Journal of Cancer Therapy
Vol.5 No.10(2014), Article ID:49466,18 pages
DOI:10.4236/jct.2014.510097
Expression of C-Terminal Modified Serine Palmitoyltransferase-1 Alters Chemosensitivity of Inflammation-Associated Human Cancer Cell Lines
Tokunbo Yerokun
Department of Biology, Spelman College, Atlanta, Georgia, USA
Email: doctortoks@gmail.com
Copyright © 2014 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 June 2014; revised 20 July 2014; accepted 15 August 2014
ABSTRACT
Background: The human serine palmitoyltransferase-1, SPTLC1, subunit is emerging as a stress responsive protein with putative role in modulating cellular stress response behavior. When compared to the parental cell line, recombinant Glioma cells expressing C-terminal modified SPTLC1 are found to show resistance to the cytotoxic effect of polycyclic hydrocarbons, PHs, including the environmental contaminant 3-methylcholanthrene. This novel functional association of SPTLC1 expression with proliferative capacity is thought to be due, in part, to its ability for crosstalk with protein regulators of different biological processes. Whether the effect of SPTLC1 on sensitivity to PHs extends to therapeutic drugs and the progression of the malignant phenotype is of research interest. Methods: In the current study, sub-cellular localization was by immunostaining for SPTLC1 in untreated and chemical treated cells and detection with confocal microscopy. The effect expressing C-terminal modified SPTLC1, in cancer cell lines of the inflammation-associated type, has on chemosensitivity and gene expression was also assessed. Parent Glioma LN18 and SKN-SH cells and their SPTLC1 recombinants were each treated with Glutamate, an excitatory neurotransmitter that can participate in both neuronal and excitotoxic signaling. In addition to the Glioma and SKN-SH cells, the PC3 prostate cancer and 647V bladder cancer cell lines were also treated with Celecoxib, a potent inhibitor of cyclooxygenase 2, COX-2, and an anti-inflammatory drug recently found to have anti-neoplastic activity against several malignancies. Results: Confocal microscopy revealed that Celecoxib mediates both rapid and enhanced redistribution of SPTLC1 and COX-2, to focal adhesion sites. In cell viability assay, SPTLC1 recombinant cells exhibited differential but dose-dependent resistance to excitotoxic levels of Glutamate. Drug co-treatment with a non-lethal dose of the potent kinase inhibitor, Sulfasalazine, increased the anti-proliferation effect of Celecoxib in a dose-dependent manner for all the cell lines tested. Conclusions: The effect of SPTLC1 expression on cellular chemosensitivity seen in the present study further highlights possible role of a C-terminal modified SPTLC1 variant in the biologic modulation of cellular behavior in response to therapeutic anticancer drugs.
Keywords:Cancer Cell Line, Serine Palmitoyltransferase-1, Glutamate, Celecoxib, Sulfasalazine

1. Introduction
1.1. Novel Signaling Role for SPTLC1
Structurally, the protein encoded by the long chain base 1 of SPTLC1 lacks an active site, but it has a membrane tethering role which is essential for the catalytic activity of the SPTLC1/SPTLC2 heterocomplex [1] . Mutations in the gene have been linked to hereditary sensory neuropathy disease (HSN), while its perturbation leads to abnormal morphology and behavior of cultured cells [2] [3] . Immunocytochemical studies show that the expression of SPTLC1 is enhanced in normal proliferating cells undergoing rapid morphological changes, such as the lung, stomach and intestinal epithelium, in stromal fibroblasts surrounding malignant epithelial tissues, as well as, in cells in in vitro model of wound healing [3] -[5] . Using antibody-mediated “pull down” approaches and colocalization studies, an increasing number of SPTLC1 binding partners including the 90 kiloDalton heat shock protein, Hsp90, continue to emerge [3] [6] [7] . Such interaction may have biological importance, particularly in the case of the molecular chaperone, Hsp90, which plays a critical role in the functional activation of many signaling proteins in diverse processes particularly cellular homeostasis and transcriptional expression of stress response genes. The complexity of changes in cellular morphology and behavior that accompany SPTLC1 gene mutations under experimental conditions with cultured cells may be, in part, attributable to protein-protein interaction characteristics and not only due to qualitative or quantitative changes in metabolic derivatives [8] . Hence, the temporal-spatial localization, crosstalk and interaction of SPTLC1 with proteins in stress response signaling modules regulating multiple biological processes [9] is likely to have implications for cell behavior under pathologic in vivo conditions. Evidence from the current report supports an emerging role for SPTLC1 in altered pattern of proliferation following exposure of inflammation-associated cancer cell lines to different toxic compounds. In addition to the expression of SPTLC1 seen in cytoplasm of cultured cells, it is also expressed in cells growing as anchorage independent colonies, a condition of increased COX-2 expression [10] [11] . Increasingly, SPTLC1 expression is found to influence cellular morphology and chemosensitivity in terms of non-enzymatic contribution to regulating important aspects of cell physiology, cell growth, inflammation, as well as, stress response mechanisms.
1.2. Human SPTLC1 and Chemosensitivity of Cancer Cells
There is overlap in aspects of the molecular mechanisms for cellular response to environmental stress, inflammation and neurodegeneration particularly with regards to cellular processes of signal transduction. The stoichiometry of the human SPTLC1 protein is now best appreciated as that of a dynamic, tissue specific fragment with the capacity to form catalytic heterocomplex [12] , modulate transport [7] or stress response [3] activities of interacting partners via direct “off-target” effects or indirect “pathway cross-talk” effects. Using cell proliferation as index of drug sensitivity, the current study shows recombinant SPTLC1 expression modulates dose dependent sensitivity to Glutamate by the resistant Glioblastoma, Glioma LN18 and the highly susceptible Neuroblastoma SKN-SH cell lines. Glioma cells in culture are known to release relatively high levels of Glutamate [13] , causing widespread excitotoxicity and may promote the growth of malignant Gliomas [14] . Glutamate release from Gliomas involves system x(c)(-) also referred to as xCT, a heterodimeric antiporter subunit member of the SCL7A11 family of proteins whose over-expression under hypoxic conditions is associated with increased Glutamate secretion that leads to neuronal cell death [15] [16] . Furthermore, the expression of xCT is linked to a wide range of other CNS functions including oxidative protection, the operation of the blood-brain barrier, neurotransmitter release, synaptic organization, viral pathology, drug addiction, chemosensitivity and chemoresistance, and brain tumor growth [17] . The release of Glutamate by the antiporter activity of xCT is accompanied by Cystine import, the amino acid used for the synthesis of glutathione (g-glutamyl-cysteinylglycine; GSH) and a major endogenous cellular antioxidant. Therefore, perturbing the Glutamate/Cysteine cycle may impact pharmacological response to therapeutic drugs or toxicological response to environmental agents. Oxidative Glutamate toxicity triggers programmed cell death [18] and the associated stress condition is implicated in the pathophysiology of neurological diseases like Alzheimer’s and Parkinson’s disease, and ischemic stroke [19] . In cancer cells of other tissue sources, elevated xCT activity is associated with growth, acquisition of the drug resistance phenotype and poor prognosis, while in others including lymphoma, prostate and breast cancers, xCT disruption significantly inhibits growth [20] . These observations suggest that the xCT antiporter activity can connect antioxidant defense with neurotransmission, as well as, cellular growth and death [17] . Glioma cells express detectable levels of SPTLC1 and xCT proteins. Although the ability of SPTLC1 variants to interact with and/or modulate the activity of proteins in the xCT transport system has implications for cancer cell growth and survival, such interaction has not been reported.
1.3. Cancer Treatment with Anti-Inflammation/Proliferation Drug
The over-expression of cyclooxygenase 2 (COX-2) seen in different cancer types and its association with increased tumor grade suggests it may have important functions in both tumorigenesis and cellular response to therapeutic interventions [21] -[24] . The expression of COX-2 found increased under the stress conditions of hypoxia also support a role in cancer survival and progression [25] . The use of anti-inflammatory drugs that also kill cancer cells is an attractive therapeutic option against cancers, particularly of the inflammation associated cancer types. Celecoxib (CBX) is a highly selective COX-2 inhibitor used as an inflammatory drug that has been found to reduce the number of adenomatous colorectal polyps in patients with familial adenomatous polyposis [26] [27] , as well as in those with sporadic colorectal adenomas [28] . At pharmacological concentrations for topical application on patients with less severe basal cell carcinoma (BCC) of the skin, CBX is reported to decrease the development of new BCCs by about 50% (<15 BCCs at base-line), compared to about 20% with placebo treatment [29] . Either alone or in combination with several regimens used in metastatic, adjuvant or neoadjuvant settings, CBX has also demonstrated anticancer effects in established invasive lung carcinoma and breast cancer [30] -[32] . Although found to have anti-proliferation effect in the PC3 cell line, this effect has not been reported in prostate cancer patients [33] [34] .
1.4. Pharmacokinetics and Mechanism of CBX Anti-Proliferation Action
The anti-proliferation effect of CBX is thought to be mediated through direct drug action or its metabolic derivative, acting on multiple signaling mechanisms. CBX is extensively metabolized in the liver primarily through methyl hydroxylation to form hydroxycelecoxib [35] . Based on serum clearance in subjects, the cytochrome p450, Cyp2C9 pathway is identified as playing a key role in CBX metabolism and disposition, although CYP3A4 is also found to play a minor role [27] [29] [35] [36] . Elevated level of Cyp3A4 is reported to concomitantly increase the Cyp2C9-mediated disposition of the methyl hydroxyl derivative of CBX [37] , suggesting that the metabolic conversion of CBX by different Cyp450 variants including Cyp3A4, may have different consequences on the anti-proliferation activity of CBX. The transcriptional expression of Cyp450s plays a limiting role in functional expression of the catalytic protein variants, thus, the ability of compounds used in cotreatment assays to induce or inhibit transcriptional expression of the Cyp450s may further affect the anti-proliferation activity of CBX. The transcriptional expression of the Cyp4501A1, 1A2 and 3A4 was examined in the Glioma cells used in the current study, knowing that Glioma cells are capable of constitutive and inducible expression of Cyp450 mRNA species [38] . The polycyclic aromatic hydrocarbon, 3-methylcholanthrene (3-MC) is a potent inducer of Cyp1A1 and 1A2 [39] , while Geldanamycin inhibits Cyp450 transcriptional expression [40] . Both compounds are thought to act through interaction with the molecular chaperone, Hsp90, in which transcriptional expression of Cyp1A1 or 1A2 is via the arylhydrocarbon receptor (AhR) [41] , or for CYP3A4 and CYP2 or CYP4 variants, through members of the “orphan” nuclear receptor superfamily such as, constitutive androstane receptor (CAR), pregnane X receptor (PXR), retinoid X receptor (RXR) and the peroxisome proliferator activated receptor, PPAR [42] . Generally, interaction with the Hsp90 molecular chaperone is essential to the proper folding of the proteins and their assembly into modular functional complexes [43] . Incidentally, SPTLC1 is an Hsp90 binding partner, an interaction that has been implicated in altered transcriptional expression of Cyp1A1 and metabolism of cognate AhR ligands including 3-MC [6] [44] .
The anti-cancer effect of CBX on cell growth and responsiveness to radiotherapy is considered to be mainly independent of the inhibition of COX-2 activity or protein expression. At pharmacological concentrations of about 10 - 35 μmol/L, CBX is reported to inhibit phosphoinositide-dependent kinase-1/Akt signaling and cyclindependent kinase activity in cultured cells [45] . The PI3K/AKT signaling pathway regulates several biological processes including migration, survival and differentiation [31] , while perturbation of the signaling pathways is implicated in cancer development, progression and resistance to chemotherapy. Thus, it has been suggested that signaling through mitogen-activated kinase-dependent activation of transcription factors and reduction of the anti-apoptotic cell survival protein, Akt, may be the more relevant mechanism for the anti-proliferative activity of CBX [46] [47] . In the cell survival signaling pathway, growth factor binding events to receptor tyrosine kinase result in activation of PI3K, followed by activated PI3K generating membrane-bound second messengers and subsequent translocation of Akt protein to the membrane. It is not known if binding of SPTLC1 occurs with any of the proteins in this signaling pathway or how it can modulate cell survival behavior of cells exposed to CBX. Furthermore, the use of CBX against glioblastoma and prostate carcinomas has seen mixed results with regards to treatment of advanced cancers and drug resistance is reported to be developed in patients undergoing treatment [48] . Recent research studies and clinical trials show that the use of CBX in combination drug treatment with inhibitors of PI3K may help circumvent acquired drug resistance by cancer cells [48] -[50] . The role of SPTLC1 in the acquisition of a drug resistance phenotype may be of interest in the design of therapeutic regimen.
1.5. CBX and Sulfasalazine (SAS) Drug Combination Treatment
The disruption of the xCT antiporter system significantly inhibits the growth of cancer cells including Gliomas. Similar to excess Glutamate, SAS targets the antioxidant xCT-system and the resulting pharmacologic effect is reported to improve antitumor efficacy of COX inhibitors in in vivo models of tumorigenesis [51] [52] . As a clinically approved xCT inhibitor [53] , SAS is reported to inhibit GSH formation or the activation of AKT and focal adhesion kinases [54] [55] . SAS has been used as an effective inhibitor of tumor growth and tumor-associated seizures, particularly for the inflammatory brain tumor [56] . At a dose of 250 µM treatment with SAS is a potent xCT blocker exerting anti-proliferation activity against Glioma cells in culture and tumor mass [57] . In prostate cancer, SAS is a potent system xCT inhibitor [58] and the induced cystine starvation seen in the same study is reported to markedly inhibit growth of the prostate cancer cell line DU-145 in vitro and PC-3 xenografts in nude mice without major toxicity to hosts. The pretreatment of cultured cells with 50 μM SAS has been used to elucidate the intracellular mechanisms involved in the growth-inhibitory effects in Glioma cells by the nitric oxide-releasing diazeniumdiolate, JS-K, [55] . Therefore, the use of SAS at 10 uM in the current study is at a non-lethal dose, thus the increased CBX anti-proliferation effect may be due to drug-drug interaction or inhibition of multiple signal effectors (kinases).
2. Materials and Methods
2.1. Cell Culture
The human brain tumor, Glioma LN18, Neuroblastoma, SKN-SH and prostate, PC3, cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA). They were grown in Dulbecco’s modified Eagle’s medium (DMEM)/F12, supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin and 100 µg/ml streptomycin. Cells were grown at 37˚C in a humidified atmosphere of 5% CO2. Cells were passaged by exposure to trypsin/EDTA and plated at densities of 4 × 105 cells per well of a 6-well plate for transfection with Lipofectamine (Invitrogen) or at 5 × 104 per well of a 96-well plate for proliferation assay.
2.2. Chemicals and Cell Treatment
Glutamate, Celecoxib and Sulfasalazine were all purchased from Sigma-Aldrich Co. (St. Louis, MO). For RNA and protein extraction, overnight cultures of 105 cells in each of 6-well plates were rinsed and incubated with DMEM supplemented with select compound. Following treatment, cells were lyzed and RNA or protein extracted with the TRIzol (Invitrogen) reagent. For proliferation assay, cells were seeded in 96-well plates and processed as described below.
2.3. Confocal Immunofluorescence Microscopy
Confocal Fluorescence Microscopy: Cells grown in culture slides were fixed with 3% paraformaldehyde in PBS for 15 min. After three washes with PBS, cells were permeabilized with 0.1% Saponin in PBS, soaked in blocking solution (1% bovine serum albumin in PBS) then incubated with primary antibody at 1:500 fold-dilutions for 1 h at room temperature. After 2 washes an alexafluor-conjugated secondary antibody (Invitrogen) at 1:1000 dilution was used for 1 hour to amplify signals, while Phalloidin 596 (Invitrogen) at 1:200 dilution was used for F-actin cytoskeleton tagging, for 1 h at room temperature, then rinsed with 1X PBS. Stained cells were mounted with coverslip and samples were examined using the Zeiss LSB 700 confocal microscope (Carl Zeiss, Germany).
2.4. Constructing SPTLC1 Recombinant Cell Lines
Briefly, 105 cells growing near confluence in 6-well plates were rinsed with phosphate-buffered saline, PBS, followed by incubation with 1 µg of the vector in Lipofecatmine Transfection mix as instructed by the manufacturer (Invitrogen, Carlsbad, CA). Treated cells were incubated in complete medium overnight. Stable transfectants were selected with Geneticin (G418) by culturing in DMEM supplemented with 300 µg/ml of the antibiotic. Cells were cultured in the selective medium for at least 10 days and surviving cells harvested and seeded in new flasks with complete culture medium. For validation of the SPTLC1 recombinants, RNA was isolated using the Trizol reagent (Life Technologies, Grand Island, NY) and SPTLC1 expression detected by reverse transcription-PCR.
2.5. Proliferation Assay
For proliferation assay, cells growing near confluence were harvested, re-suspended in DMEM supplemented with FCS and seeded into 96-well at 5 × 104 cells per well. Prior to chemical treatment, cells seeded overnight, in order to adhere and acclimatize, were rinsed with 1X PBS and then re-suspended in serum-free DMEM medium for 1 h. The medium was removed and cells were treated with pre-determined concentration of the test compound in at least triplicate wells. The survival/proliferation assay was performed using a modification of the microculture tetrazolium (MTT) assay, with the reagent WST-1 (Roche Diagnostics, Indianapolis, USA). The metabolic cleavage of a tetrazolium salt to a water-soluble formazan is accompanied by color which directly correlates with the number of metabolically active cells in culture. This allows for quantitative colorimetric assessment of cellular viability and proliferation following treatment. After the incubation period, absorbance in each well was measured with a microplate reader at 450 nm. The absorbance by formazan product at this wavelength, after correcting for background from control wells, was used as an index of viability/cell proliferation. Percentage of viable cells was computed as follows:
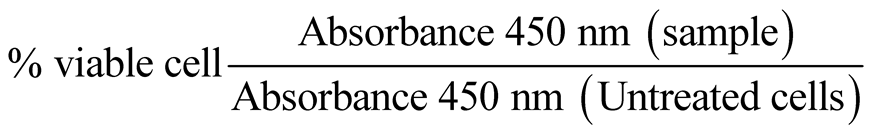
2.6. Cell Lysis, Immunoprecipitation and PAGE
Protein Isolation, Immunoprecipitation and PAGE: Cells were lysed with Immunoprecipitation RIPA buffer (Pierce Fisher Thermo Scientific, Rockford, IL), then crude lysate was subjected to 10 min 10,000 rpm centrifugation at 4˚C. Supernatant was collected as total cell lysate, supplemented with protease inhibitor cocktail and stored frozen until use. For immunoprecipitation, 200 µL lysate was incubated with 5 µg primary antibody in a microfuge tube and allowed to incubate on a rocking platform with gentle shaking at 4˚C for 1 hour. Then 50 µL of protein G Magnetic bead slurry was added to the immunoprecipitate mix and incubated on a rocking platform with gentle shaking at 4˚C for 1 hour. The tube was then centrifuged at 10,000 rpm for 30 seconds to pellet the beads. The supernatant was carefully removed and the beads washed 3 times with 500 µL of lysis buffer. After the final wash and aspiration of supernatant, the pelleted immunoprecipitate was mixed with 30 µL of Laemmli sample buffer. The mix was heated at 90˚C for 5 minutes, centrifuged at 10,000 rpm for 1 minute. The supernatant was carefully aspirated and loaded onto the gel, using See Blue Plus Pre-stained protein marker (Invitrogen). Gel was run until the dye front was at the bottom of the unit, removed and stained for visual analysis of immuno-precipitated protein with Gel Code Blue (Invitrogen).
2.7. Statistical Analysis
All experiments were repeated at least three times. Results where applied are shown as the mean ± SEM (Standard Error of Mean computed from raw data). Where there is no overlap in error bar, statistical comparisons between data sets were further analyzed with two-tailed Student’s t-test. P < 0.05 was taken to indicate statistical significance.
3. Results and Discussions
Confocal microscopy was used both for sub-cellular localization of SPTLC1 and to confirm its translocation from cytoplasm to focal adhesion sites in cells responding to the stress of chemical treatment.
The SPTLC1 protein, as shown in Figure 1, is detectable by immunostaining in the cytoplasm of all the cell lines examined in the current study. The expression of a green fluorescent SPTLC1 chimera is also seen in the cytoplasm of cells from live imaging as shown in the bottom panel (right) of Figure 1.
Immunostaining for comparative sub-cellular localization or co-localization was to the cytoskeletal F-actin or Paxillin protein, the images of Figure 2 in the top panel support the localization of SPTLC1 in the cytoplasm, which appears as green fluorescence, surrounded by red fluorescing F-actin stress fibers. In the bottom panel of Figure 2, localization of either the SPTLC1 or Paxillin protein is at focal adhesion sites in individual images and they correspond to each other in the merged image.
As Paxillin is a known focal adhesion protein, the observed translocation of SPTLC1 from the cytoplasm to focal adhesion sites of stressed cells seen in the images of Figure 3 support a novel stress responsive nature of the human SPTLC1 protein [3] [6] to CBX. The focal adhesion localization of COX-2 mediated by CBX has not been reported before and is of interest that it corresponds with the localization of the other stress induced focal adhesion proteins in this study.
Anchorage independent colonies represent in vitro correlates of tumor mass with cells in the center under hypoxic condition. Immunostaining images of Figure 4 show that SPTLC1 is expressed in cells aggregated as anchorage independent colonies. It is known that hypoxic regions where oxygen-deprived cells in tumors thrive

Table 1 . Effect of SPTLC1 on 48 h Cytotoxicity Index CI50 for CBX.
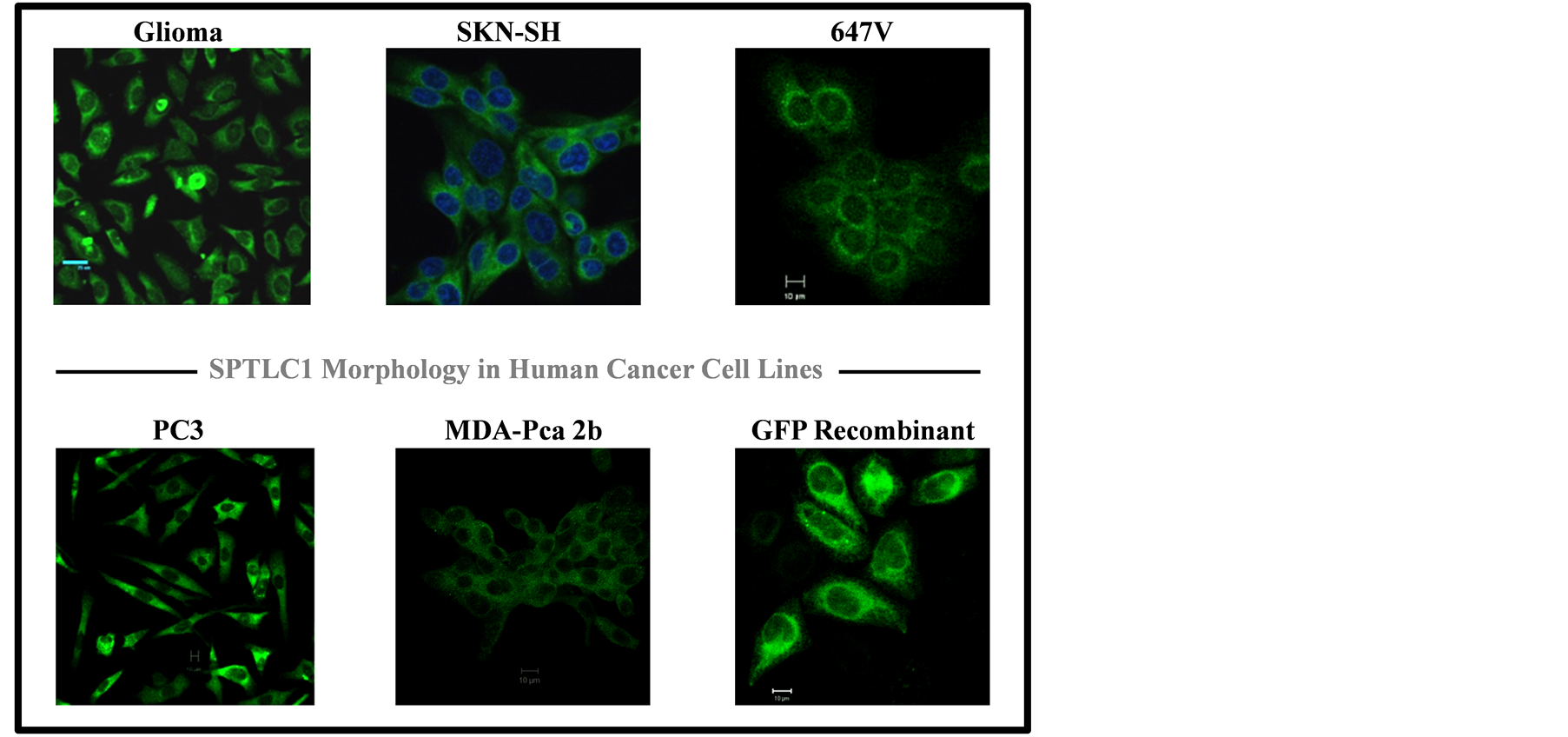
Figure 1. Immunostaining pattern of SPTLC1 in inflammation-associated human cancer cell lines. Images of formaldehyde-fixed cells on glass slides were taken using a Zeiss 700 confocal microscope (Zeiss, Germany), with a 40× objective lens. The SPTLC1 protein is in Green fluorescence localized to the cytoplasm. Nuclei staining are in Blue. Horizontal line is 10 micrometer scale.

Figure 2. Co-localization of SPTLC1 with cytoskeletal protein. Images of formaldehyde-fixed cells on glass slides were taken using a Zeiss 700 confocal microscope (Zeiss, Germany), with a 40× objective lens. Target SPTLC1 protein is in Green fluorescence and actin filaments are in red. Horizontal line is 10 micrometer scale.
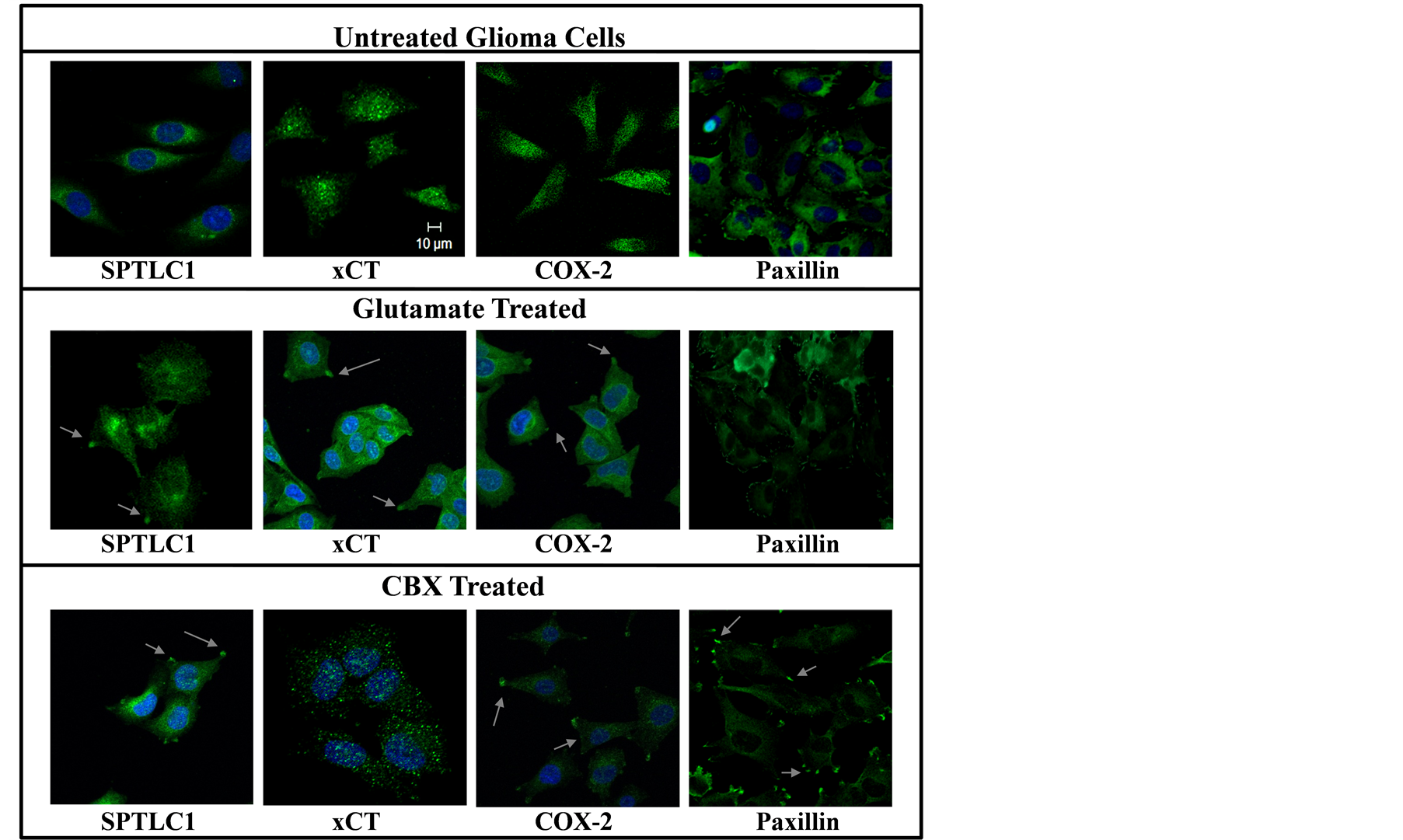
Figure 3. Focal adhesion localization of SPTLC1, xCT and COX-2 in Chemical treated cells. Glioma LN18 cells cultured on cover slide were treated with 10 uM Glutamate or Celecoxib in culture medium without supplements, for 3 hours. Images of formaldehyde-fixed cells immunostained for SPTLC1, xCT, COX-2 or Paxillin, were taken with a confocal microscope, 40× objective lens. Target proteins are seen as green fluorescent signals, nuclei are in blue. Horizontal line is 10 micrometer scale.
have altered gene expression, in a manner that contributes to cell survival and is thought to be important in the acquisition of a multidrug resistance phenotype.
Glioblastomas are resistant to Glutamate toxicity, but release relatively high levels of Glutamate in the brain, that can cause widespread Glutamate-mediated excitotoxicity [13] [14] , that is known to lead to neuronal cell death [15] . For both the SKN and Glioma cell lines, in the data of Figure 5, the recombinant cell constructs transfected with C-terminal modified SPTLC1 (-CSPT) show dose-dependent decrease in their sensitivity to the cytotoxic effect of Glutamate.
Implicated in chemical mediated anti-proliferation effect, particularly for Glutamate in other tissues, is the inhibition of the xCT antiporter system leading to lowered levels of protective anti-oxidant intracellular GSH species [20] . In the left panel of Figure 6, both parental and SPTLC1 recombinant Glioma cells exhibit similar dose-dependent sensitivity to CBX alone. In the right panel, the anti-proliferation activity of SAS alone is seen only at drug doses greater than 50 μM in both 24 h and 48 h proliferation assays. The slight increase in the number of viable cells assessed at the high SAS dose treatment could be due to compensatory recovery of cell proliferation over the 48 hour time period.
Exposure to the environmental contaminant, 3-MC, can adversely affect wildlife and human health. The compound is a potent inducer, through AhR, of the transcriptional expression of Cyp1A1 [39] . Although SPTLC1 can affect transcriptional expression of Cyp1A1 [6] , panel C of Figure 7 indicates that the presence of 3-MC did not significantly modulate chemosensitivity to CBX. Geldanamycin is known to inhibit the transcriptional expression of Cyp450 through binding to Hsp90 and hence disrupting AhR receptor-mediated transcriptional expression of Cyp1A1 [59] . This disruption is also known to deplete many oncogenic client proteins and has been implicated in the antitumor activity of Geldanamycin. In panel D of Figure 7, increased chemosensitivity to combination drug treatment with Geldanamycin is seen with the Glio-CSPT recombinant cells, when compared to the parental or recombinant -Iso cell line. This contrasts sharply with the CBX treatment alone in panel A, where all the Glioma cell lines exhibit similar dose-dependent chemosensitivity to CBX.
In tissues such as the prostate and bladder, CBX acts through the angiogenic endothelium [45] which may affect the bioavailability of the drug. However, drug treatment of cells in culture reflects direct effect on proliferation as no barrier to bioavailability is encountered. As indicated in the graphs of Figure 7 and Figure 8, depending on the cell type, there are differences in the dose-dependent cellular chemosensitivity to CBX. Although SPTLC1 expression appears to modify sensitivity of cells to CBX, however, there are differences in this modulating effect, also probably cell line dependent.
The computation of CI50 values allow for comparative analysis of dose-dependent cellular response to drug

Figure 4. Localization of SPTLC1 in cells undergoing anchorage independent growth. Panel (A) is light microscopy image of cells growing as anchorage independent colonies on soft agarose and those transferred unto culture plate re-attaching. In panel (B), distinct SPTLC1 expression in cell aggregates formed during anchorage independent growth is seen in green. Green fluorescent immuno-labeled SPTLC1 is seen in cells of panel (C) containing cells from colonies undergoing re-attachment on poly-lysine coated culture plate. Panel (C) nuclei stain (top) is in blue and F-actin staining is in red (bottom).
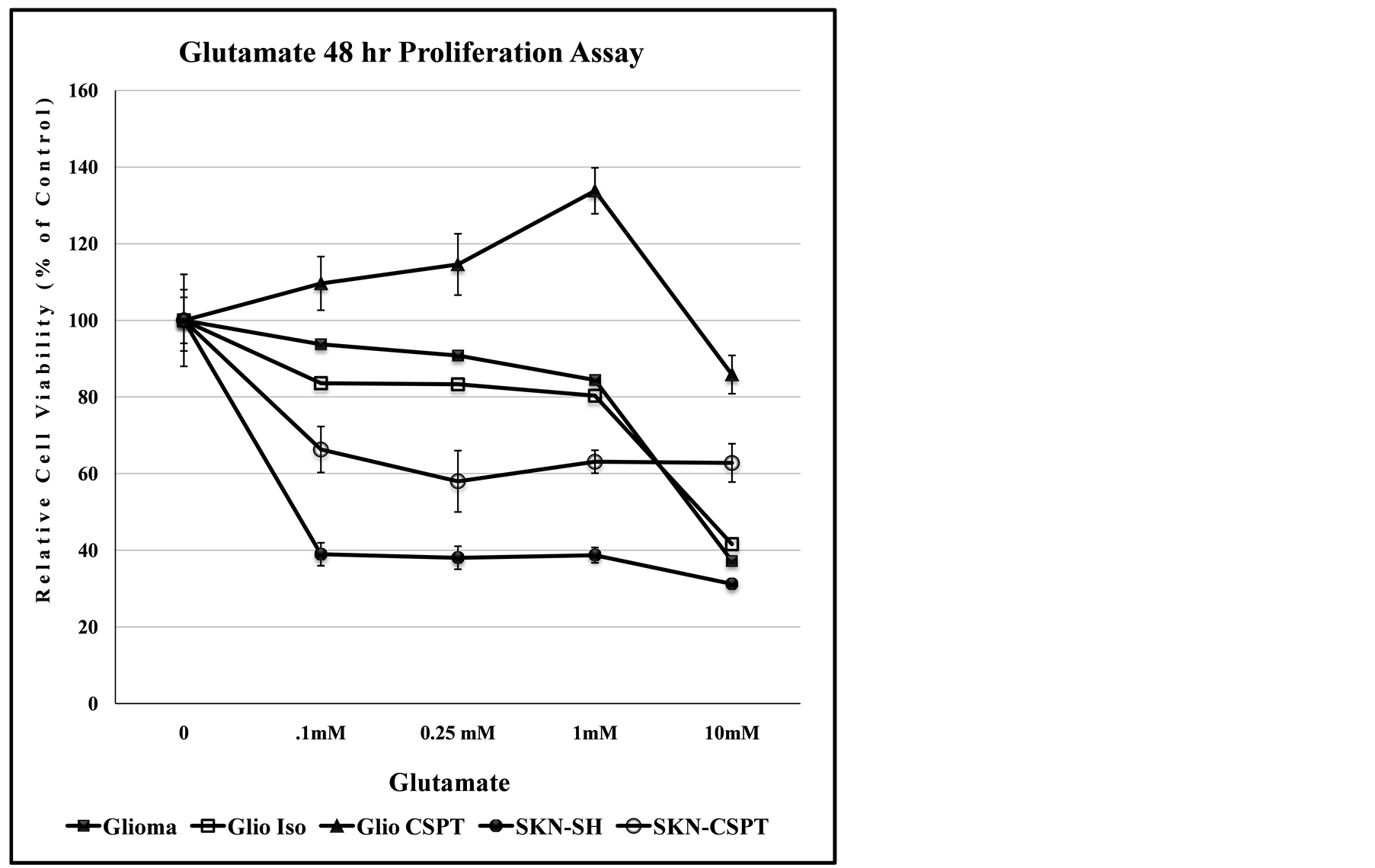
Figure 5. Recombinant SPTLC1 expression modulates dose-dependent anti-proliferation effect of glutamate. The Glioma LN18 and SKN-SH cell lines are differentially sensitive to the toxicity of Glutamate. Cells growing in 96-well culture plate were exposed to increasing concentration of Glutamate and assessed for proliferation after 48 hours. Data points represent relative cell viability compared to untreated control for each cell line.

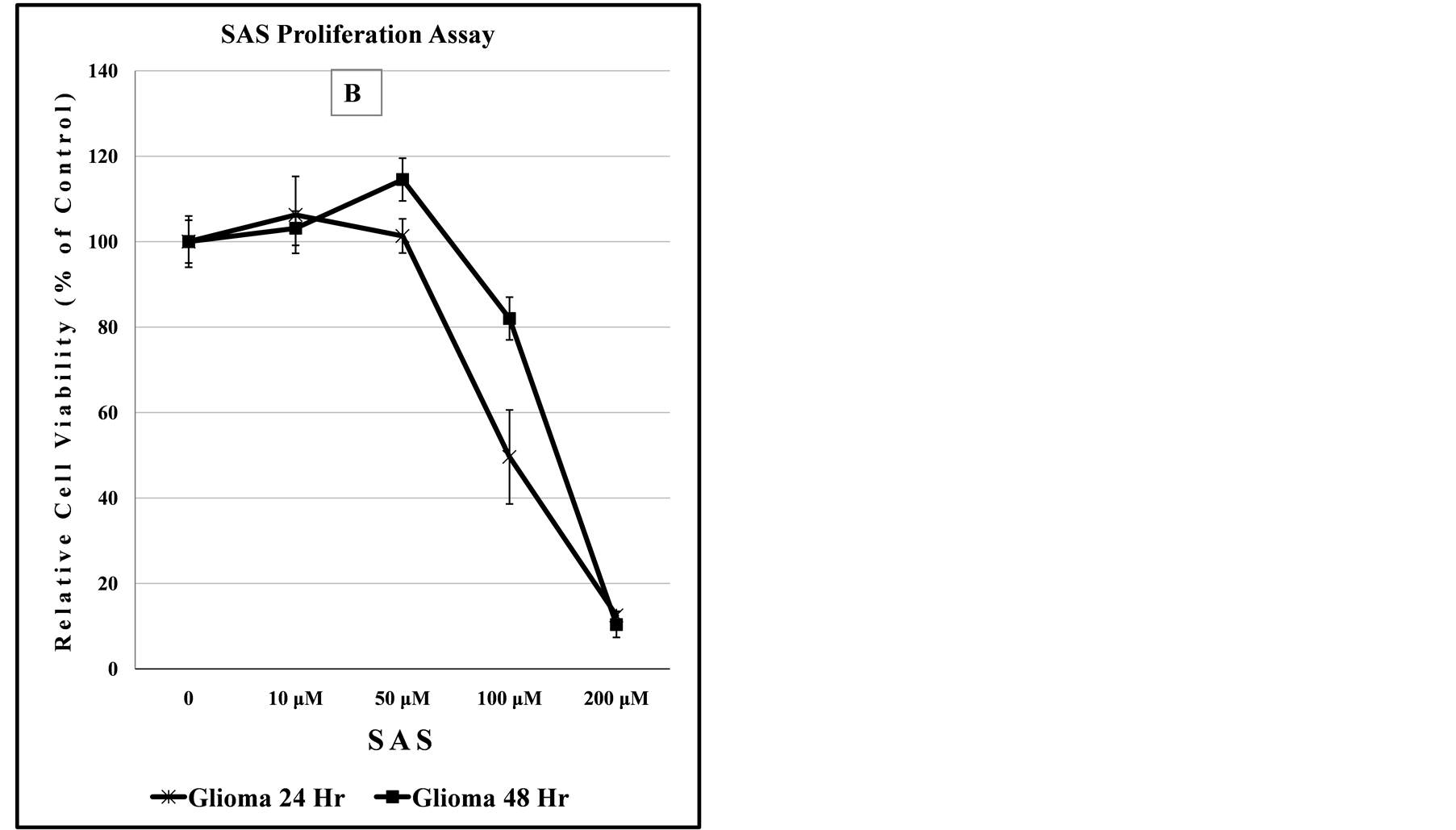
Figure 6. Recombinant SPTLC1 expression does not modulate anti-proliferation effects of CBX or SAS on Glioma cells. Cells growing in 96-well culture plate were exposed to increasing concentration of CBX (panel A) of SAS (panel B). Relative cell viability was assessed after 48 hours. Data points represent relative cell viability compared to untreated control for each cell line.
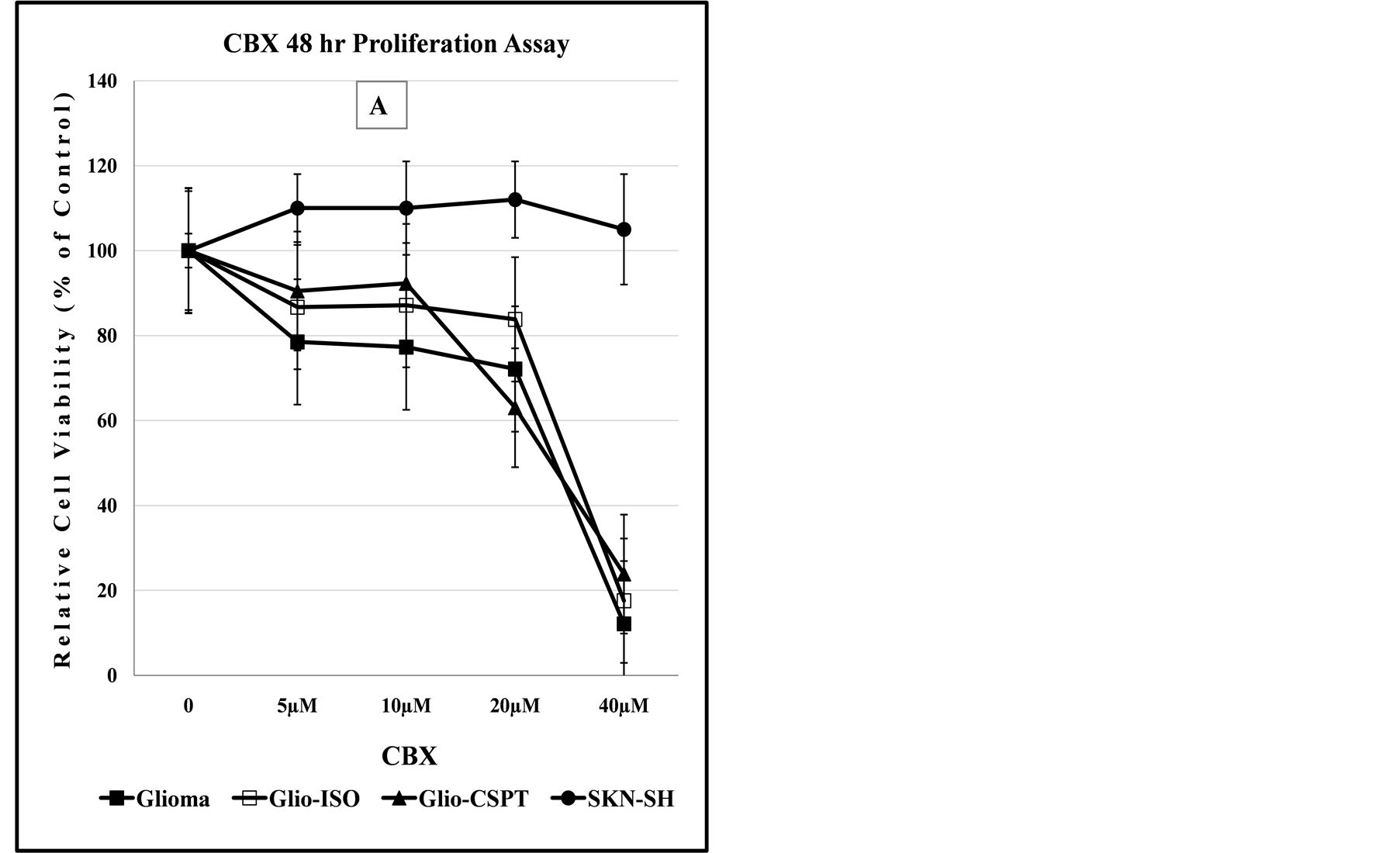
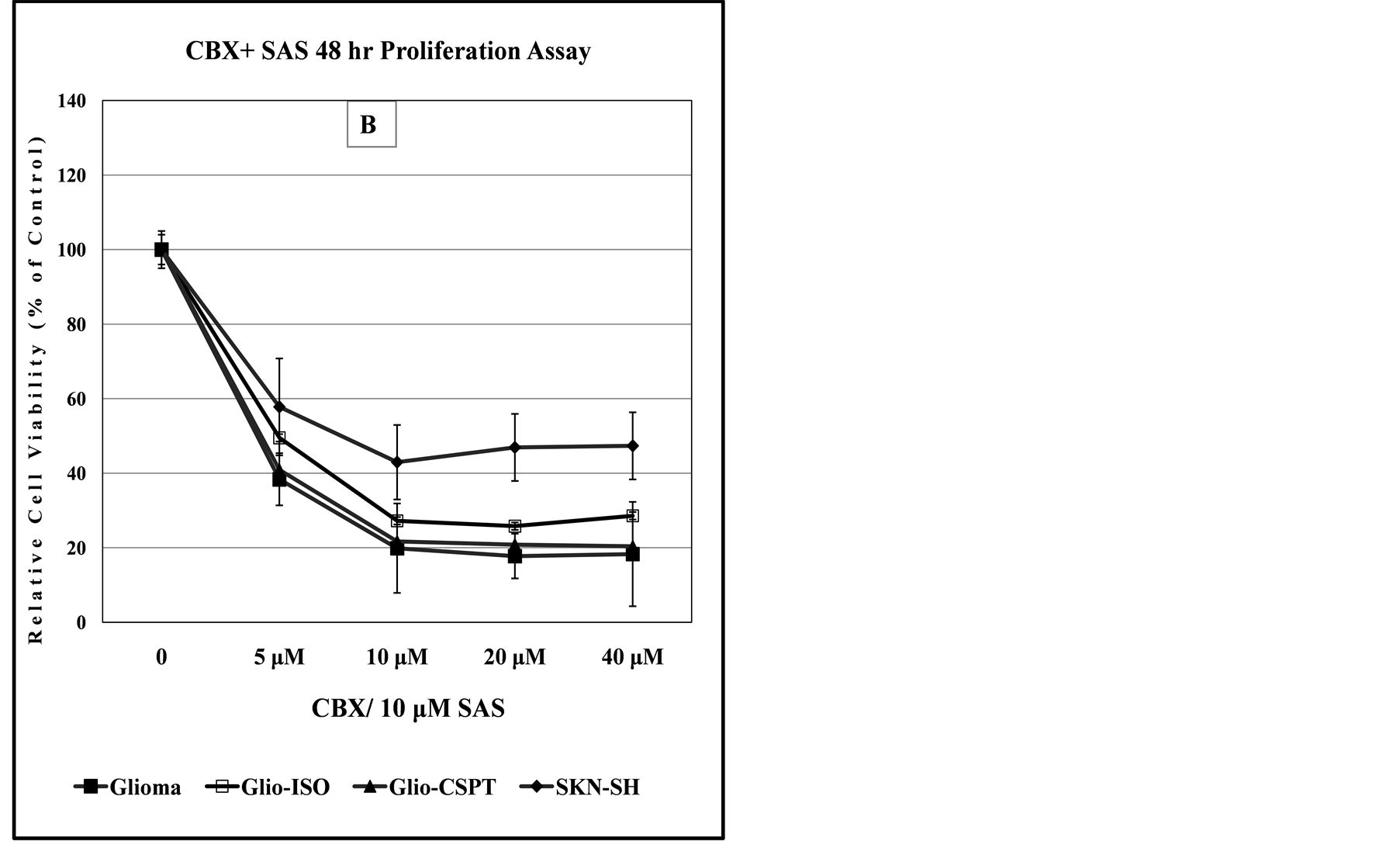

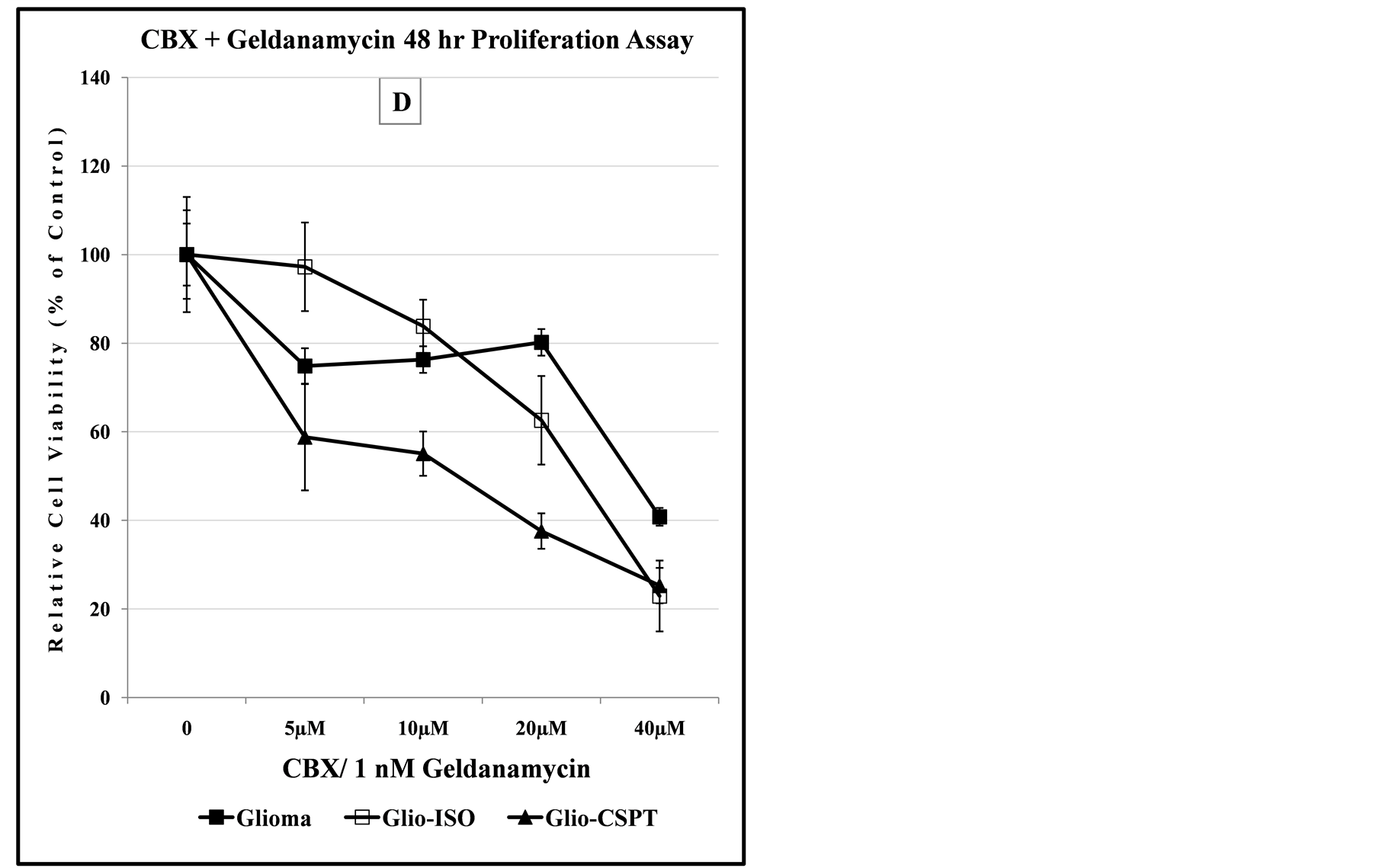
Figure 7. Dose-dependent anti-proliferation effect of CBX alone or in combination with SAS, 3-MC or Geldanamycin. Panels (A) and (B) indicate that SKN-SH cells display less sensitivity to the cytotoxic effect of CBX or CBX/SAS co-treatment compared to Glioma cells. Co-treatment with a non-lethal dose of SAS increases the anti-proliferation effect of CBX for the SKN-SH, Glioma and SPTLC1 recombinant Glioma cell lines tested. In panel (C), all cell lines co-treated with CBX and 3-MC, show similarity in the dose-dependent sensitivity to the chemical treatment. In contrast, the graph of panel (D) indicates that cells transfected with the C-terminal modified SPTLC1 recombinant vector (-CSPT) are significantly more sensitive than the parental Glioma or -Iso cell lines, when treated with the combination of CBX and Geldanamycin.
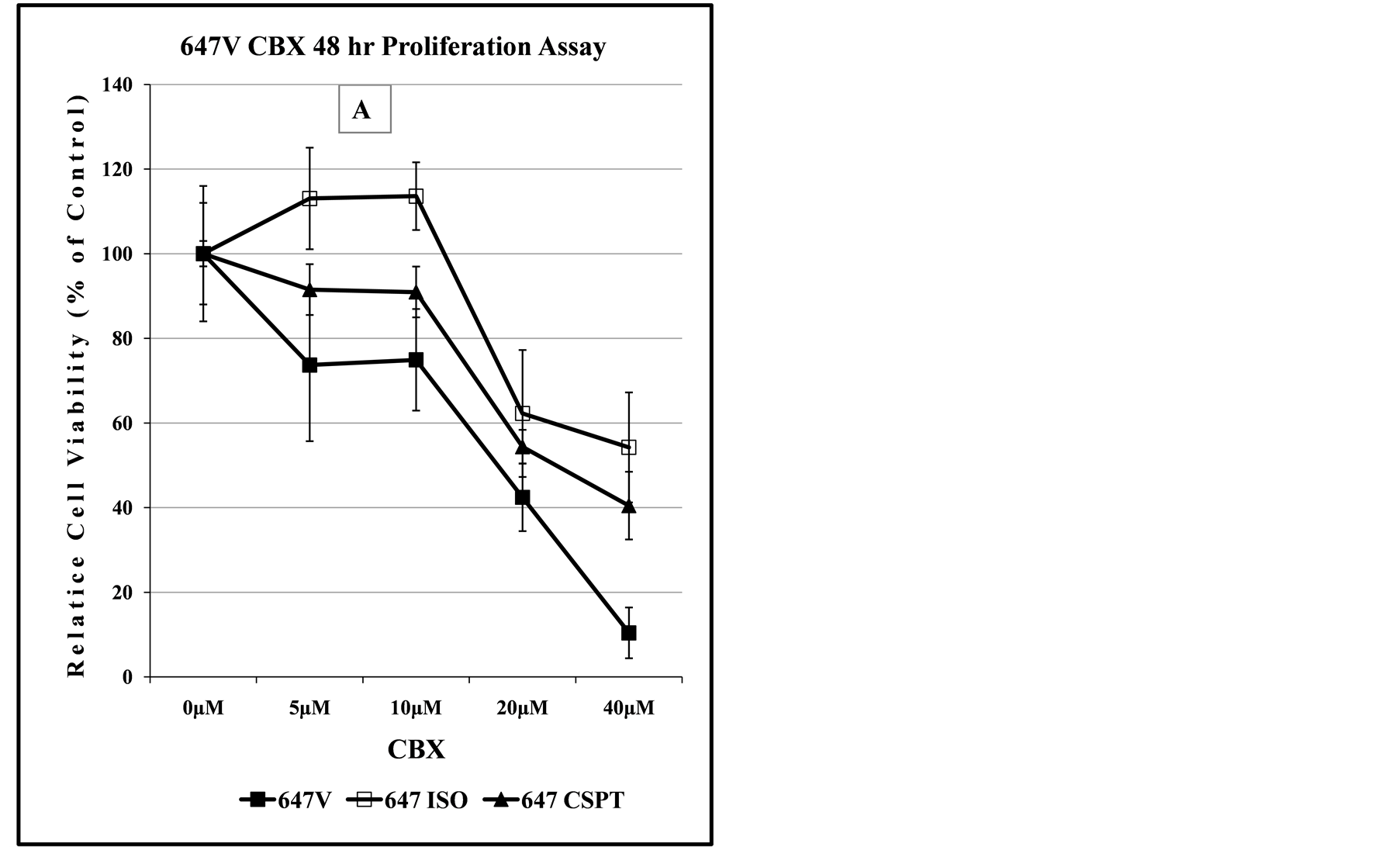
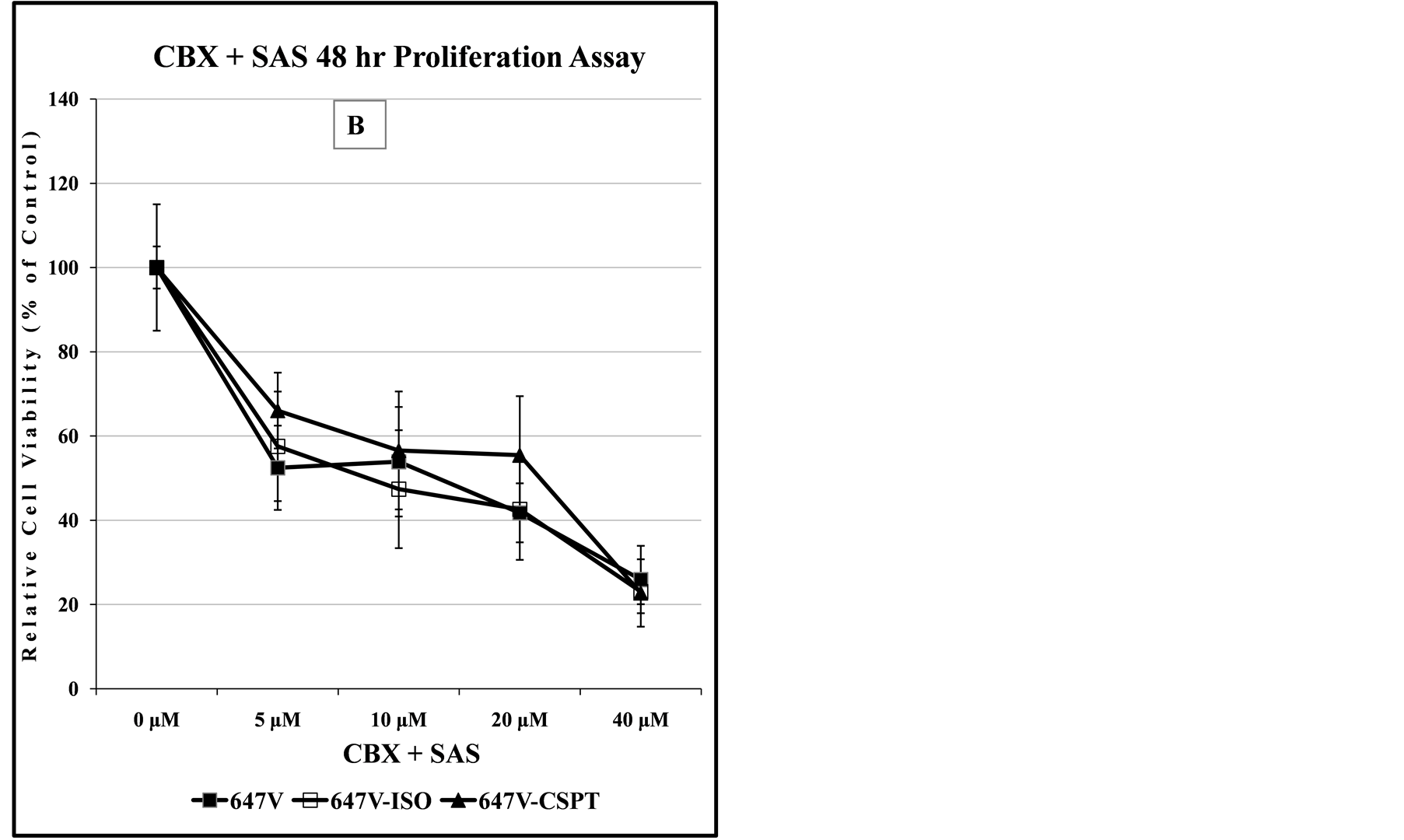
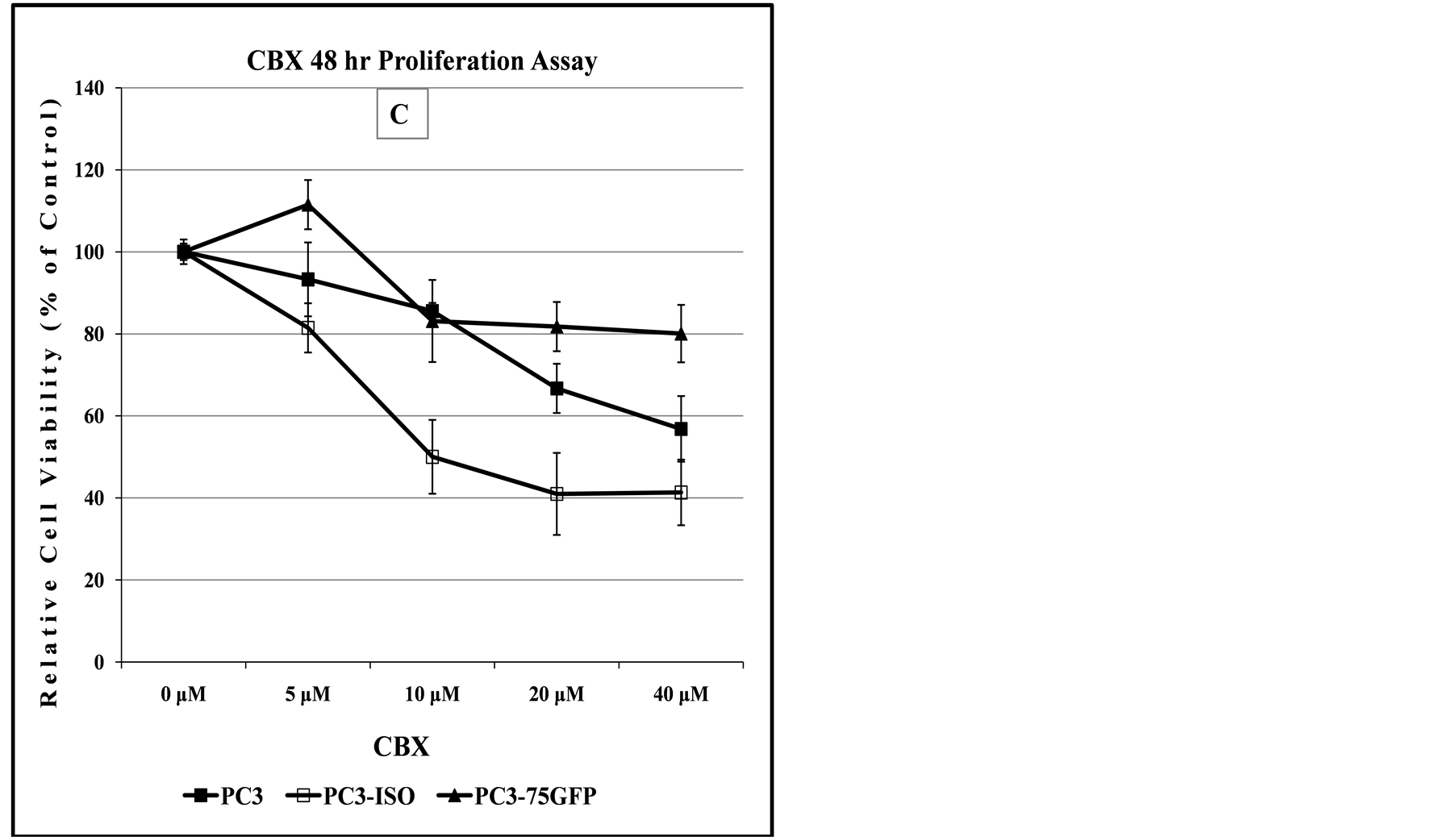
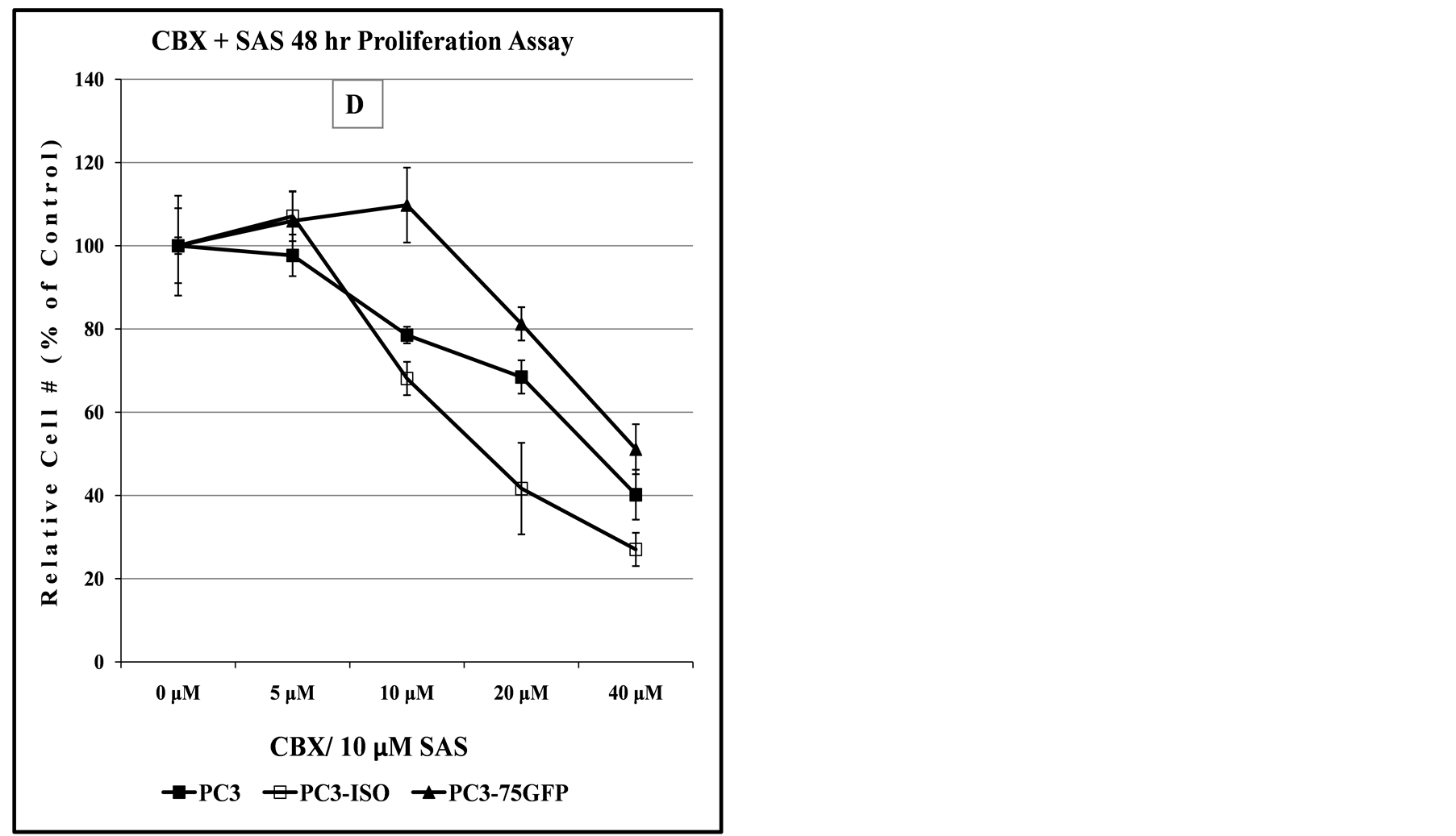
Figure 8. Comparison of dose-dependent anti-proliferation effect of CBX treatment alone or in combination with SAS in prostate and bladder cancer cells. The graph in panel (A) indicates the dose-dependent response of 647V bladder cancer cell lines to the cytotoxic effect of CBX, suggesting that at doses up to 10 µM, the -Iso recombinant 647V cells are resistant to the anti-proliferation effect of CBX, when compared to the parental or -CSP counterparts. As shown in panel (B) this difference in chemosensitivity to CBX does not exist for the cell lines when they are treated with a combination of CBX and a non-lethal dose of SAS. In panel (C), the graph shows that at all dose treatments used, the -Iso recombinant 647V cells exhibit greater sensitivity, than the parental or the recombinant -75GFP counterparts, to the dose-dependent anti-proliferation effect of CBX, either alone or when used in combination with a non-lethal dose of SAS. The PC3-75GFP recombinant is a C-terminal modified SPTLC1 recombinant containing the GFP sequence, similar to the -CSPT construct except it is also missing 75 nucleotides at the C-terminus, which are present in -CSPT.
treatment. For Table 1, an increase in the CI50 value from 25 μM to 40 μM for CBX/3-MC treated Glio-CSPT cells is indicative of decreased chemosensitivity to CBX. In contrast, a decrease of the CI50 value from 25 μM to 12 μM for Glio-CSPT cells treated with CBX/Geldanamycin is indicative of increase in chemosensitivity to CBX. Coincidentally, the Glio-CSPT cells are reported to have attenuated transcriptional expression of Cyp1A1 when treated with 3-MC [6] . In Figure 9, similar to decreased transcriptional expression of Cyp1A1 in 3-MC treated cells, the transcriptional expression of Cyp3A4 is also attenuated in Glio-CSPT recombinant cells when treated with CBX. The Cyp3A4, but not Cyp1A1 variant is considered to play a greater role in the metabolism of CBX [35] [37] .
As shown in Table 2, at 48 hours, co-treatment of all the Glioma cell lines with CBX and SAS resulted in similar reduction of the CI50 to about 5 µM. When all the cell lines examined are compared, the data of Table 2 also indicates that the 647V-Iso recombinant Bladder cancer cell line, with about 8-fold increase in CI50 appears to be the most sensitive to the CBX-SAS combination treatment.
Although the Cyp2C9 variant is known to play a key role in the metabolism of CBX (27), Cyp3A4 is considered to play a minor role and so it was of interest to determine the Cyp450 expression in cells treated with CBX. The data of Figure 9 shows the relative levels of transcriptional expression of the three Cyp450 variants. In all samples tested, Cyp3A4 transcript level in CBX treated cells is markedly decreased in Glio-CSPT recombinant Glioma cells, compared to the other cell lines, consistent with the ability of the C-terminal modified SPTLC1 variant to alter Cyp450 gene transcript level (6). Together, the data in Figure 7 and Figure 9, as well as Table 1, show that in only the Glio-CSPT cell construct, SPTLC1 expression alters CBX mediated transcriptional ex
Table 2 Effect of SPTLC1 on 48 h Cytotoxicity Index CI50 for CBX treated inflammation associated cancer cell lines and their SPTLC1 recombinants.
*Cytotoxicity Index values (CI50) shown here in µM is the drug dose required to kill 50% of treated cells, which is extrapolated from individual dose response curve. Values were computed from the proliferation curve generated with cells exposed to either CBX alone, in combination with 3-MC (5 µM) or with Geldanamycin (1 µM), in Dulbecco’s MEM/F12 culture medium for 48 h at 37˚C. ND indicates “not detectable” value for the CI50 as this could not be computed from the dose response curve.
*Δ represents change in chemosensitivity: this was estimated from fold change in CI50 value computed from the CBX treatment divided by that of the CBX/SAS treatment. The assignment of “Increase” in chemosensitivity is based on computed ratio greater than 2 and “Decrease” based on computed ratio less than 0.5. Values not meeting the thresholds are assigned “None”.
pression of Cyp3A4 and concomitantly, the chemosensitivity of these cell construct to combination drug treatment with CBX and Geldanamycin.
The protein electrophoresis conducted in this study was only for qualitative purpose since the variant of SPTLC1 expressed, but not the quantity of SPTLC1 in cells is considered to be more likely responsible for the modulating effect on cellular chemosensitivity. The gel image of Figure 10 is both representative of the presence of SPTLC1 in cells and is in agreement with the positive immuno-stain for SPTLC1 in all the cell lines used in the current study.
4. Conclusions
The positive immuno-labeling of SPTLC1 in the cytoplasm of the cancer cell lines of different tissue origin used in the current study corresponds with its broad tissue expression. The similarity seen in the focal adhesion localization of SPTLC1 with that of COX-2, xCT System and Paxillin, is consistent with the behavior of signaling

Figure 9. SPTLC1 modulates Cyp450 transcriptional expression in CBX treated cells. 1 = Glioma; 2 = CBX treated Glioma; 3 = Glio-Iso; 4 = CBX treated Glio-Iso Cyp1A1; 5 = Glio-CSPT; 6 = CBX treated Glio-CSPT. Total RNA was harvested from samples in triplicate wells of untreated and treated cells. Following reverse transcription of about 1 µg RNA, cDNA generated were subjected to quantitative PCR with primers specific for each of the three -1A1, -1A2 and -3A4 Cytochrome p450 variants.
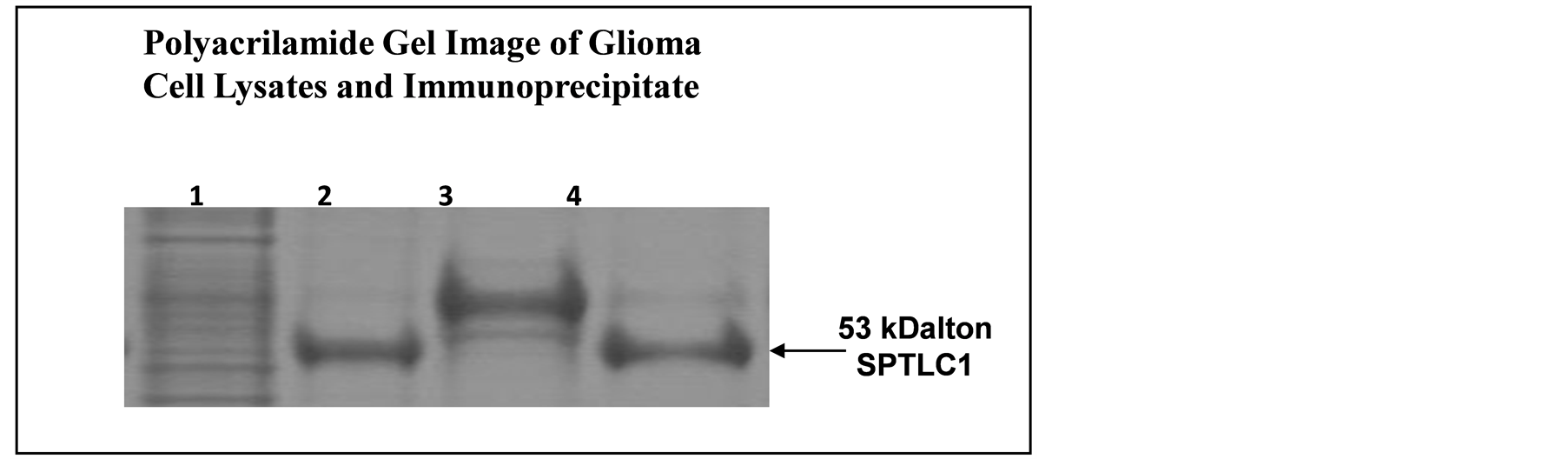
Figure 10. Immunoprecipitation-PAGE analysis of SPTLC1 protein expression in cells growing attached and as colonies. 1 = Glioma Cell Lysate; 2 = Immunoprecipitate of Lysate; 3 = Anchorage Independent Colony Lysate; 4 = Immunoprecipitate of Colony Lysate. SPTLC1 protein band seen in the immunoprecipitate sample lanes (2 and 4) correspond to the full length SPTLC1 fragment reported at the 53 kDalton band region. The gel image is from loading immunoprecipitate generated from 50 µg of total protein extracted from attached cells and anchorage independent colonies, using antibody against SPTLC1. Band intensity was assessed using the ImageJ analysis program.
proteins in cells responding to diverse environmental stress stimuli [60] . This localization of SPTLC1 also places it in sub-cellular domains where there is the opportunity for crosstalk with proteins involved in pro-survival signaling, but the extent of all the possible stress response proteins it interacts with and the mechanisms by which it functions in the biological process of survival remain to be fully determined. In addition, SPTLC1 expression is seen in cell colonies. Highly proliferative Glioma tumor cells uniquely display anchorage independent growth in inert matrices (e.g. soft agar and agarose) and are highly tumorigenic as xenografts in nude mice [61] . How the presence of SPTLC1 seen in anchorage independent colonies in the present study may contribute to the drug resistance phenotype or the process of metastasis remains to be investigated.
Stress induced cellular response triggered by toxic compounds such as CBX, may lead to the death of cancer cells of different tissue origins [28] [62] . Generally, in chemosensitivity assay of the current study, recombinant cells expressing SPTLC1 (-Iso or -CSPT) are found to exhibit some of the traits associated with adaptive response to toxic insults including chemoresistance to Glutamate or CBX alone and the ability for anchorage independent growth in soft agarose. The use of SAS at 10 μM in the current study is at a non-lethal dose, but may still elicit signaling response. Therefore increase in anti-proliferation mediated by CBX when cells are co-treated cannot be attributed to the toxicity of SAS, but may be due to drug-drug interaction or the consequence of inhibiting multiple signal effectors (kinases). Drug-drug interaction, including with SAS, can modulate therapeutic effect through multiple mechanisms. For example, SAS potentiates the anti-bleeding effect of the anticoagulant, warfarin, commonly used to control and prevent thromboembolic disorders or avoid hemorrhagic complications [63] [64] , but loss of blood coagulation is also associated with SAS co-treatment, thought to be possibly through drug-drug interaction causing resistance to blood coagulation.
Similarly, exposure to 3-MC or Geldanamycin, which are potent modifiers of transcriptional expression of members of the Cytochrome p450 gene family can affect therapeutic efficacy of drugs. The expression of C-terminal modified SPTLC1 in Glio-CSPT cells significantly increases chemosensitivity to CBX in the presence of Geldanamycin, as evidenced by a change in value from a CI50 of 25 µM to 12.5 µM. Interestingly, it is also only in the Glio-CSPT cells that CBX-induced transcriptional expression of Cyp3A4 is found to be significantly attenuated, when compared to its parental or -Iso recombinant. Geldanamycin is metabolized by Cyp3A4 and the hydroquinone derivative is implicated in its cytotoxicity although a much lower dose than that required for cytotoxicity of Geldanamycin was used in the present study. It is possible that the modulation of CBX-mediated transcriptional expression of Cyp3A4 by SPTLC1, via a mechanism-based inhibition effect, combined with systemic metabolic inhibition due to Geldanamycin, may be contributing to the increased chemosensitivity exhibited by the Glio-CSPT recombinant cells used in the current study. This observation corresponds to a previous report implicating SPTLC1 expression in mechanism-based inhibition of the transcriptional expression of Cyp1A1 in 3-MC treated cells [6] .
While data from the current study suggest that SPTLC1 expression may affect sensitivity to drug treatment, differences in chemosensitivity of the cell lines suggest that this effect depends on chemical, cell and tissue type. However, taken together, the results of this study further support the idea that expressing the C-terminal modified SPTLC1 variant, probably through crosstalk with proteins in signaling modules, is capable of modulating cellular stress response behavior. This is of particular interest to human health with regards to therapeutic response, the acquisition of a multidrug resistance phenotype which promotes cell survival and malignant progression.
Acknowledgements
Dr. Johnnella Butler, the Provost and Vice President for Academic Affairs, Dr. Carmen Sidbury, Associate Provost of Research, Spelman College, made available the Startup Fund and Title III Program fund for the purchase of equipments and reagents used in the research study reported here.
References
- Yasuda, S. Nishijima, M. and Hanada, K. (2003) Localization, Topology and Function of the LCB1 Subunit of Serine Palmitoyltransferase in Mammalian Cells. The Journal of Biological Chemistry, 278, 4176-4183. http://dx.doi.org/10.1074/jbc.M209602200
- Dawkins, J.L., Hulme, D.J., Brahmbhatt, S.B., Auer-Grumbach, M. and Nicholson, G.A. (2001) Mutations in SPTLC1, Encoding Serine Palmitoyltransferase, Long Chain Base Subunit-1, Cause Hereditary Sensory Neuropathy Type I. Nature Genetics, 27, 309-312. http://dx.doi.org/10.1038/85879
- Wei, J. Yerokun, T., Leipelt, M., Haynes, C., Radhakrishna, H., Merrill, A.H., et al. (2009) Serine Palmitoyltransferase Subunit 1 Is Present in the Endoplasmic Reticulum, Nucleus and Focal Adhesions, and Functions in Cell Morphology. Biochimica et Biophysica Acta, 1791, 746-756. http://dx.doi.org/10.1016/j.bbalip.2009.03.016
- Carton, J.M., Uhlinger, D.J., Bartheja, A.D., Derian, C., Ho, G., Argenteri, D. and D’Andrea, M.R. (2003) Enhanced Serine Palmitoyltransferase Expression in Proliferating Fibroblasts, Transformed Cell Lines and Human Tumors. Journal of Histochemistry Cytochemistry, 51, 715-726. http://dx.doi.org/10.1177/002215540305100603
- Bartheja, A.D., Uhlinger, D.J., Canton, J.M., Ho, G. and D’Andrea, M.R. (2003) Characterization of Serine Palmitoyltransferases in Normal Human Tissues. Journal of Histochemistry Cytochemistry, 51, 687-696. http://dx.doi.org/10.1177/002215540305100514
- Yerokun, T. and Stewart, J. (2006) Novel Functional Association of Serine Palmitoyltransferase 1—A Peptide in Sphingolipid Metabolism with Cytochrome p4501a1 Transactivation and Proliferative Capacity of the Human Glioma LN18 Brain Tumor Cell Line. International Journal of Environmental Research and Public Health, 3, 252-261. http://dx.doi.org/10.3390/ijerph2006030030
- Tamehiro, N., Zhou, S., Okuhira, K., Benita, Y., Brown, C.E., Zhuang, D.Z., Latz, E., Hornemann, T., von Eckardstein, A., Xavier, R.J., Freeman, M.W. and Fitzgerald, M.L. (2008) SPTLC1 Binds ABCA1 to Negatively Regulate Trafficking and Cholesterol Efflux Activity of the Transporter. Biochemistry, 47, 6138-6147. http://dx.doi.org/10.1021/bi800182t
- Penno, A., Reilly, M.M., Houlden, H., Laurá, M., Rentsch, K., Niederkofler, V., Stoeckli, E.T., Nicholson, G., Eichler, F., Brown Jr., R.H., von Eckardstein, A. and Hornemann, T. (2010) Hereditary Sensory Neuropathy Type 1 Is Caused by the Accumulation of Two Neurotoxic Sphingolipids. The Journal of Biological Chemistry, 285, 11178-11187. http://dx.doi.org/10.1074/jbc.M109.092973
- Dedov, V.N., Dedov, N. and Nicholson, G.A. (2004) Hypoxia Causes Aggregation of Serine Palmitoyltransferase Followed by Non-Apoptotic Death of Human Lymphocytes. Cell Cycle, 3, 1271-1277. http://dx.doi.org/10.4161/cc.3.10.1163
- Ravenna, L., Sale, P., Di Vito, M., Russo, A., Salvatori, L., Tafani, M., Mari, E., Sentinelli, S., Petrangeli, E., Gallucci, M., Di Silverio, F. and Russo, M.A. (2009) Up-Regulation of the Inflammatory-Reparative Phenotype in Human Prostate Carcinoma. Prostate, 69, 1245-1255. http://dx.doi.org/10.1002/pros.20966
- Tafani, M., Russo, A., Di Vito, M., Sale, P., Pellegrini, L., Schito, L., Gentileschi, S., Bracaglia, R., Marandino, F., Garaci, E. and Russo, M.A. (2010) Up-Regulation of Proinflammatory Genes as Adaptation to Hypoxia in MCF-7 Cells and in Human Mammary Invasive Carcinoma Microenvironment. Cancer Science, 101, 1014-1023. http://dx.doi.org/10.1111/j.1349-7006.2010.01493.x
- Hornemann, T., Wei, Y. and von Eckardstein, A. (2007) Is the Mammalian Serine Palmitoyltransferase a High-Molecular-Mass Complex? Biochemical Journal, 405, 157-164.
- Ye, Z.C. and Sonteimer, H. (1999) Glioma Cells Release Excitotoxic Concentrations of Glutamate. Cancer Research, 59, 4383-4391.
- Takano, T., Lin, J.H., Arcuino, G., Gao, Q., Yang, J. and Nedergaard, M. (2001) Glutamate Release Promotes Growth of Malignant Gliomas. Nature Medicine, 7, 1010-1015. http://dx.doi.org/10.1038/nm0901-1010
- Shih, A.Y., Erb, H., Sun, X., Toda, S., Kalivas, P.W. and Murphy, T.H. (2006) Cystine/Glutamate Exchange Modulates Glutathione Supply for Neuroprotection from Oxidative Stress and Cell Proliferation. The Journal of Neuroscience, 26, 10514-10523. http://dx.doi.org/10.1523/JNEUROSCI.3178-06.2006
- Sonnewald, U., Qu, H. and Aschner, M. (2002) Pharmacology and Toxicology of Astrocyte-Neuron Glutamate Transport and Cycling. Journal of Pharmacology and Experimental Therapeutics, 301, 1-6. http://dx.doi.org/10.1124/jpet.301.1.1
- Bridges, R.J., Natale, N.R. and Patel, S.A. (2012) System xc-Cystine/Glutamate Antiporter: An Update on Molecular Pharmacology and Roles within the CNS. British Journal of Pharmacology, 165, 20-34. http://dx.doi.org/10.1111/j.1476-5381.2011.01480.x
- Murphy, T.H., Miyamoto, M., Sastre, A., Schnaar, R.L. and Coyle, J.T. (1989) Glutamate Toxicity in a Neuronal Cell Line Involves Inhibition of Cystine Transport Leading to Oxidative Stress. Neuron, 2, 1547-1558. http://dx.doi.org/10.1016/0896-6273(89)90043-3
- Praticò, D. (2002) Alzheimer’s Disease and Oxygen Radicals: New Insights. Biochemical Pharmacology, 63, 563-567. http://dx.doi.org/10.1016/S0006-2952(01)00919-4
- Huang, Y., Dai, Z., Barbacioru, C. and Sadée, W. (2005) Cystine-Glutamate Transporter SLC7A11 in Cancer Chemosensitivity and Chemoresistance. Cancer Research, 65, 7446-7454. http://dx.doi.org/10.1158/0008-5472.CAN-04-4267
- Joki, T., Heese, O., Nikas, D.C., Bello, L., Zhang, J., Kraeft, S.K., Seyfried, N.T., Abe, T., Chen, L.B., Carroll, R.S. and Black, P.M. (2000) Expression of Cyclooxygenase 2 (COX-2) in Human Glioma and in Vitro Inhibition by a Specific COX-2 Inhibitor, NS-398. Cancer Research, 60, 4926-4931.
- Deininger, M.H., Weller, M., Streffer, J., Mittelbronn, M. and Meyermann, R. (1999) Patterns of Cyclooxygenase-1 and -2 Expression in Human Gliomas in Vivo. Acta Neuropathologica, 98, 240-244. http://dx.doi.org/10.1007/s004010051075
- Panagopoulos, A.T., Lancellotti, C.L., Veiga, J.C., de Aguiar, P.H. and Colquhoun, A. (2008) Expression of Cell Adhesion Proteins and Proteins Related to Angiogenesis and Fatty Acid Metabolism in Benign, Atypical, and Anaplastic Meningiomas. Journal of Neuro-Oncology, 89, 73-87. http://dx.doi.org/10.1007/s11060-008-9588-3
- Annabi, B., Laflamme, C., Sina, A., Lachambre, M.P. and Béliveau, R. (2009) A MT1-MMP/NF-κB Signaling axis as a Checkpoint Controller of COX-2 Expression in CD133(+) U87 Glioblastoma Cells. Journal of Neuroinflammation, 6, 8. http://dx.doi.org/10.1186/1742-2094-6-8
- Ravenna, L., Sale, P., Di Vito, M., Russo, A., Salvatori, L., Tafani, M., Mari, E., Sentinelli, S., Petrangeli, E., Gallucci, M., Di Silverio, F. and Russo, M.A. (2009) Up-Regulation of the Inflammatory-Reparative Phenotype in Human Prostate Carcinoma. Prostate, 69, 1245-1255. http://dx.doi.org/10.1002/pros.20966
- Arber, N., Eagle, C.J., Spicak, J., Rácz, I., Dite, P., Hajer, J., et al. (2006) Celecoxib for the Prevention of Colorectal Adenomatous Polyps. The New England Journal of Medicine, 355, 885-895. http://dx.doi.org/10.1056/NEJMoa061652
- Davies, N.M., McLachlan, A.J., Day, R.O. and Williams, K.M. (2000) Clinical Pharmacokinetics and Pharmacodynamics of Celecoxib: A Selective Cyclo-Oxygenase-2 Inhibitor. Clinical Pharmacokinetics, 38, 225-242. http://dx.doi.org/10.2165/00003088-200038030-00003
- Zhu, J., Huang, J.W., Tseng, P.H., Yang, Y.T., Fowble, J., Shiau, C.W., Shaw, Y.J., Kulp, S.K. and Chen, C.S. (2004) From the Cyclooxygenase-2 Inhibitor Celecoxib to a Novel Class of 3-Phosphoinositide-Dependent Protein Kinase-1 Inhibitors. Cancer Research, 64, 4309-4318. http://dx.doi.org/10.1158/0008-5472.CAN-03-4063
- Tang, C., Shou, M., Rushmore, T.H., Mei, Q., Sandhu, P., Woolf, E.J., Rose, M.J., et al. (2001) In-Vitro Metabolism of Celecoxib, a Cyclooxygenase-2 Inhibitor, by Allelic Variant Forms of Human Liver Microsomal Cytochrome P450 2C9: Correlation with CYP2C9 Genotype and in-Vivo Pharmacokinetics. Pharmacogenetics, 11, 223-235.
- Ma, H.I., Chiou, S.H., Hueng, D.Y., Tai, L.K., Huang, P.I., Kao, C.L., Chen, Y.W. and Sytwu, H.K. (2011) Celecoxib and Radioresistant Glioblastoma-Derived CD133+ Cells: Improvement in Radiotherapeutic Effects. Journal of Neurosurgery, 114, 651-662. http://dx.doi.org/10.3171/2009.11.JNS091396
- Miller, T.W., Rexer, B.N., Garrett, J.T. and Arteaga, C.L. (2011) Mutations in the Phosphatidylinositol 3-Kinase Pathway: Role in Tumor Progression and Therapeutic Implications in Breast Cancer. Breast Cancer Research, 13, 224. http://dx.doi.org/10.1186/bcr3039
- Perroud, H.A., Rico, M.J., Alasino, C.M., Queralt, F., Mainetti, L.E., Pezzotto, S.M., Rozados, V.R. and Scharovsky, O.G. (2013) Safety and Therapeutic Effect of Metronomic Chemotherapy with Cyclophosphamide and Celecoxib in Advanced Breast Cancer Patients. Future Oncology, 9, 451-462. http://dx.doi.org/10.2217/fon.12.196
- Katkoori, V.R., Manne, K., Vital-Reyes, V.S., Rodríguez-Burford, C., Shanmugam, C., Sthanam, M., Manne, U., Chatla, C., Abdulkadir, S.A. and Grizzle, W.E. (2013) Selective COX-2 Inhibitor (Celecoxib) Decreases Cellular Growth in Prostate Cancer Cell Lines Independent of p53. Biotechnic & Histochemistry, 88, 38-46. http://dx.doi.org/10.3109/10520295.2012.724713
- James, N.D., Sydes, M.R., Mason, M.D., Clarke, N.W., Anderson, J., Dearnaley, D.P., et al. (2012) Celecoxib Plus Hormone Therapy versus Hormone Therapy Alone for Hormone-Sensitive Prostate Cancer: First Results from the STAMPEDE Multiarm, Multistage, Randomized Controlled Trial. The Lancet Oncology, 13, 549-558. http://dx.doi.org/10.1016/S1470-2045(12)70088-8
- Paulson, S.K., Hribar, J.D., Liu, N.W., Hajdu, E., Bible Jr., R.H., Piergies, A. and Karim, A. (2000) Metabolism and Excretion of [(14)C]Celecoxib in Healthy Male Volunteers. Drug Metab Dispos., 28, 308-314.
- Sandberg, M., Yasar, ü., Stromberg, P., Hoog, J.O. and Eliasson, E. (2002) Oxidation of Celecoxib by Polymorphic Cytochrome P450 2C9 and Alcohol Dehydrogenase. British Journal of Clinical Pharmacology, 54, 423-429. http://dx.doi.org/10.1046/j.1365-2125.2002.01660.x
- Rodrigues, A.D. (2005) Impact of CYP2C9 Genotype on Pharmacokinetics: Are All Cyclooxygenase Inhibitors the Same? Drug Metabolism and Disposition, 33, 1567-1575. http://dx.doi.org/10.1124/dmd.105.006452
- Vasquez, H.G. and Strobel, H. (1998) Identification of Cytochrome P450s in Human Glioma Cell Line. International Journal of Oncology, 12, 1291-1294.
- Denison, M.S. and Whitlock Jr., J.P. (1995) Xenobiotic-Inducible Transcription of Cytochrome P450 Genes. Journal of Biological Chemistry, 270, 18175-18178. http://dx.doi.org/10.1074/jbc.270.31.18175
- Guo, W., Reigan, P., Siegel, D. and Ross, D. (2008) Enzymatic Reduction and Glutathione Conjugation of Benzoquinone Ansamycin Heat Shock Protein 90 Inhibitors: Relevance for Toxicity and Mechanism of Action. Drug Metabolism and Disposition, 36, 2050-2057. http://dx.doi.org/10.1124/dmd.108.022004
- Nebert, D.W., Wikvall, K. and Miller, W.L. (2013) Human Cytochromes P450 in Health and Disease. Philosophical Transactions of the Royal Society B: Biological Sciences, 368, Article ID: 20120431. http://dx.doi.org/10.1098/rstb.2012.0431
- Guengerich, F.P. (1991) Molecular Advances for the Cytochrome P-450 Superfamily. Trends in Pharmacological Sciences, 12, 281-283. http://dx.doi.org/10.1016/0165-6147(91)90574-C
- Young, J.C., Barral, J.M. and Hartl, F.U. (2003) More than Binding: Localized Functions of Cytosolic Chaperones. Trends in Biochemical Sciences, 28, 541-547. http://dx.doi.org/10.1016/j.tibs.2003.08.009
- Merrill Jr., A.H., Nikolova-Karakashian, M., Schmelz, E.M., Morgan, E.T. and Stewart, J. (1999) Regulation of Cytochrome P450 Expression by Sphingolipids. Chemistry and Physics of Lipids, 102, 131-139. http://dx.doi.org/10.1016/S0009-3084(99)00081-X
- Lin, H.P., Kulp, S.K., Tseng, P.H., Yang, Y.T., Yang, C.C. and Chen, C.S. (2004) Growth Inhibitory Effects of Celecoxib in Human Umbilical Vein Endothelial Cells Are Mediated through G1 Arrest via Multiple Signaling Mechanisms. Molecular Cancer Therapeutics, 3, 1671-1680.
- Zhang, G.S., Liu, D.S., Dai, C.W. and Li, R.J. (2006) Antitumor Effects of Celecoxib on K562 Leukemia Cells Are Mediated by Cell-Cycle Arrest, Caspase-3 Activation, and Downregulation of Cox-2 Expression and Are Synergistic with Hydroxyurea or Imatinib. American Journal of Hematology, 81, 242-255. http://dx.doi.org/10.1002/ajh.20542
- Gr?sch, S., Maier, T.J., Schiffmann, S. and Geisslinger, G. (2006) Cyclooxygenase-2 (COX-2)-Independent Anticarcinogenic Effects of Selective COX-2 Inhibitors. Journal of the National Cancer Institute, 98, 736-747. http://dx.doi.org/10.1093/jnci/djj206�
- Kang, K.B., Zhu, C., Yong, S.K., Gao, Q. and Wong, M.C. (2009) Enhanced Sensitivity of Celecoxib in Human Glioblastoma Cells: Induction of DNA Damage Leading to p53-Dependent G1 Cell Cycle Arrest and Autophagy. Molecular Cancer, 8, 66. http://dx.doi.org/10.1186/1476-4598-8-66
- Lin, M.T., Lee, R.C., Yang, P.C., Ho, F.M. and Kuo, M.L. (2001) Cyclooxygenase-2 Inducing Mcl-1-Dependent Survival Mechanism in Human Lung Adenocarcinoma CL1.0 Cells. Involvement of Phosphatidylinositol 3-Kinase/Akt Pathway. The Journal of Biological Chemistry, 276, 8997-9002. http://dx.doi.org/10.1074/jbc.M107829200
- Ki, K., Gerelchuluun, A., Hong, Z., Sun, L., Zenkoh, J., Moritake, T. and Tsuboi, K. (2013) Celecoxib Enhances Radiosensitivity of Hypoxic Glioblastoma Cells through Endoplasmic Reticulum Stress. Neuro-Oncology, 15, 1186-1199.
- Balza, E., Castellani, P., Delfino, L., Truini, M. and Rubartelli, A. (2013) The Pharmacologic Inhibition of the xc-Antioxidant System Improves the Antitumor Efficacy of COX Inhibitors in the in Vivo Model of 3-MCA Tumorigenesis. Carcinogenesis, 34, 620-626. http://dx.doi.org/10.1093/carcin/bgs360
- Nabeyama, A., Kurita, A., Asano, K., Miyake, Y., Yasuda, T., Miura, I., Nishitai, G., Arakawa, S., Shimizu, S., Wakana, S., Yoshida, H. and Tanaka, M. (2010) xCT Deficiency Accelerates Chemically Induced Tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America, 107, 6436-6441. http://dx.doi.org/10.1073/pnas.0912827107
- Gout, P.W., Buckley, A.R., Simms, C.R. and Bruchovsky, N. (2001) Sulfasalazine, a Potent Suppressor of Lymphoma Growth by Inhibition of the xc-Cystine Transporter: A New Action for an Old Drug. Leukemia, 15, 1633-1640. http://dx.doi.org/10.1038/sj.leu.2402238
- de Groot, J. and Sontheimer, H. (2011) Glutamate and the Biology of Gliomas. Glia, 59, 1181-1189. http://dx.doi.org/10.1002/glia.21113
- Weyerbrock, A., Osterberg, N., Psarras, N., Baumer, B., Kogias, E., Werres, A., Bette, S., Saavedra, J.E., Keefer, L.K., and Papazoglou, A. (2012) JS-K, a Glutathione S-Transferase-Activated Nitric Oxide Donor with Antineoplastic Activity in Malignant Gliomas. Neurosurgery, 70, 497-510. http://dx.doi.org/10.1227/NEU.0b013e31823209cf
- Sontheimer, H. and Bridges, R.J. (2012) Sulfasalazine for Brain Cancer Fits. Expert Opinion on Investigational Drugs, 21, 575-578. http://dx.doi.org/10.1517/13543784.2012.670634
- Lyons, S.A., Chung, W.J., Weaver, A.K., Ogunrinu, T. and Sontheimer, H. (2007) Autocrine Glutamate Signaling Promotes Glioma Cell Invasion. Cancer Research, 67, 9463-9471. http://dx.doi.org/10.1158/0008-5472.CAN-07-2034
- Doxsee, D.W., Gout, P.W., Kurita, T., Lo, M., Buckley, A.R., Wang, Y., Xue, H., Karp, C.M., Cutz, J.C., Cunha, G.R., and Wang, Y.Z. (2007) Sulfasalazine-Induced Cystine Starvation: Potential Use for Prostate Cancer Therapy. The Prostate, 67, 162-171. http://dx.doi.org/10.1002/pros.20508
- Lang, W., Caldwell, G.W., Li, J., Leo, G.C., Jones, J.J. and Masucci, J.A. (2007) Biotransformation of Geldanamycin and 17-Allylamino-17-Demthoxygeldanamycin by Human Liver Microsomes: Reduction versus Oxidative Metabolism and Implications. Drug Metabolism and Disposition, 35, 21-29. http://dx.doi.org/10.1124/dmd.106.009639
- Turner, C.E. (2000) Paxillin Interactions. Journal of Cell Science, 113, 4139-4140.
- Gao, C.F., Xie, Q., Su, Y.L., Koeman, J., Khoo, S.K., Gustafson, M., Knudsen, B.S., Hay, R., Shinomiya, N. and Woude, G.F.V. (2005) Proliferation and Invasion: Plasticity in Tumor Cells. Proceedings of the National Academy of Sciences of the United States of America, 102, 10528-10533. http://dx.doi.org/10.1073/pnas.0504367102
- Kardosh, A., Golden, E.B., Pyrko, P., Uddin, J., Hofman, F.M., Chen, T.C., Louie, S.G., Petasis, N.A. and Sch?nthal, A.H. (2008) Aggravated Endoplasmic Reticulum Stress as a Basis for Enhanced Glioblastoma Cell Killing by Bortezomib in Combination with Celecoxib or Its Non-Coxib Analogue, 2,5-Dimethyl-celecoxib. Cancer Research, 68, 843-851. http://dx.doi.org/10.1158/0008-5472.CAN-07-5555�
- Teefy, A.M., Martin, J.E. and Kovacs, M.J. (2000) Warfarin Resistance Due to Sulfasalazine. Annals of Pharmacotherapy, 34, 1265-1268. http://dx.doi.org/10.1345/aph.10076
- Hall, S. and Ridone, J.P. (2001) A Case of Sulphasalazine Potentiating the Hypoprothombinemic Effect of Warfarin Resulting in Bleeding. Journal of Clinical Pharmacy and Therapeutics, 36, 246-248. http://dx.doi.org/10.1111/j.1365-2710.2010.01173.x