Journal of Intelligent Learning Systems and Applications
Vol.08 No.01(2016), Article ID:62271,14 pages
10.4236/jilsa.2016.81002
Accurate Plant MicroRNA Prediction Can Be Achieved Using Sequence Motif Features
Malik Yousef1,2*, Jens Allmer3,4, Waleed Khalifa1,2
1Computer Science, The College of Sakhnin, Sakhnin, Israel
2The Institute of Applied Research, The Galilee Society, Shefa-’Amr, Israel
3Molecular Biology and Genetics, Izmir Institute of Technology, Urla, Turkey
4Bionia Incorporated, IZTEKGEB A8, Urla, Turkey
![]()
Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 7 November 2015; accepted 25 December 2015; published 28 December 2015
ABSTRACT
MicroRNAs (miRNAs) are short (~21 nt) nucleotide sequences that are either co-transcribed during the production of mRNA or are organized in intergenic regions transcribed by RNA polymerase II. In animals, Drosha, and in plants DCL1 recognize pre-miRNAs which set themselves apart by their characteristic stem loop (hairpin) structure. This structure appears important for their recognition during the process of maturation leading to functioning mature miRNAs. A large body of research is available for computational pre-miRNA detection in animals, but less within the plant kingdom. For the prediction of pre-miRNAs, usually machine learning approaches are employed. Therefore, it is necessary to convert the pre-miRNAs into a set of features that can be calculated and many such features have been described. We here select a subset of the previously described features and add sequence motifs as new features. The resulting model which we called MotifmiRNAPred was tested on known pre-miRNAs listed in miRBase and its accuracy was compared to existing approaches in the field. With an accuracy of 99.95% for the generalized plant model, it distinguishes itself from previously published results which reach an average accuracy between 74% and 98%. We believe that our approach is useful for prediction of pre-miRNAs in plants without per species adjustment.
Keywords:
MicroRNA Prediction, Plant, Bioinformatics, Machine Learning, Sequence Motifs

1. Introduction
MicroRNAs (miRNAs) are short RNA sequences that form a hairpin structure which harbors one or more mature miRNAs of about 21 nucleotides in length [1] . Mature miRNAs, when incorporated into RISC, provide a template sequence for the recognition of their target mRNAs which are then either degraded or whose translation is reduced [2] . Since their discovery by Lee and colleagues [3] , they have received increasing attention and it is now clear that in case of animals they are also involved in many diseases [4] and in case of plants play essential roles in regulation, development, response to cold stress and nutrient deprivation [5] . MicroRNAs are found in multicellular organisms ranging from sponges [6] to human, but the plant miRNA pathway may have evolved distinctly from the animal one [7] .
The experimental study of miRNAs is quite involved and complicated by the fact that the miRNA and their targets have to be expressed at the same time in the same cell to lead to a measurable effect. For this reason, computational detection of miRNAs and their targets is important [8] [9] . Different approaches to computational miRNA detection have been applied, but most approaches are based on feature extraction followed by machine learning [10] [11] . The so called ab initio miRNA detection methodology is well established in animal models for which abundant learning data are available for example in miRBase [12] .
Most studies which report new ab initio approaches to pre-miRNA prediction have used different data sets which make it difficult to compare the results. Additionally, various computational approaches (apart from machine learning) have been employed for example based on sequence conservation and/or structural similarity [13] - [17] . However, most detrimental for a true comparison of methodologies is that there is no fully annotated genome available, which would allow a proper accuracy assessment on real data. For these reasons, accuracies and other measures reported in the studies below cannot be compared directly, but can provide a general idea.
NOVOMIR [18] uses a series of filter steps and a statistical model to discriminate a pre-miRNA from other RNAs and reports a sensitivity of 80% at a specificity of 99%. MiRenSVM an algorithm combining three SVM achieved a sensitivity of 93% at a specificity of 97% [19] . Xue and colleagues trained a support vector machine on human data (93% sensitivity at 88% specificity) but interestingly also achieved high accuracies of up to 90% in other species [20] . Jiang and colleagues [21] added a P-value and minimum free energy to the classification parameters of Xue and colleagues and using Random Forrest, a different classification algorithm, achieved a sensitivity of 95% at a specificity of 98%. A recent study by Zeller and coworkers employed structure/sequence conservation, homology to known microRNAs, and phylgenetic footprinting [22] . Others have used homology searches for revealing paralog and ortholog miRNAs [14] [23] - [26] . Additionally, Wang and others [27] developed a method based on sequence and structure alignment for miRNA identification. Finally, Hertel and Stadler included multiple sequence alignment for microRNA detection [28] .
Many algorithms for miRNA gene prediction are based on machine learning strategies. In general, these algorithms need a sufficient number of positive as well as negative examples. Although many miRNA genes seem to be unique in any organism, positive training examples can easily be found whereas negative examples are hard to come by [19] [29] - [31] . Some negative examples that were picked in studies, for example mRNA sequences [32] are dubious since to our current knowledge miRNAs can originate from any part of a pri-miRNA. Thus, defining the negative class is a major challenge in training machine learning algorithms for miRNA discovery. For this reason, one-class machine learning which only needs positive examples has been tried [20] [31] .
As pointed out above, plant miRNAs may have evolved distinct from animal ones and thus the approaches for miRNA detection introduced so far may need to be adapted when applied to plant miRNA detection. It has been found that plant miRNAs are more variable in size and very heterogeneous, but usually larger than animal miRNAs. Also their base pairing propensity (bonds in the stem) seems to be more extensive and their length is close to 21 nucleotides [33] . Billoud and colleagues predicted miRNAs in brown algae, which are different from both land plants and animals using a set of normalized features like Shannon entropy that have previously been used for detection of miRNAs in plants and animals [34] . Other studies also use tools developed for miRNA detection in animals for studies in plants [18] [35] [36] . PlantMiRNAPred achieved an accuracy of more than 90% when used with multiple plant species [36] . One study shows that generalized training using multiple plant data as input for training a decision tree leads to sensitivity of 84% at a specificity of 99% [37] . This may be due to their concurrent usage of structural features and targeting parameters for miRNA prediction which is beneficial for the accuracy of miRNA prediction [38] . In Arabidopsis thaliana, one approach searched for all complementary pairs of sequences within its transcriptome of the expected size of a miRNA-mRNA duplex and then successfully filtered the results according to divergence patterns [39] .
We should note in passing that high-throughput methods for sequencing isolated small RNAs provides a new tool for discovering novel microRNA species [40] [41] and that such information for plants is available in PMRD [5] . Another new method for amplifying low-concentration microRNAs allows easier testing of predictions [42] . These tools are equally important for plant and animal models. However, this study is interested in the ab inito detection of miRNAs from genomic rather than from transcriptomic data.
Compared to animals, less effort for computational detection of miRNAs and their targets has been exerted since it was thought to be simple, but it has become clear that miRNA regulation in plants is more complex than anticipated [43] . It is difficult to differentiate between miRNAs and short interfering RNAs in plants [44] , but this is beyond the scope of this study. Here, we aimed to improve upon current methodologies for plant pre- miRNA prediction. To achieve this, we pursued two routes for the ab inito prediction of miRNAs. Like many other studies, we employed features describing hairpins but included many more than usual (~700) of which we selected the 100 most discriminative. This strategy led to a prediction accuracy of 98%, which is comparable to previous studies. The second approach describes miRNAs solely based on motifs. This novel approach is also of comparable accuracy (90%) to previous studies in itself. Employing a hybrid approach using the best of both descriptors led to an accuracy of 99.48% which is the best result reported for plants today.
2. Materials and Methods
2.1. Data
We downloaded microRNAs from different plant species available on miRBase (Release 20 and 21).We considered Brassicaceae with 699 pre-miRNAs, that consists of Arabidopsis lyrata (205 precursors), Arabidopsis thaliana (298 precursors), Brassica napus (90 precursors), Brassica oleracea (10 precursors), and Brassica rapa (96 precursors). We also included the data published on the web server PlantMiRNAPred [36] whose training dataset consist of 980 real pre-miRNAs and 980 pseudo pre-miRNAs (we refer to this data as PlantMiRNAPred data in the following). Our negative data pool of the 980 pseudo pre-miRNAs in the PlantMiRNAPred dataset.
2.2. Parameters for Machine Learning
2.2.1. Motif Parameters
Here a sequence motif is a short stretch of nucleotides that is widespread among plant hairpins. Motif discovery in turn is the process of finding short sequences within a larger sequence; here motifs in plant hairpins.
The MEME (Multiple EM for Motif Elicitation) [45] suite web server is used in our study to discovery sequence motifs from our input data which consist of plant pre-microRNA (positive sequences) and plant pseudo hairpins (negative sequences). The MEME algorithm for motif discovery is based on [46] which works by searching for repeated, ungapped sequence motifs that occur in the DNA or protein sequences. MEME provides the results as regular expressions (Table 1). Nucleotides within brackets represent alternatives for the given position in the sequence; without brackets only the given nucleotide occurs abundantly within all collected sequences representing the motif. More visual representations of such motifs are sequence logos (Figure 1). MEME was instructed to generate 20 motifs, each of which must appear in at least 10 sites to be an acceptable motif.

Figure 1. Motif Construction. The sequence logo corresponding to one of the motifs discovered in this study. Size of letters in stacks represents their frequencies while the height of the stack represents the information content. Not all options in the profile may be incorporated into the corresponding regular expression: [GA]A[GAC][AC][GC]A[AG]A[CG][AG][GA][ACG][AC][AGC][AC][CG][GAC][AGC]AAA.

Table 1. Match score calculation. Example of match score between a motif and a part of a sequence. The number of matches is 6. For the assessment the score is normalized by the length of the motif. The final match score is 6/19 = 0.31.
2.2.2. Sequence-Based and Motif Features for Plant Pre-miRNA Detection
Simple sequence-based features have been described and used for ab initio pre-miRNA detection in numerous studies (see Hairpin Feature Calculation). These simple features, also called words, k-mers, or n-grams describe a short sequence of nucleotides of length k or n. For example a 1-gram over the alphabet {A,T,C,G} can produce the words A,T,C,G; while a 2-gram over {A,U,C,G} can generate: AA, AC, AG, AU, CA, CC, CG, CU, GA, GC, GG, GU, UA, UC, UG, and UU. Higher n have also been used [38] but selective for interesting 3- grams.
Motif features are different from n-grams in that they are not exact and allow some degree of error tolerance. In this study motifs are represented as regular expressions (see above). Regular expressions are widespread in approximate pattern matching and many programs allow searching with regular expressions (e.g.: most Linux tools such as grep). Here we use PatMatch [47] to analyze whether a pattern is within a hairpin (1) or not (0). The hairpin is analyzed using the following algorithm:

2.2.3. Traditional Hairpin Feature Calculation
Apart from the novel motifs discovered in this study, we also calculated conventional features which may be statistical in nature, thermodynamic, sequence-based, structural, or any combination of these. The features calculated were taken from 9 studies presenting ab initio detection of hairpins in animals [19] - [21] [32] [48] - [52] . We further added their logical extensions and normalizations, for example normalization based on stem or hairpin length. While it is outside of the scope of this work, some of the features are further explained in Saçar and Allmer [10] . All features were implemented using Java and the calculations were distributed over a 200 core HTCondor [53] cluster at the Izmir Institute of Technology, Urla, Turkey.
2.2.4. Feature Selection
Features were ranked according to the recursive feature elimination with SVM procedure (SVM-RFE) implemented in WEKA for the motif and the traditional approach individually. SVM-RFE [54] is a SVM based model that removes features, recursively based on their contribution to the discrimination, between the two classes. The lowest scoring features by coefficient weights are removed and the remaining features are scored again and the procedure is repeated until only a few features remain. Supplementary Table S1 contains the 60 top ranked features for the three models as presented in Table 5.
2.3. Support Vector Machine Classification
Support Vector Machines (SVMs) are used in machine learning for classification [55] . In general, during training of a linear SVM examples from two classes need to be provided (positive and negative). The SVM learns a model by finding a hyperplane which best separates the two classes maximizing the margin from hyperplane to the support vectors (Figure 2).
For training a set of labeled examples E need to be provided where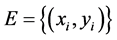 ,
, ![]() is a l dimensional vector, and
is a l dimensional vector, and ![]() defines the class of the
defines the class of the ![]() example (i.e.:
example (i.e.:![]() ;
; ![]() with p representing the posi-
with p representing the posi-
![]()
Figure 2. A support vector machine separates examples, represented by vectors with n-dimensions using a hyperplane. Positive examples (green points) and negative examples (grey doughnuts) are separated by maximising the margins which intersect with the so called support vectors.
tive (+1) and n representing the negative class (−1)). The separating hyperplane then takes the following form:
![]()
where w is the norm of the hyperplane and b defines its position in space. In order to predict a new instance given a trained model the formulation ![]() can be solved and a positive result indicates membership to the positive class and negative otherwise. If the value is zero then the example on the separating hyperplane and cannot be classified.
can be solved and a positive result indicates membership to the positive class and negative otherwise. If the value is zero then the example on the separating hyperplane and cannot be classified.
Here we used the SVM learner which has become the method of choice to solve difficult classification problems in a wide range of application domains and especially in the field of bioinformatics. In previous studies we have examined other classifiers and while there were no great differences in outcome SVM worked well consistently.
We used the WEKA software [56] for the implementation of our SVM classifier based on LibSVM [57] . The radial basis function was set to a gamma value of 0.7 and the cost parameter was chosen to be 4.0 and the normalization option set to true.
Any machine learning algorithm needs initial training and we performed five-fold cross validation during learning employing stratified random sampling (Figure 3).
Trained Models
We trained three separate models using the strategy outlined above to investigate whether motifs, or other previously described features or their combination are most successful for separating true from false plant pre- miRNAs. For training the motif-only model, we used the best 60 motifs and n-grams. For training the traditional model, we used the best 60 features. For the combined model the top 60 motifs were selected from the mixture of n-grams, traditional features, and motifs. The selected features are listed in Supplementary Table S1.
2.4. Evaluation Methods
Positive data from miRBase and negative data from PlantMiRNAPred was used to evaluate the models derived via SVM training. We calculated the performance of the classifier with the known sensitivity (SE) and specificity (SP) and accuracy (ACC) statistics as follows (TP refers to true positives, FP to false positives, TN to true negatives, and FN to false negatives):
![]() ,
, ![]() ,
, 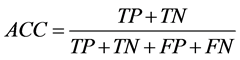
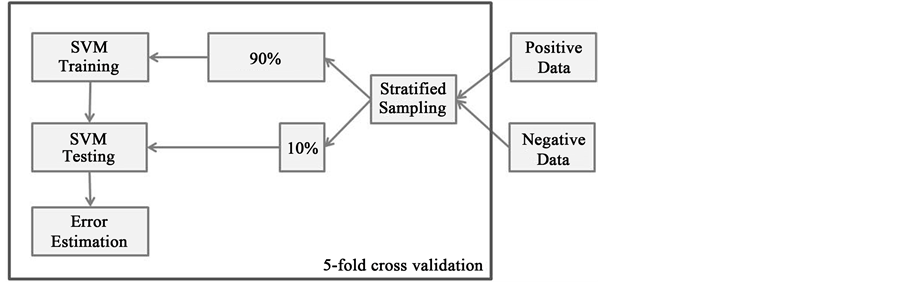
Figure 3. SVM Training. The figure depicts the workflow that was used to train the SVM classifier. Positive and negative data were combined and stratified random sampling was applied. The sampled data was split into 90% data for training and 10% for testing. This procedure was repeated 5 times.
3. Results
The PlantMiRNAPred data was divided into two parts, PlantMiRNAPred-p1 data consisting of 450 pre-miRNAs (positive data) and 450 pseudo pre-miRNAs (negative data) and PlantMiRNAPred-p2 data composed of 530 pre-miRNAs and 530 pseudo pre-miRNAs. The Brassicaceae data was also divided into two parts, first part consists of one third of the data (233 sequences; named Brassicaceae-p1) the remaining two third (named Brassicaceae-p2) contain 466 sequences. MEME software was used to discover motifs in the dataset as described in the Materials and Methods Section, and several motifs were found in all datasets as seen in Table 2. MEME was used to discover motifs in one part of the divided dataset (p1) and the same motifs were used for further experiments in the remainder of the data (p2) to ensure that the extracted motifs are meaningful and not dataset dependent.
The selected motifs and the n-grams (short nucleotide sequences; see Materials and Methods; Supplementary Table S1), were used to train a support vector machine (SVM) model for which the accuracy and other performance measures were established (Table 3). To see the impact of motifs on the classification accuracy, two models were trained for all datasets, one which uses both motifs and n-grams and one which only relies only on the latter.
Table 3 presents the average performance of our SVM classifier MotifmiRNAPred using five-fold cross validation. For the motifs extracted from PlantMiRNAPred-p1 and applied to PlantMiRNAPred-p2 we see a decrease in performance of the model by about 13% which indicates that there is some data dependency of the motifs in this case. For Brassicaceae there was no significant difference between the datasets p1 and p2 which shows that in this case stable motifs were generated that are not affected by differences in the tested datasets. When comparing the results on PlantMiRNAPred-p1 with the results achieved by PlantMiRNAPred [36] it can be seen that our methodology achieves a similar performance (Table 4). PlantMiRNAPred achieves accuracies between 92% and 100% when the data is separated into species with a trend to be more successful for smaller datasets.
In Table 4, we considered the data from PlantMiRNAPred web server [36] to perform a comparison performance with the classification results of PlantMiRNAPred, TripletSVM [20] , and microPred [58] . The data was represented by 174 features consisting of 84 n-grams and 90 motifs. From these, the top 60 selected features by SVM-RFE, feature selection method available in WEKA [56] , were considered and the performance resulting from five-fold cross validation are presented (Table 4).
The nucleotide T(U) is one of the most informative features (Supplementary Table S1) and it always appeared on the top of the selected features for each data set individually. This observation is also confirmed by the study of Zhang, Pan et al. [59] . This seems to confirm that the sequences of pre-miRNAs and mature miRNAs are slightly enriched in T(U) and T(U) plus G, respectively.
The comparison in Table 4 shows that using motifs for miRNA detection is comparably successful to using traditional features while at times even slightly more successful. Following this, we set forth and calculated the traditional features used to describe hairpins and trained a model (traditional) for pre-miRNA detection. We
Table 2. Dataset description. Dataset description and number of generated motifs per dataset.
Table 3. Classifier performance. The result of MotifmiRNAPred applied to different plant miRNA data. The first value given as performance measure refers to the model trained with both n-grams and motifs and the value following is in respect to a model trained with only the former. ROC: receiver operator characteristic.
Table 4. Performance comparison among tools. Comparison of MotifmiRNAPred with different methods. The first 4 columns taken from the PlantMiRNAPred paper. The columns below MotifmiRNAPred present our results using the same data as in the PlantMiRNAPred paper.
calculated about 700 features, but ranked them as above and selected only the top 60 features for machine learning. Additionally, we combined the traditional features with the motifs and ranked the mixture and again selected the 60 best ranked features to train a model (combined). These two models were compared to the initially learned model (motifs-only) which is only based on motifs and n-grams. The combined model performs better than the underlying models individually with an increase by about 11% and 1%, respectively (Table 5).
The best accuracy was achieved by the combined feature set. It is striking that the accuracy is even better than
Table 5. Performance comparison among feature sets. Comparison of two synergistic feature sets and their synthesis proposed in this study. The models were trained on the combined dataset including all plant miRNAs.
the best accuracy for any of the models trained on the individual plant data sets (Table 4). Since our motif-only approach is comparable in accuracy with previously published studies (Table 4), and due to the fact that the combined feature set is significantly better than the motifs-only one, we propose, that it suffices to use our feature set and create one model for miRNA detection to be applicable in even different plant species.
4. Conclusion
An abundance of features describing miRNA hairpins have been proposed which are mostly based on structural, statistical and thermodynamic features [60] . Here we showed that for plant miRNA detection, motif based features are useful and they by themselves lead to a good recognition of pre-miRNAs at an accuracy of 90% - 95%, depending on the plant species (Table 4). When using a mixture of plant pre-miRNAs to train models based on motifs and n-grams, traditional features, and their combination, it can be seen that the combination of features is most successful (Table 5). We found no great difference when comparing the performance of selected features from the domain of animal pre-miRNA detection to using sequence motifs and n-grams as features (Table 4). However, the combination of these features (Table 5) performed about 4% better, even when compared to the average performance of classifiers specifically trained for plant species (Table 4). We conclude that using motifs for the prediction of pre-miRNAs is useful and in combination with traditional features is most successful. Furthermore, we propose that it may be sufficient to use a classifier trained in this manner to detect plant pre- miRNAs at an accuracy level high enough to warrant experimental confirmation of predicted pre-miRNAs.
Acknowledgements
The work was supported by the Ministry of Science, Israel to MY and WK. and the Scientific and Technological Research Council of Turkey [113E326 to JA].
Cite this paper
MalikYousef,JensAllmer,WaleedKhalifa,11, (2016) Accurate Plant MicroRNA Prediction Can Be Achieved Using Sequence Motif Features. Journal of Intelligent Learning Systems and Applications,08,9-22. doi: 10.4236/jilsa.2016.81002
References
- 1. Erson-Bensan, A.E. (2014) Introduction to microRNAs in Biological Systems. Methods in Molecular Biology, 1107, 1-14.
http://www.ncbi.nlm.nih.gov/pubmed/24272428 - 2. Allmer, J. and Yousef, M. (2012) Computational Methods for ab Initio Detection of microRNAs. Frontiers in Genetics.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3467617&tool=pmcentrez&rendertype= abstract - 3. Lee, R.C., Feinbaum, R.L. and Ambros, V. (1993) The C. elegans Heterochronic Gene lin-4 Encodes Small RNAs with Antisense Complementarity to lin-14. Cell, 75, 843-854.
http://www.ncbi.nlm.nih.gov/pubmed/8252621 - 4. Tüfekci, K.U., Oner, M.G., Meuwissen, R.L.J. and Genc, S. (2014) The Role of microRNAs in Human Diseases. Methods in Molecular Biology, 1107, 33-50.
http://www.ncbi.nlm.nih.gov/pubmed/24272430 - 5. Zhang, Z., Yu, J., Li, D., Zhang, Z., Liu, F., Zhou, X., et al. (2010) PMRD: Plant microRNA Database. Nucleic Acids Research, 38, D806-D813.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2808885&tool=pmcentrez&rendertype= abstract - 6. Kim, V.N., Han, J. and Siomi, M.C. (2009) Biogenesis of Small RNAs in Animals. Nature Reviews Molecular Cell Biology, 10, 126-139.
http://www.ncbi.nlm.nih.gov/pubmed/19165215 - 7. Chapman, E.J. and Carrington, J.C. (2007) Specialization and Evolution of Endogenous Small RNA Pathways. Nature Reviews Genetics, Nature Publishing Group, 8, 884-896.
- 8. Allmer, J. (2014) Computational and Bioinformatics Methods for microRNA Gene Prediction. Methods in Molecular Biology, 1107, 157-175.
http://www.ncbi.nlm.nih.gov/pubmed/24272436 - 9. Hamzeiy, H., Allmer, J. and Yousef, M. (2014) Computational Methods for microRNA Target Prediction. Methods in Molecular Biology, 1107, 207-221.
http://www.ncbi.nlm.nih.gov/pubmed/24272439 - 10. Sa?ar, M.D. and Allmer, J. (2013) Comparison of Four ab Initio microRNA Prediction Tools. Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms, SciTePress—Science and and Technology Publications, Barcelona, 190-195.
http://www.scitepress.org/DigitalLibrary/Link.aspx?doi=10.5220/0004248201900195 - 11. de ON Lopes, I., Schliep, A. and de Carvalho, A.C.P. de L.F. (2014) The Discriminant Power of RNA Features for Pre-miRNA Recognition. BMC Bioinformatics, 15, 124.
http://dx.doi.org/10.1186/1471-2105-15-124
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4046174&tool=pmcentrez&rendertype= abstract - 12. Kozomara, A. and Griffiths-Jones, S. (2011) miRBase: Integrating microRNA Annotation and Deep-Sequencing Data. Nucleic Acids Research, 39, D152-D157.
http://dx.doi.org/10.1093/nar/gkq1027
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3013655&tool=pmcentrez&rendertype= abstract - 13. Lim, L.P., Glasner, M.E., Yekta, S., Burge, C.B. and Bartel, D.P. (2003) Vertebrate microRNA Genes. Science, 299, 1540. http://www.ncbi.nlm.nih.gov/pubmed/12624257
http://dx.doi.org/10.1126/science.1080372 - 14. Weber, M.J. (2005) New Human and Mouse microRNA Genes Found by Homology Search. FEBS Journal, 272, 59-73.
http://www.ncbi.nlm.nih.gov/pubmed/15634332
http://dx.doi.org/10.1111/j.1432-1033.2004.04389.x - 15. Lim, L.P., Lau, N.C., Weinstein, E.G., Abdelhakim, A., Yekta, S., Rhoades, M.W., et al. (2003) The microRNAs of Caenorhabditis elegans. Genes & Development, 17, 991-1008.
http://www.ncbi.nlm.nih.gov/pubmed/12672692
http://dx.doi.org/10.1101/gad.1074403 - 16. Lai, E.C., Tomancak, P., Williams, R.W. and Rubin, G.M. (2003) Computational Identification of Drosophila microRNA Genes. Genome Biology, 4, R42.
http://www.ncbi.nlm.nih.gov/pubmed/12844358
http://dx.doi.org/10.1186/gb-2003-4-7-r42 - 17. Grad, Y., Aach, J., Hayes, G.D., Reinhart, B.J., Church, G.M., Ruvkun, G., et al. (2003) Computational and Experimental Identification of C. elegans microRNAs. Molecular Cell, 11, 1253-1263.
http://www.ncbi.nlm.nih.gov/pubmed/12769849
http://dx.doi.org/10.1016/S1097-2765(03)00153-9 - 18. Teune, J.-H. and Steger, G. (2010) NOVOMIR: De Novo Prediction of MicroRNA-Coding Regions in a Single Plant-Genome. Journal of Nucleic Acids, 2010, Article ID: 495904.
http://www.ncbi.nlm.nih.gov/pubmed/20871826
http://dx.doi.org/10.4061/2010/495904 - 19. Ding, J., Zhou, S. and Guan, J. (2010) MiRenSVM: Towards Better Prediction of microRNA Precursors Using an Ensemble SVM Classifier with Multi-Loop Features. BMC Bioinformatics, 11, S11.
http://www.ncbi.nlm.nih.gov/pubmed/21172046
http://dx.doi.org/10.1186/1471-2105-11-s11-s11 - 20. Xue, C., Li, F., He, T., Liu, G.-P., Li, Y. and Zhang, X. (2005) Classification of Real and Pseudo microRNA Precursors Using Local Structure-Sequence Features and Support Vector Machine. BMC Bioinformatics, 6, 310.
http://www.ncbi.nlm.nih.gov/pubmed/16381612
http://dx.doi.org/10.1186/1471-2105-6-310 - 21. Jiang, P., Wu, H., Wang, W., Ma, W., Sun, X. and Lu, Z. (2007) MiPred: Classification of Real and Pseudo microRNA Precursors Using Random Forest Prediction Model with Combined Features. Nucleic Acids Research, 35, W339-W344.
http://www.ncbi.nlm.nih.gov/pubmed/17553836
http://dx.doi.org/10.1093/nar/gkm368 - 22. Keshavan, R., Virata, M., Keshavan, A. and Zeller, R.W. (2010) Computational Identification of Ciona intestinalis microRNAs. Zoological Science, 27, 162-170.
http://www.ncbi.nlm.nih.gov/pubmed/20141421
http://dx.doi.org/10.2108/zsj.27.162 - 23. Lagos-Quintana, M., Rauhut, R., Lendeckel, W. and Tuschl, T. (2001) Identification of Novel Genes Coding for Small Expressed RNAs. Science, 294, 853-858.
http://www.ncbi.nlm.nih.gov/pubmed/11679670
http://dx.doi.org/10.1126/science.1064921 - 24. Lau, N.C., Lim, L.P., Weinstein, E.G. and Bartel, D.P. (2001) An Abundant Class of Tiny RNAs with Probable Regulatory Roles in Caenorhabditis elegans. Science, 294, 858-862.
http://www.ncbi.nlm.nih.gov/pubmed/11679671
http://dx.doi.org/10.1126/science.1065062 - 25. Lee, R.C. and Ambros, V. (2001) An Extensive Class of Small RNAs in Caenorhabditis elegans. Science, 294, 862-864.
http://dx.doi.org/10.1126/science.1065329 - 26. Pasquinelli, A.E., Reinhart, B.J., Slack, F., Martindale, M.Q., Kuroda, M.I., Maller, B., et al. (2000) Conservation of the Sequence and Temporal Expression of Let-7 Heterochronic Regulatory RNA. Nature, 408, 86-89.
http://www.ncbi.nlm.nih.gov/pubmed/11081512
http://dx.doi.org/10.1038/35040556 - 27. Wang, X., Zhang, J., Li, F., Gu, J., He, T., Zhang, X., et al. (2005) MicroRNA Identification Based on Sequence and Structure Alignment. Bioinformatics, 21, 3610-3614.
http://www.ncbi.nlm.nih.gov/pubmed/15994192
http://dx.doi.org/10.1093/bioinformatics/bti562 - 28. Hertel, J. and Stadler, P.F. (2006) Hairpins in a Haystack: Recognizing microRNA Precursors in Comparative Genomics Data. Bioinformatics, 22, 197-202.
http://www.ncbi.nlm.nih.gov/pubmed/16873472
http://dx.doi.org/10.1093/bioinformatics/btl257 - 29. Ritchie, W., Gao, D. and Rasko, J.E.J. (2012) Defining and Providing Robust Controls for microRNA Prediction. Bioinformatics, 28, 1058-1061.
http://www.ncbi.nlm.nih.gov/pubmed/22408193
http://dx.doi.org/10.1093/bioinformatics/bts114 - 30. Wu, Y., Wei, B., Liu, H., Li, T. and Rayner, S. (2011) MiRPara: A SVM-Based Software Tool for Prediction of Most Probable microRNA Coding Regions in Genome Scale Sequences. BMC Bioinformatics, 12, 107.
http://www.ncbi.nlm.nih.gov/pubmed/21504621
http://dx.doi.org/10.1186/1471-2105-12-107 - 31. Yousef, M., Jung, S., Showe, L.C. and Showe, M.K. (2008) Learning from Positive Examples When the Negative Class Is Undetermined—microRNA Gene Identification. Algorithms for Molecular Biology, 3, 2.
http://www.ncbi.nlm.nih.gov/pubmed/18226233
http://dx.doi.org/10.1186/1748-7188-3-2 - 32. Sewer, A., Paul, N., Landgraf, P., Aravin, A., Pfeffer, S., Brownstein, M.J., et al. (2005) Identification of Clustered microRNAs Using an ab Initio Prediction Method. BMC Bioinformatics, 6, 267.
http://www.ncbi.nlm.nih.gov/pubmed/16274478
http://dx.doi.org/10.1186/1471-2105-6-267 - 33. Gomes, C.P.C., Cho, J.-H., Hood, L., Franco, O.L., Pereira, R.W. and Wang, K. (2013) A Review of Computational Tools in microRNA Discovery. Frontiers in Genetics, 4, 81.
http://dx.doi.org/10.3389/fgene.2013.00081
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3654206&tool=pmcentrez&rendertype= abstract - 34. Billoud, B., Nehr, Z., Le Bail, A. and Charrier, B. (2014) Computational Prediction and Experimental Validation of microRNAs in the Brown Alga Ectocarpus siliculosus. Nucleic Acids Research, 42, 417-429.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3874173&tool=pmcentrez&rendertype= abstract
http://dx.doi.org/10.1093/nar/gkt856 - 35. Oliveira, J.S., Mendes, N.D., Carocha, V., Graca, C., Paiva, J.A. and Freitas, A.T. (2013) A Computational Approach for MicroRNA Identification in Plants: Combining Genome-Based Predictions with RNA-Seq Data. Journal of Data Mining in Genomics & Proteomics, 4, 130.
http://www.omicsonline.org/2153-0602/2153-0602-4-130.php?aid=14889
http://dx.doi.org/10.4172/2153-0602.1000130 - 36. Xuan, P., Guo, M., Liu, X., Huang, Y., Li, W. and Huang, Y. (2011) PlantMiRNAPred: Efficient Classification of Real and Pseudo Plant Pre-miRNAs. Bioinformatics, 27, 1368-1376. http://www.ncbi.nlm.nih.gov/pubmed/21441575
http://dx.doi.org/10.1093/bioinformatics/btr153 - 37. Williams, P.H., Eyles, R. and Weiller, G. (2012) Plant MicroRNA Prediction by Supervised Machine Learning Using C5.0 Decision Trees. Journal of Nucleic Acids, 2012, Article ID: 652979.
http://dx.doi.org/10.1155/2012/652979
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3503367&tool=pmcentrez&rendertype= abstract - 38. Cakir, M.V. and Allmer, J. (2010) Systematic Computational Analysis of Potential RNAi Regulation in Toxoplasma gondii. Proceedings of the 5th International Symposium on Health Informatics and Bioinformatics, Ankara, 20-22 April 2010, 31-38.
http://dx.doi.org/10.1109/hibit.2010.5478909 - 39. Adai, A., Johnson, C., Mlotshwa, S., Archer-Evans, S., Manocha, V., Vance, V., et al. (2005) Computational Prediction of miRNAs in Arabidopsis thaliana. Genome Research, 15, 78-91.
http://dx.doi.org/10.1101/gr.2908205 - 40. Rajagopalan, R., Vaucheret, H., Trejo, J. and Bartel, D.P. (2006) A Diverse and Evolutionarily Fluid Set of microRNAs in Arabidopsis thaliana. Genes & Development, 20, 3407-3425.
http://dx.doi.org/10.1101/gad.1476406
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1698448&tool=pmcentrez&rendertype= abstract - 41. Jain, M., Chevala, V.V.S.N. and Garg, R. (2014) Genome-Wide Discovery and Differential Regulation of Conserved and Novel microRNAs in Chickpea via Deep Sequencing. Journal of Experimental Botany, 65, 5945-5958.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4203128&tool=pmcentrez&rendertype= abstract
http://dx.doi.org/10.1093/jxb/eru333 - 42. Berezikov, E., Cuppen, E. and Plasterk, R.H.A. (2006) Approaches to microRNA Discovery. Nature Genetics, 38, 2-7.
http://www.ncbi.nlm.nih.gov/pubmed/16736019
http://dx.doi.org/10.1038/ng1794 - 43. Dai, X., Zhuang, Z. and Zhao, P.X. (2011) Computational Analysis of miRNA Targets in Plants: Current Status and Challenges. Briefings in Bioinformatics, 12, 115-121.
http://www.ncbi.nlm.nih.gov/pubmed/20858738
http://dx.doi.org/10.1093/bib/bbq065 - 44. Kurtoglu, K.Y., Kantar, M., Lucas, S.J. and Budak, H. (2013) Unique and Conserved microRNAs in Wheat Chromosome 5D Revealed by Next-Generation Sequencing. PLoS ONE, 8, e69801.
http://dx.doi.org/10.1371/journal.pone.0069801
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3720673&tool=pmcentrez&rendertype= abstract - 45. Bailey, T.L., Boden, M., Buske, F.A., Frith, M., Grant, C.E., Clementi, L., et al. (2009) MEME SUITE: Tools for Motif Discovery and Searching. Nucleic Acids Research, 37, W202-W208.
http://dx.doi.org/10.1093/nar/gkp335
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2703892&tool=pmcentrez&rendertype= abstract - 46. Bailey, T.L. and Elkan, C. (1994) Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. Proceedings of the International Conference on Intelligent Systems for Molecular Biology, 2, 28-36.
http://www.ncbi.nlm.nih.gov/pubmed/7584402 - 47. Yan, T., Yoo, D., Berardini, T.Z., Mueller, L.A., Weems, D.C., Weng, S., et al. (2005) PatMatch: A Program for Finding Patterns in Peptide and Nucleotide Sequences. Nucleic Acids Research, 33, W262-W266.
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1160129&tool=pmcentrez&rendertype= abstract
http://dx.doi.org/10.1093/nar/gki368 - 48. van der Burgt, A., Fiers, M.W.J.E., Nap, J.-P. and van Ham, R.C.H.J. (2009) In Silico miRNA Prediction in Metazoan Genomes: Balancing between Sensitivity and Specificity. BMC Genomics, 10, 204.
http://www.biomedcentral.com/1471-2164/10/204/
http://dx.doi.org/10.1186/1471-2164-10-204 - 49. Bentwich, I. (2008) Identifying Human microRNAs. Current Topics in Microbiology and Immunology, 320, 257-269.
http://dx.doi.org/10.1007/978-3-540-75157-1_12 - 50. Nam, J.-W., Shin, K.-R., Han, J., Lee, Y., Kim, V.N. and Zhang, B.-T. (2005) Human microRNA Prediction through a Probabilistic Co-Learning Model of Sequence and Structure. Nucleic Acids Research, 33, 3570-3581.
http://www.ncbi.nlm.nih.gov/pubmed/15987789
http://dx.doi.org/10.1093/nar/gki668 - 51. Nam, J.-W., Kim, J., Kim, S.-K., Zhang, B.-T. (2006) ProMiR II: A Web Server for the Probabilistic Prediction of Clustered, Nonclustered, Conserved and Nonconserved microRNAs. Nucleic Acids Research, 34, W455-W458.
http://www.ncbi.nlm.nih.gov/pubmed/16845048
http://dx.doi.org/10.1093/nar/gkl321 - 52. Ng, K.L.S. and Mishra, S.K. (2007) De Novo SVM Classification of Precursor microRNAs from Genomic Pseudo Hairpins Using Global and Intrinsic Folding Measures. Bioinformatics, 23, 1321-1330.
http://www.ncbi.nlm.nih.gov/pubmed/17267435
http://dx.doi.org/10.1093/bioinformatics/btm026 - 53. Thain, D., Tannenbaum, T. and Livny, M. (2005) Distributed Computing in Practice: The Condor Experience. Concurrency and Computation: Practice and Experience, 17, 2-4.
http://dx.doi.org/10.1002/cpe.938
http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.6.3035 - 54. Guyon, I., Weston, J., Barnhill, S. and Vapnik, V. (2002) Gene Selection for Cancer Classification Using Support Vector Machines. Machine Learning, 46, 389-422.
http://link.springer.com/article/10.1023%2FA%3A1012487302797
http://dx.doi.org/10.1023/A:1012487302797 - 55. Vapnik, V.N. (1995) The Nature of Statistical Learning Theory. Springer-Verlag, New York.
http://dl.acm.org/citation.cfm?id=211359
http://dx.doi.org/10.1007/978-1-4757-2440-0 - 56. Gewehr, J.E., Szugat, M. and Zimmer, R. (2007) BioWeka—Extending the Weka Framework for Bioinformatics. Bioinformatics, 23, 651-653.
http://www.ncbi.nlm.nih.gov/pubmed/17237069
http://dx.doi.org/10.1093/bioinformatics/btl671 - 57. Chang, C.-C. and Lin, C.-J. (2011) LIBSVM. ACM Transactions on Intelligent Systems and Technology, 2, 1-27.
http://dl.acm.org/citation.cfm?doid=1961189.1961199
http://dx.doi.org/10.1145/1961189.1961199 - 58. Batuwita, R. and Palade, V. (2009) microPred: Effective Classification of Pre-miRNAs for Human miRNA Gene Prediction. Bioinformatics, 25, 989-995.
http://www.ncbi.nlm.nih.gov/pubmed/19233894
http://dx.doi.org/10.1093/bioinformatics/btp107 - 59. Zhang, B.H., Pan, X.P., Cox, S.B., Cobb, G.P. and Anderson, T.A. (2006) Evidence That miRNAs Are Different from Other RNAs. Cellular and Molecular Life Sciences, 63, 246-254.
http://dx.doi.org/10.1007/s00018-005-5467-7 - 60. Sacar, M.D. and Allmer, J. (2014) Machine Learning Methods for microRNA Gene Prediction. Methods in Molecular Biology, 1107, 177-187.
http://www.ncbi.nlm.nih.gov/pubmed/24272437
http://dx.doi.org/10.1007/978-1-62703-748-8_10
Supplementary
Table S1. The 60 best features were selected from the set of traditional features and motifs, individually (first two columns). The combination of traditional and motif features was again ranked and the 60 best features were selected (column 3). More information about the traditional features can be found in references [10] and [60] .
NOTES

*Corresponding author.