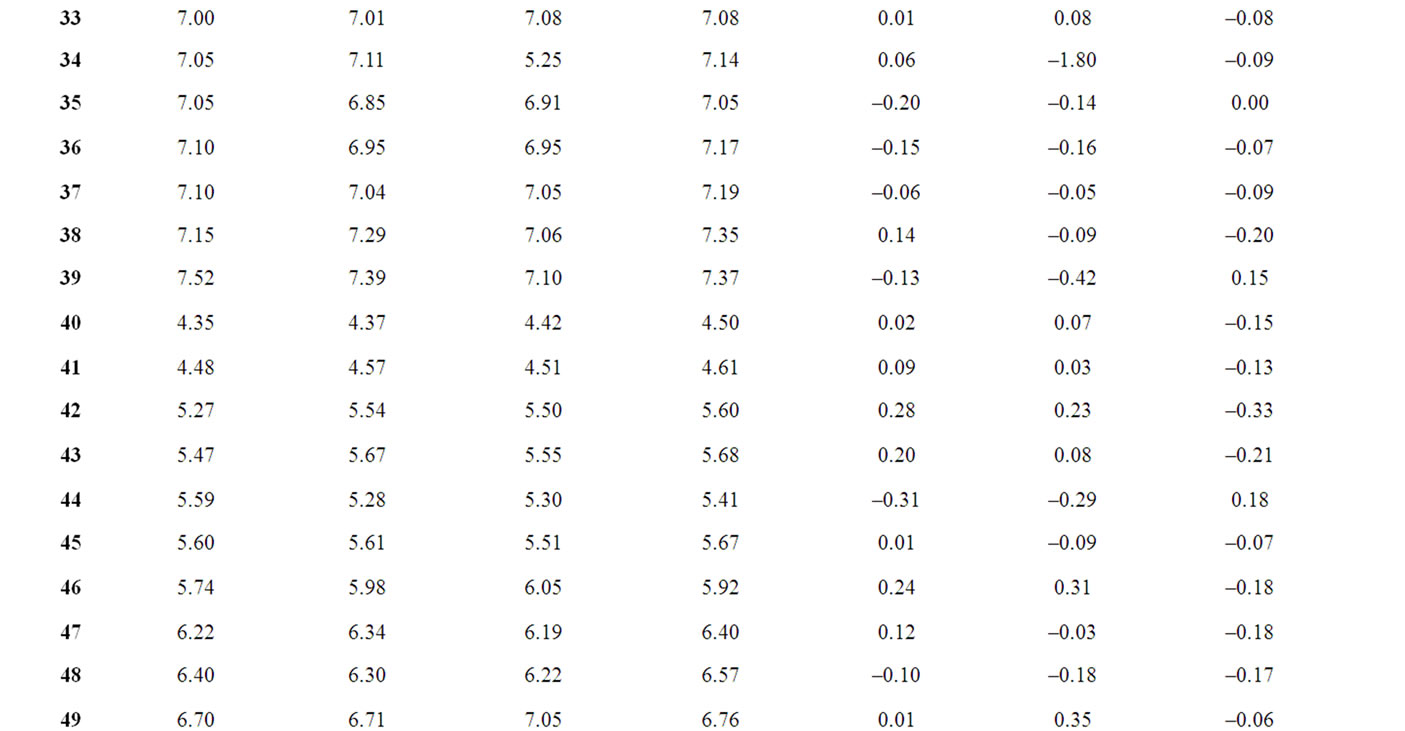
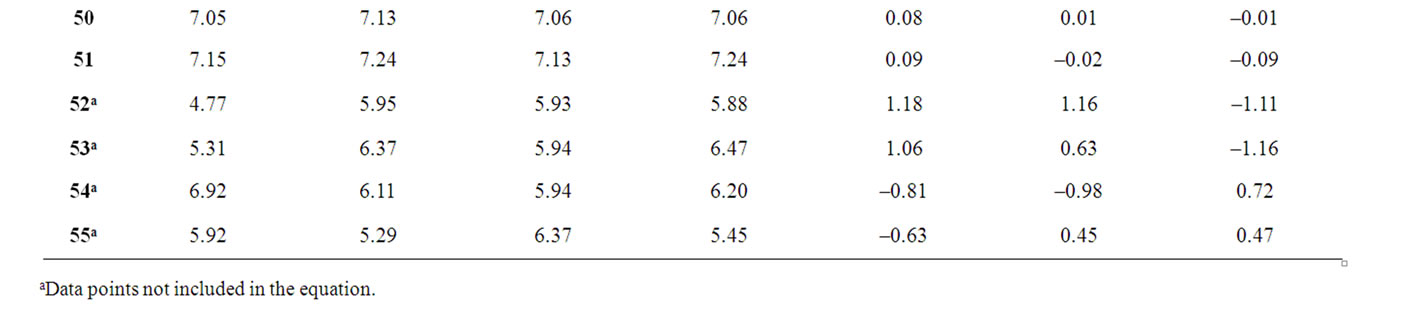
Table 3. Observed and calculated anti-HIV-1 activity (pIC50) of dihydroalkoxybenzyloxopyrimidines (DABO) derivatives (training set).
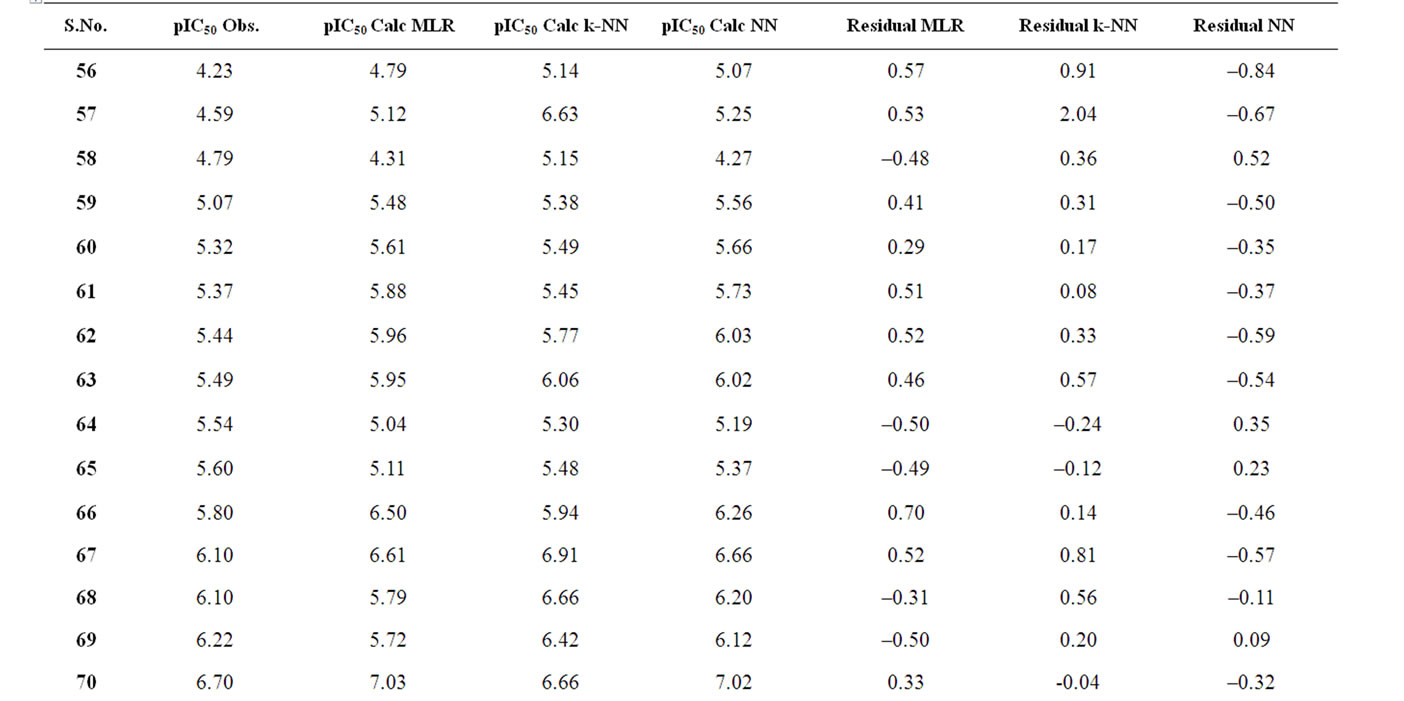

Table 4. Observed and calculated anti-HIV-1 activity (pIC50) of dihydroalkoxybenzyloxopyrimidines (DABO) derivatives (test set).
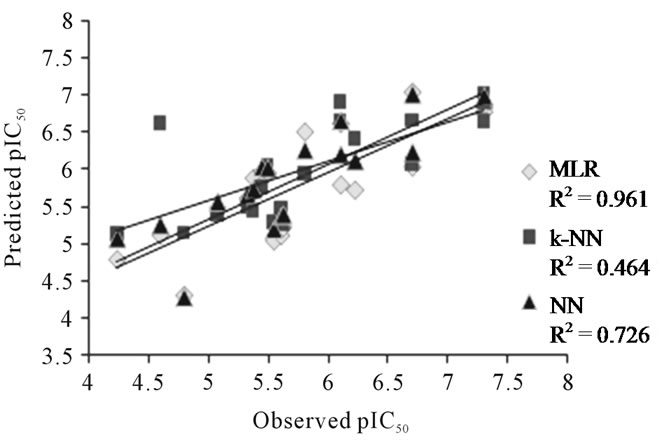
Figure 2. Graph between observed and calculated pIC50 values for test set.
0.645) was lower in comparison to the best non-linear model developed using NN (r2 = 0.726) but that of k-NN non-linear model gave very low value of correlation coefficient (r2 = 0.464). Also the degree of relatedness could be visualized by the difference between the observed and predicted values of pIC50 i.e. from the residual values. Thus, the above results account for some non-linear relationship between the pIC50 and E-State Indices.
4. CONCLUSIONS
From the above study related to Dihydroalkoxybenzyloxopyrimidines derivatives following conclusions can be drawn: The 2D-QSAR studies indicated that the anti-HIV-1 activity of DABO derivatives was strongly dependent on the nature of the substituents at N1, N3, C2, C4, C5, and C6 of the pyrimidine ring. Three models developed here viz. MLR, NN and k-NN has different level of robustness for the complete set of compounds. Among the three methods, NN was found to be more successful than the linear model MLR, reflecting that the relationship between descriptors and antiviral activity of DABO derivatives being nonlinear. Though, MLR regression is a simple but powerful method to obtain a subset of significant input variables but it does not account for non-linear relationships while NN plays a significant role depicting non-linearity in building structure activity relationships. Also the degrees of freedom were found to be 40 less than the data points i.e. 51 which proves that the model fits the general trend. Our studies have proven the feasibility of the combination of the E-State indices and the NN method to build successful QSAR models.
The most relevant structural conclusions of this study are the following: 1) replacement of pyrimidine nucleus with pyridine nucleus enhance the activity; 2) the presence of bulkiness at the C2 position would lead to higher activity compounds; 3) presence of oxygen group at C4 position in the parent compound is favourable for the activity; 4) DABOs are active with hydrogen in the fifth position comparable with compounds having methyl group.
5. ACKNOWLEDGEMENTS
Authors thank to MP council of Science and Technology (MPCST) R&D/C-14/08-09 and Dr. P. K. Sen, Director, SGSITS, Indore for providing necessary facilities.
REFERENCES
- Lam, S.N., Acharya, P., Wyatt, R., Kwong, P.D. and Bewley, C.A. (2008) Tyrosine-sulfate isosteres of CCR5 N-terminus as tools for studying HIV-1 entry. Bioorganic Medicinal Chemistry, 16, 10113-10120. doi:10.1016/j.bmc.2008.10.005
- AIDS Epidemic Update (2008). http://data.unaids.org/pub/Report/2009/JC1796_Outlook_en.pdf
- Adamson, C.S. and Freed, E.O. (2010) Novel approaches to inhibiting HIV-1 replication. Antiviral Research, 85, 119-141. doi:10.1016/j.antiviral.2009.09.009
- Sarafianos, S.G., Marchand, B., Das, K., Himmel, D.M., Parniak, M.A., Hughes, S.H. and Arnold, E. (2009) Structure and function of HIV-1 reverse transcriptase: Molecular mechanisms of polymerization and inhibition. Journal of Molecular Biology, 385, 693-713. doi:10.1016/j.jmb.2008.10.071
- Sluis-Cremer, N. and Tachedjian, G. (2002) Modulation of the oligomeric structures of HIV-1 retroviral enzymes by synthetic peptides and small molecules. European Journal of Biochemistry, 269, 5103-5111. doi:10.1046/j.1432-1033.2002.03216.x
- Sahlberg, C. and Zhou, X.-X. (2008) Development of non-nucleoside reverse transcriptase inhibitors for antiHIV therapy. Anti-Infective Agents in Medicinal Chemistry, 7, 101-117.
- Jäger, J. Smerdon, S.J., Wang, J. Boisvert, D.C. and Steitz, T.A. (1994) Comparison of three different crystal forms shows HIV-1 reverse transcriptase displays an internal swivel motion. Structure, 2, 869-876.
- Kopp, E.B., Miglietta, J.J., Shrutkowski, A.G., Shih, C.K., Grob, P.M. and Skoog, M.T. (1991) Steady state kinetics and inhibition of HIV-1 reverse transcriptase by a nonnucleoside dipyridodiazepinone, BI-RG-587, using a heteropolymeric template. Nucleic Acids Research, 19, 3035-3039. doi:10.1093/nar/19.11.3035
- Cihlar, T. and Ray, A.S. (2010) Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Research, 85, 39-58. doi:10.1016/j.antiviral.2009.09.014
- Jochmans, D., Vingerhoets, J., Arnoult, E., Geeraert, L. and Guillemont, J. (2009) Antiviral research: Strategies in antiviral drug discovery. Chapter 3, human immunodeficiency virus type 1 non-nucleoside reverse transcriptase inhibitors. ASM Press, Washington, 33-50.
- Spence, R.A., Kati, W.M., Anderson, K.S. and Johnson, K.A. (1995) Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Science, 267, 988-993. doi:10.1126/science.7532321
- Kohlstaedt, L.A., Wang, J., Friedman, J.M., Rice, P.A. and Steitz, T.A. (1992) Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science, 256, 1783-1790. doi:10.1126/science.1377403
- Tambuyzer, L., Azijn, H., Rimsky, L.T., Vingerhoets, J., Lecocq, P., Kraus, G., Picchio, G. and De Bethune, M.P. (2009) Compilation and prevalence of mutations associated with resistance to non-nucleoside reverse transcriptase inhibitors. Antiviral Therapy, 14, 103-109.
- Baba, M., Tanaka, H., De Clercq, E., Pauwels, R., Balzarini, J., Schols, D., Nakashima, H., Perno, C.F., Walker, R. T. and Miyasaka, T. (1989) Highly specific inhibition of human immunodeficiency virus type 1 by a novel 6-substituted acyclouridine derivative. Biochemical and Biophysical Research Communities, 165, 1375-1381. doi:10.1016/0006-291X(89)92756-3
- Miyasaka, T., Tanaka, H., Baba, M., Hayakawa, H., Walker, R.T., Balzarini, J. and De Clercq, E. (1989) A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxy ethoxy)methyl]-6-(phenylthio)thymine. Journal of Medicinal Chemistry, 32, 2507-2509. doi:10.1021/jm00132a002
- Debyser, Z., Pauwels, R., Andries, K., Desmyter, J., Kukla, M., Janssen, P.A. and De Clercq, E. (1991) An antiviral target on reverse transcriptase of human immunodeficiency virus type 1 revealed by tetrahydroimidazo-[4,5,1-jk][1,4]benzodiazepin-2(1H)-one and -thione derivatives. Proceedings of National Academy of Sciences USA, 88, 1451-1455. doi:10.1073/pnas.88.4.1451
- Pauwels, R., Andries, K., Desmyter, J., Schols, D., Kukla, M.J., Breslin, H.J., Raeymaeckers, A., Van Gelder, J., Woestenborghs, R., Heykants, J., Schellekens, K., Janssen, M.A.C., De Clercq E., Janssen, P.A.J. (1990) Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature, 343, 470-474. doi:10.1038/343470a0
- De Bethune, M.P. (2010) Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989-2009). Antiviral Research, 85, 75-90. doi:10.1016/j.antiviral.2009.09.008
- Dickinson, L., Khoo, S. and Back, D. (2010) Pharmacokinetics and drug-drug interactions of anti-retrovirals: An update. Antiviral Research, 85, 176-189. doi:10.1016/j.antiviral.2009.07.017
- Sapre, N.S., Gupta, S., Pancholi, N. and Sapre, N. (2009) A group center overlap based approach for “3D QSAR” studies on TIBO derivatives. Journal of Computational Chemistry, 30, 922-933. doi:10.1002/jcc.21114
- Sapre, N.S., Pancholi, N., Gupta, S., Sikarwar, A. and Sapre, N. (2007) Molecular Modelling studies on 2- Amino 6-aryl-Sulphonylbenzonitriles as non-nucleoside reverse transcriptase inhibitors of HIV-1: A QSPR approach. Journal of Chemical Sciences, 119, 625-630. doi:10.1007/s12039-007-0078-5
- Sapre, N.S., Gupta, S., Pancholi, N. and Sapre, N. (2008) Molecular docking studies on tetrahydroimidazo-[4,5,1-jk] [1,4]-benzodiazepinone (TIBO) derivatives as HIV-1 NNRT inhibitors. Journal of Computer Aided Molecular Design, 22, 69-80. doi:10.1007/s10822-007-9161-8
- Sapre, N.S., Gupta, S. and Sapre, N. (2008) Assessing ligand efficiencies using template-based molecular docking and tabu-clustering on tetrahydroimidazo-[4,5,1-jk] [1,4]-benzodiazepin-2(1H)-one-thione (TIBO) derivatives as HIV-1RT inhibitors. Journal of Chemical Science, 120, 395-404. doi:10.1007/s12039-008-0063-7
- Botta, M., Artico, M., Massa, S., Gambacorta, A., Marongiu, M.E., Pani, A. and La Colla, P. (1992) Synthesis, antimicrobial and antiviral activities of isotrimethoprim and some related derivatives. European Journal of Medicinal Chemistry, 27, 251-257. doi:10.1016/0223-5234(92)90009-P
- Costi, R., Di Santo, R., Artico, M., Massa, S., Lavecchia, A., Marceddu, T., Sanna, L., La Colla, P. and Marongiu, M.E. (2000) Structure-activity relationship studies on potential non-nucleoside DABO-like inhibitors of HIV-1 reverse transcriptase. Antiviral Chemistry and Chemotherapy, 11, 117-133.
- Sbardella, G., Mai, A., Artico, M., Chimenti, P., Massa, S., Loddo, R., Marongiu, M.E., La Colla, P. and Pani, A. (2001) Structure-activity relationship studies on new DABOS: Effect of substitutions at pyrimidine C-5 and C-6 positions on anti-HIV-1 activity. Antiviral Chemistry and Chemotherapy, 12, 37-50.
- Yin, L.-Q., Yu, S.-W., Yao, L.-F., He, Y.-P. and Xie, X.-G., (2008) QSAR studies on 6-(1-naphthylmethyl) substituted S-DABO derivatives as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Chinese Journal of Structural Chemistry, 27, 1214-1222.
- De Brito, M.A., Rodrigues, C.R., Cirino, J.J., De Alencastro, R.B., Castro, H.C. and Albuquerque, M.G. (2008) 3D-QSAR CoMFA of a series of DABO derivatives as HIV-1 reverse transcriptase non-nucleoside inhibitors. Journal of Chemical Information and Modelling, 48, 1706-1715. doi:10.1021/ci8001217
- Sudbeck, E.A., Mao, C., Vig, R., Venkatachalam, T.K., Tuel-Ahlgren, L. and Uckun, F.M. (1998) Structure-based design of novel dihydroalkoxybenzyloxopyrimidine derivatives as potent non-nucleoside inhibitors of the human immunodeficiency virus reverse transcriptase. Antimicrobial Agents and Chemotherapy, 42, 3225-3233.
- Mai, A., Sbardella, G., Artico, M., Ragno, R., Massa, S., Novellino, E., Greco, G., Lavecchia, A., Musiu, C., La Colla, M., Murgioni, C., La Colla, P. and Loddo, R., (2001) Structure-based design, synthesis, and biological evaluation of conformationally restricted novel 2-alkyl thio-6-[1-(2,6-difluorophenyl)alkyl]-3,4-dihydro-5-alkyl-pyrimidin-4 (3H)-ones as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Journal of Medicinal Chemistry, 44, 2544-2554. doi:10.1021/jm010853h
- Tramontano, E., Marongiu, M.E., de Montis, A., Loi, A. G., Artico, M., Massa, S., Mai, A. and La Colla, P. (1994) Characterization of the anti-HIV-1 activity of 3,4-dihydro-2-alkoxy-6-benzyl-4-oxopyrimidines (DABOs), new non-nucleo side reverse transcriptase inhibitors. New Microbiologica, 17, 269-279.
- Mai, A., Artico, M. Ragno, R., Sbardella, G., Massa, S., Musiu, C., Mura, M., Marturana, F., Cadeddu, A., Maga, G., and La Colla, P. (2005) 5-Alkyl-2-alkyl amino-6- (2,6-difluorophenylalkyl)-3,4-di hydro pyrimidin-4(3H)- ones, a new series of potent, broad-spectrum non-nucleoside reverse transcriptase inhibitors belonging to the DABO family. Bioorganic Medicinal Chemistry, 13, 2065-2077. doi:10.1016/j.bmc.2005.01.005
- Mai, A., Artico, M., Sbardella, G., Massa, S., Loi, A. G., Tramontano, E., Scano, P. and La Colla, P. (1995) Synthesis and anti-HIV-1 activity of thio analogues of Dihydroalkoxybenzyloxopyrimidines. Journal of Medicinal Chemistry, 38, 3258-3263. doi:10.1021/jm00017a010
- Mai, A., Artico, M., Sbardella, G., Massa, S., Novellino, E., Greco, G., Loi, A.G., Tramontano, E., Marongiu, M.E. and La Colla, P. (1999) 5-Alkyl-2-(alkylthio)-6-(2,6-di halophenylmethyl)-3,4-dihydropyrimidin-4(3H)-ones: Novel potent and selective dihydro-alkoxy-benzyl-oxopyrimidine derivatives. Journal of Medicinal Chemistry, 42, 619-627. doi:10.1021/jm980260f
- Qin, H., Liu, C., Guo, Y., Wang, R., Zhang, J., Ma, L., Zhang, Z., Wang, X., Cui, Y. and Liu, J. (2010) Synthesis and biological evaluation of novel C5 halogen-functionalized S-DABO as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorganic Medicinal Chemistry, 18, 3231-3237. doi:10.1016/j.bmc.2010.03.025
- Deeb, O., Hemmateenejad, B., Jaber, A., Garduno-Juarez, R. and Miri, R. (2007) Effect of the electronic and physicochemical parameters on the carcinogenesis activity of some sulfa drugs using QSAR analysis based on genetic-MLR and genetic-PLS. Chemosphere, 67, 2122- 2130. doi:10.1016/j.chemosphere.2006.12.098
- Shen, Q., Shi, W.M., Yang, X.P. and Ye, B.X. (2006) Particle swarm algorithm trained neural network for QSAR studies of inhibitors of platelet-derived growth factor receptor phosphorylation. European Journal of Pharmaceutical Science, 28, 369-376. doi:10.1016/j.ejps.2006.04.001
- Kier, L.B. and Hall, L.H. (1990) An electrotopological-state index for atoms in molecules. Pharmaceutical Research, 7, 801-807. doi:10.1023/A:1015952613760
- Hall, L.H. and Kier, L.B., (1995) Electrotopological state indices for atom types: A novel combination of electronic, topological, and valence state information. Journal of Chemical. Information and Computer Science, 35, 1039- 1045.
- Kier, L.B. and Hall, L.H., (1999) Molecular structure description: The electrotopological state. Academic Press, San Diego.
- VLife Sciences Technologies: MDS 3.5 (2010) Molecular Design Suite, Pvt Ltd. Pune, India.
- E-Calc version (1999) 1.1.
- Sapre, N.S., Pancholi, N. and Gupta, S. (2008) Computational modeling of substitution effect on HIV-1 non-nucleoside reverse transcriptase inhibitors with Kier-Hall electrotopological state (E-State) indices. Internet Electronic Journal of Molecular Design, 7, 55-67.
- Sapre, N.S., Pancholi, N., Gupta, S. and Sapre, N. (2008) Computational modeling of tetrahydroimidazo-[4,5,1-jk] [1,4]-benzodiazepinone derivatives: An atomistic drug design approach using Kier-Hall electrotopological state (E-State) indices. Journal of Computational Chemistry, 29, 1699-1706. doi:10.1002/jcc.20931
- Wang, H.Y., Li, Y., Ding, J., Wang, Y. and Chang, Y.Q. (2008) Prediction of binding affinity for estrogen receptor alpha modulators using statistical learning approaches. Journal of Molecular Diversity, 12, 93-102.
- Ragno, R., Mai, A., Sbardella, G., Artico, M., Massa, S., Musiu, C., Mura, M., Marturana, F., Cadeddu, A. and La Colla, P. (2004) Computer-aided design, synthesis, and anti-HIV-1 activity in vitro of 2-alkylamino-6-[1-(2,6-di fluorophenyl)alkyl]-3,4-dihydro-5-alkylpyrimidin-4(3H)- ones as novel potent non-nucleoside reverse transcriptase inhibitors, also active against the Y181C variant. Journal of Medicinal Chemistry, 47, 928-934. doi:10.1021/jm0309856
- Sun, G.F., Kuang, Y.Y., Chen, F.E., De Clercq, E., Balzarini, J. and Pannecouque, C. (2005) Non-nucleoside HIV reverse transcriptase inhibitors, Part 6: Synthesis and anti-HIV activity of novel 2-[(arylcarbonylmethyl) thio]-6- arylthio DABO analogues. Archive der Pharmazie, 338, 457-461. doi:10.1002/ardp.200400961
- Petricci, E., Mugnaini, C., Radi, M., Togninelli, A., Bernardini, C., Manetti, F., Parlato, M.C., Renzulli, M.L., Alongi, M., Falciani, C., Corelli, F. and Botta, M., (2006) Towards new methodologies for the synthesis of biologically interesting 6-substituted pyrimidines and 4(3H)- pyrimidinones. Arkivoc, 7, 452-478.
- Petricci, E., Botta, M., Corelli, F. and Mugnaini, C., (2002) An improved synthesis of solid-supported reagents (SSRs) for selective acylation of amines by microwave irradiation. Tetrahedron Letter, 43, 6507-6509. doi:10.1016/S0040-4039(02)01478-8
- Guha, R. and Jurs, P.C. (2005) Interpreting computational neural network QSAR models: A measure of descriptor importance. Journal of Chemical Information and Modelling, 45, 800-806. doi:10.1021/ci050022a
- McCulloch, W.S. and Pitts, W. (1943) A logical calculus of the ideas immanent in nervous activity. Bulletin of Mathematical Biology, 5, 115-133.
- Hoskins, J.C. and Himmelbau, D.M. (1988) Artificial neural network models of knowledge representation in chemical engineering. Computers and Chemical Engineering, 12, 881-890. doi:10.1016/0098-1354(88)87015-7
- Cover, T. and Hart, P. (1967) Nearest neighbour pattern classification. IEEE Transactions Information Theory, 13, 21-27. doi:10.1109/TIT.1967.1053964
- Hand, D., Mannila, H. and Smyth, P. (2001) Principles of data mining. The MIT Press, Cambridge.