Journal of Cancer Therapy
Vol. 4 No. 6A1 (2013) , Article ID: 33925 , 13 pages DOI:10.4236/jct.2013.46A1002
Anti-Cancer Activities and Interaction of Imiquimod and Flex-Het, SHetA2, in Melanoma and Ovarian Cancer
![]()
1Department of Surgery, University of Oklahoma, Tulsa, USA; 2Department of Biostatistics and Epidemiology, University of Oklahoma Health Sciences Center, Oklahoma City, USA; 3Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, USA; 4Department of Obstetrics and Gynecology, University of Oklahoma Health Sciences Center, Oklahoma City, USA; 5Biochemistry and Molecular Biology, University of Oklahoma Health Sciences Center, Oklahoma City, USA.
Email: *doris-benbrook@ouhsc.edu
Copyright © 2013 Mark F. Naylor et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 23rd, 2013; revised June 20th, 2013; accepted June 27th, 2013
Keywords: Melanoma; Ovarian Cancer; Heteroarotinoids; Flex-Hets; SHetA2; Apoptosis
ABSTRACT
New strategies are needed to treat cancers that do not respond to chemotherapy or resist chemotherapy after responding initially. The objective of this study was to evaluate non-cytotoxic drugs against two of these cancers, melanoma and ovarian cancer. Imiquimod is an immune stimulant that induces apoptosis in cancer cells. Flexible-Heteroarotinoids (Flex-Hets) are small molecules that regulate growth, differentiation and apoptosis in cancer cells with reduced effects on normal cells. Both imiquimod and SHetA2 inhibited growth and induced apoptosis in the B16 melanoma cell line and cisplatin-sensitive A2780 and cisplatin-resistant OVCAR-3 ovarian cancer cell lines. The growth inhibition was additive in A2780 and B16, and synergistic in OVCAR-3. Both compounds inhibited endothelial tube branching in vitro and exerted an additive effect when combined. Various combinations of imiquimod and SHetA2 did not cause significant differences in the overall survival in the syngeneic B16 murine melanoma model. SHetA2 induced complete tumor regression and a melanoma-free natural life-span in two mice. These cures occurred in one of ten mice treated with oral SHetA2 and one of ten mice intratumorally-injected with SHetA2. Exploratory modeling of the distribution of survival times suggested that the two surviving mice represent rare events. Histologic evaluation of the tumors revealed that imiquimod induced necrosis, SHetA2 induced differentiated architecture and increased cytoplasm, both agents reduced mitotic indices and angiogenesis and neither agent counteracted the effects of the other. No overt toxicities were observed. In conclusion, imiquimod and SHetA2 exhibit complementary anti-cancer activity in vitro and SHetA2 has promise as a single agent.
1. Introduction
Immune response modifiers (IRMs) are immunotherapy agents that mimic, augment, or require participation of host immune cells for optimal effectiveness [1]. Efficacy of IRMs for cancer therapy requires that the tumor is capable of eliciting an immune response. Two of the most immunogenic tumor types are melanoma and ovarian cancer [2,3]. Malignant melanoma is the most lethal skin cancer [4]. The standard of care for melanoma is surgery followed by adjuvant therapy [5]. Because melanoma does not respond to cytotoxic chemotherapies, a number of different adjuvant therapies are being investigated including IRMs, however interferon (IFN) α2B is the only agent currently approved by the United States (US) Food and Drug Administration (FDA) for melanoma [5]. The most promising class of agents being evaluated in melanoma clinical trials consists of drugs that inhibit the BRAF kinase [6]. Current clinical trials are studying the combination IRMs with B-RAF inhibitors [7,8]. IRMs are also being investigated in ovarian cancer [3]. Because of the lack of early detection methods, ovarian cancer most frequently presents at an advanced stage and is one of the most lethal cancers [9]. New drugs with singleagent activity in recurrent ovarian cancer have been developed, however an international cooperative trial revealed that response rates or survival were not improved by combination of these drugs with cytotoxic chemotherapies [10].
Imiquimod is the first low molecular weight compound classified as an IRM that stimulates local release of cytokines and immunological responses against tumors. Topically applied imiquimod is currently approved by the US FDA for treatment of warts and non-melanoma skin cancer, and is an experimental agent for treatment of melanoma [11-17]. Topical imiquimod is also an effective treatment for infantile hemangiomas [18-20] and can inhibit vascular development in tumors [21]. In a case of melanoma metastases, treatment with imiquimod was demonstrated to alter transcription of several molecules involved in regulation of angiogenesis and metastasis [11]. In irradiated mice, imiquimod inhibited angiogenesis via an interleukin (IL)-18 dependent pathway [22]. However, topical administration cannot address solid tumors that would otherwise be good candidates for IRM therapy, including ovarian cancer. Oral administration of an IRM can provoke generalized cytokine release, which is poorly tolerated [23], and the response to intravenous (IV) administration has so far not proved efficacious [24]. Regional (as opposed to systemic) administration of these drugs is anticipated to enhance their ratio of therapeutic to toxic effects and to produce the strongest immunological responses against the cancer. Intraperitoneal (IP) administration is a rationale strategy to deliver imiquimod to ovarian tumors, because IP administration of cytotoxic chemotherapy to enhance regional exposure to ovarian tumors improved ovarian cancer patient survival by over 15 months in comparison to IV administration in a Phase III trial [25].
The effects of IRMs, interferons and interleukins have been shown to be enhanced by all-trans and 13-cis isomers of retinoic acid and synthetic analogs (retinoids). Topical retinoids are commonly combined with imiquimod cream for treatment of warts, molluscum, and actinic keratoses. The oral retinoid, acetretin, has been combined with topical imiquimod to treat difficult cases of skin cancer [26,27]. Retinoic acid isomers potentiate the effects of interferons in acute promyelocytic leukemia and metastatic melanoma [28,29]. In vitro studies suggest that tumor specific complementation of interferon by retinoic acid isomers is due to increased expression of HLA class II immune components in malignant glioblastoma cells [30], sensitivity to MHC class I-restricted killing in uveal melanoma cells [31], regulation of the Jak-STAT signaling pathway in promyelocytic leukemia cells [32], and enhancement of apoptosis in a variety of other cancer cell types [33]. Combining IRMs with drugs that synergize with them may limit their toxicity by permitting their use at doses that are less inflammatory and, therefore, less likely to produce local dose limiting side effects such as pain, fibrosis, and adhesions. A phase II study of IL-2 and 13-cis-retinoic acid as maintenance therapy in metastatic colorectal cancer demonstrated a low toxicity profile, clinical benefit and improved lymphocyte and natural killer cell count [34].
Retinoids are promising as cancer chemoprevention agents because of their abilities to regulate growth, differentiation, apoptosis, angiogenesis, metastasis, and immune function in a wide spectrum of cancers [35-40]. The 13-cis-retinoic acid (acetretin) is used to treat leukemia and neuroblastoma, but the clinical utility of retinoids is limited by toxicity and the development of resistance [40]. Both the efficacies and toxicities of retinoids are mediated through activation of nuclear retinoid receptors that act as transcription factors [41]. Early efforts to improve the therapeutic ratio of retinoids involved constraining the retinoic acid double-bonds, by inclusion of an aromatic ring of chemical structures called arotinoids. The first arotinoids developed were potent but considerably more toxic than retinoic acid [42-45]. To reduce the toxicity of arotinoids, we incorporated oxygen or sulfur heteroatoms in the cyclic ring of arotinoids. The resulting compounds, called Heteroarotinoids (Hets), exhibited similar biological activities to retinoic acid and arotinoids [42,47], but significantly less toxicity [42,46]. The clinical application of a Het called Tazarotene (produced by Allergan) for treatment of psoriasis, has confirmed the predicted improvement in therapeutic ratio for compounds with heteroatoms [48].
A significant advancement in the development of Hets occurred with the synthesis of Hets containing threeatom urea or thiourea linkers, which increased the flexibility of their conformations [49,50]. These flexible Hets (Flex-Hets) are potent inducers of apoptosis in vitro and in vivo, while not harming normal cells or tissues [50-56]. The lead Flex-Het, SHetA2, inhibited growth of ovarian cancer xenografts without producing evidence of in vivo toxicity, skin irritancy, or teratogenicity [51,57]. Because SHetA2 does not activate the retinoic acid receptors, it is not classified as a retinoid and does not induce toxicities associated with retinoids [49,51,55,57,58]. SHetA2 can be classified as a Retinoid-Related Molecule (RRM), which exert similar biological effects as retinoids, such as inhibition of cell growth and induction of differentiation, but also exert additional effects, such as apoptosis [59]. Retinoids and RRMs have demonstrated some activity in melanoma and ovarian cancer [5,60,61]. The efficacy and lack of toxicity prompted pre-clinical development of SHetA2 in the US National Cancer Institute (NCI) RAID and RAPID programs, which demonstrated lack toxicity of SHetA2 at doses 50 fold above the dose required to reduce xenograft tumor growth [62-65].
The objective of this study was to evaluate the anticancer activities of imiquimod and SHetA2 as single agents and in combination. Ovarian cancer and melanoma were studied because they are both immunogenic tumors that have the potential to be treated with regional therapy.
2. Materials and Methods
Cell Culture: A2780 (cisplatin-sensitive) and OVCAR-3 (cisplatin-resistant) ovarian cancer cell lines were cultured in RPMI media supplemented with 10% fetal bovine serum (FBS). The B16 mouse melanoma cell line was cultured in Dulbecco’s Modified Eagle’s medium (DMEM) and supplemented with 4% FBS, 5 µg/ml insulin, 5 µg/ml transferring, and 5 ng/ml sodium selenite. EAhy.926 cells, a gift of Cora-Jean S. Edgell, Ph.D., were formed by fusing human umbilical vein cells (HUVECs) with a human carcinoma cell line [66], and were cultured in DMEM with 10% FBS, and HUVECs in specialized Endothelial Growth Media (EGM-Clonetics).
Drugs: A liposomal preparation of imiquimod was prepared and stored and at 4˚C. Empty liposomes were used as controls because they do not affect ovarian cancer cell growth or endothelial vessel formation at concentrations used to deliver effective doses of imiquimod. SHetA2 (synthesized by K. Darrell Berlin, Oklahoma State University, Stillwater, OK, USA) was dissolved in dimethyl sulfoxide (DMSO), stored in aliquots at −80˚C, and manipulated under subdued lighting to avoid photooxidation. Final concentrations of DMSO in treated and control cultures were less than 0.1%, a dose that does not affect growth or differentiation.
Cytotoxicity Assay: Cells were seeded onto microtiter plates at concentrations of 1000 cells per well. The next day, the plates were treated in triplicate by the addition of various drug concentrations. Control cultures were treated with equal volumes of solvent or empty liposomes. Three days later, growth was assayed using the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2Htetrazolium inner salt) assay (Promega, Madison, WI, USA). Metabolically active cells reduced the MTS compound to an aqueous soluble formazan resulting in an optical density (OD) that can be measured at 490 nm. The growth index was derived for each treated culture by dividing its average OD by the average OD of untreated control cultures. These proportions were used to construct isobolograms, which suggest whether the drugs’ effects on cell survival are additive, synergistic, or antagonistic.
Apoptosis: The annexin-V-FITC assay was used for this study because it quantitatively differentiates between apoptosis and necrosis. After 24 or 48 hours of treatment with drug combinations or, in the control cultures, with solvent or liposomes, cells were trypsinized, resuspended in 1 ml of PBS, and incubated with Annexin-V-FITC. Flow cytometric analysis was performed at an excitation of 488 nm and an emission of 518 nm. Co-incubation with propidium idodide (PI) permitted a 4-channel evaluation to differentiate necrosis from apoptosis.
In Vitro Endothelial Tube Formation Assay: Endothelial tube formation assays were performed with slight modifications from published methods [67,68]. The EAhy.926 endothelial cells were plated onto Matrigelcoated wells and grown for 20 hours in the presence various concentrations of imiquimod and SHetA2 alone and in combination. The number of tube branches was counted under a microscope, and photomicrographs were made to record the cultures’ characteristics under various treatment conditions. The individual who counted vessel branches was blind as to the treatments, which were performed in triplicate. Differences in the mean number of vessel branches were evaluated using a single factor analysis of variance, and with assessments of preplanned contrasts.
Animal Model: A survival study of mice treated with these agents singly and in combination was carried out in a B16 melanoma model. B16 cells were trypsinized, rinsed in PBS and resuspended in DMEM. 50,000 cells were injected subcutaneously into the upper back of C57BL/6J black mice. After 7 days (before tumors were evident), the mice were arbitrarily divided into 6 treatment groups of 10 mice each. SHetA2 was dissolved in polyethylene glycol (PEG400) and imiquimod was dissolved in a liposome preparation. Treatments were administered on Mondays, Wednesdays, and Fridays for four weeks or until sacrifice. Half of the untreated control mice were injected with liposomes and the other half were gavaged with polyethylene glycol (PEG400). All 10 mice in each of the other five groups were treated by 1) gavage with 60 mg/kg/day SHetA2; 2) intratumoral injection with 9.25 mg/kg SHetA2; 3) intratumoral injection of imiquimod, 250 µg; 4) a combination of oral SHetA2 and intratumoral imiquimod; 5) intratumoral injection of both SHetA2 (9.25 mg/kg) and imiquimod (250 µg).
Mice were euthanized when their tumors reached 10% of their body weight, and their tumors were excised, fixed in formalin, and embedded in paraffin. Five-micron sections were stained with hematoxylin and eosin (H&E), Ki-67 (Biogenix, Freemont, CA, USA), a marker of proliferation and CD34 (Cell Marque, Rocklin, CA, USA), a marker of endothelial cells. The secondary antibody detection system used was multilink horseradish peroxidase DAB (Biogenix). An experienced pathologist (S.L.), unaware of treatment assignment, reviewed the histology and counted the number of Ki-67-positive cells per high powered field. The mean number of positive cells in ten sections was used to calculate “mitotic indices.” These were compared between the six groups in a single factor analysis of variance, followed by tests of multiple comparisons (Student-Newman-Keuls) that ensured an overall probability of Type I error (alpha) of less than 0.05. Kaplan Meier estimates of group-specific survival (time to euthanasia) were calculated and compared using a log rank test. The overall distribution of survival times was also modeled as a Weibull distribution to estimate probability of specific durations of survival.
3. Results
3.1. Growth Inhibition
Cytotoxicity assays demonstrated that imiquimod treatment caused dose-responsive inhibition of cisplatin resistant (OVCAR-3) and cisplatin-sensitive (A2780) ovarian cancer cell lines and the B16 melanoma cell line (Figure 1). Simultaneous treatment with SHetA2 enhanced the growth inhibition (Figure 1). Isobolograms revealed that the interaction between the two drugs was synergistic in the OVCAR-3 cell line and additive in the A2780 and B16 cell lines (Figure 1).
3.2. Apoptosis
Induction of apoptosis by imiquimod in ovarian cancer cell lines was demonstrated using the annexin-V assay. The exact amount of apoptosis could not be quantified because the liposomes in the imiquimod formulation interfered with counting of individual cells in the live quadrant only. However, the pattern of Annexin-V-positive cells in imiquimod treated cultures is different than in SHetA2-treated cultures and the combination appears to be at least additive (Figure 2).
3.3. Endothelial Tube Formation
The ability of SHetA2 and imiquimod to inhibit blood vessel formation was evaluated with the matrigel endothelial tube formation assay (Figure 3). Both agents exhibited a dose-responsive inhibition of endothelial tube branching. The effect on vessel branching of combining SHetA2 with the lower concentration (2 ug/ml) of imiquimod was additive. In statistical analysis, a preplanned contrast failed to reject the null hypothesis that the SHetA2 treatment combined with the lower imiquimod concentration in an additive matter (p = 0.1237). However, the effect of combining SHetA2 with the higher concentration (11 ug/ml) of imiquimod was more than additive (p < 0.0001).
3.4. B16 Syngeneic Mouse Melanoma Model
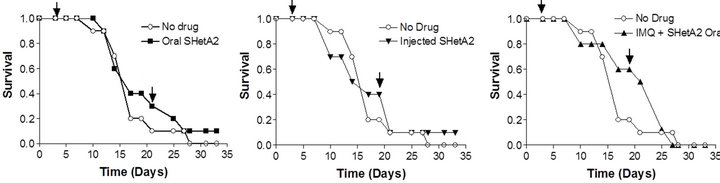
In vivo activity of SHetA2 and imiquimod was evaluated in the B16 synegenic mouse model (Figure 4). Animals were randomized into 6 different treatment groups: untreated control, oral SHetA2, SHetA2 injected into the tumor, imiquimod, combination of oral SHetA2 and imiquimod and combination of tumorally-injected SHetA2 and imiquimod. In all treatment groups, imiquimod was injected into the tumor. No overt toxicities were observed in any of the treatment groups. Log rank tests detected no difference among the Kaplan-Meier estimates of the groups’ survival distributions. The tumors could not be measured because they were highly necrotic and loose. Amazingly, one of the ten mice in each of the groups treated with SHetA2 survived to live a natural life-span.
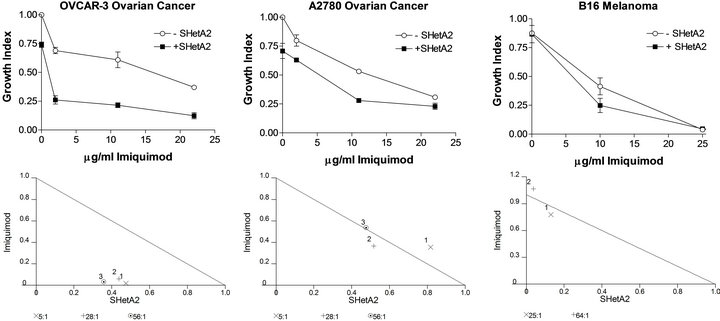
Figure 1. Growth inhibition by SHetA2 and imiquimod is synergistic in OVCAR3 and additive in A2780 and B16 cultures. Cultures were incubated for 72 hours at various concentrations of SHetA2 (in μM) in dimethyl sulfoxide (DMSO) or IMQ (in μM/ml) in liposomes. MTS dye was added for the last 4 hrs of treatment and then solubilized for OD reading. The growth index was determined by dividing the OD of treated cells by the OD of cells treated with DMSO and empty liposomes. Isobolograms (lower panels) plot the effective dose (ED) needed to reduce survival by 50% (ED50) for imiquimod versus SHetA2. The drugs’ combined effect is additive if the combination data point falls on the diagonal line, synergistic if the point falls on the lower left, and antagonistic if the point falls on the upper right.
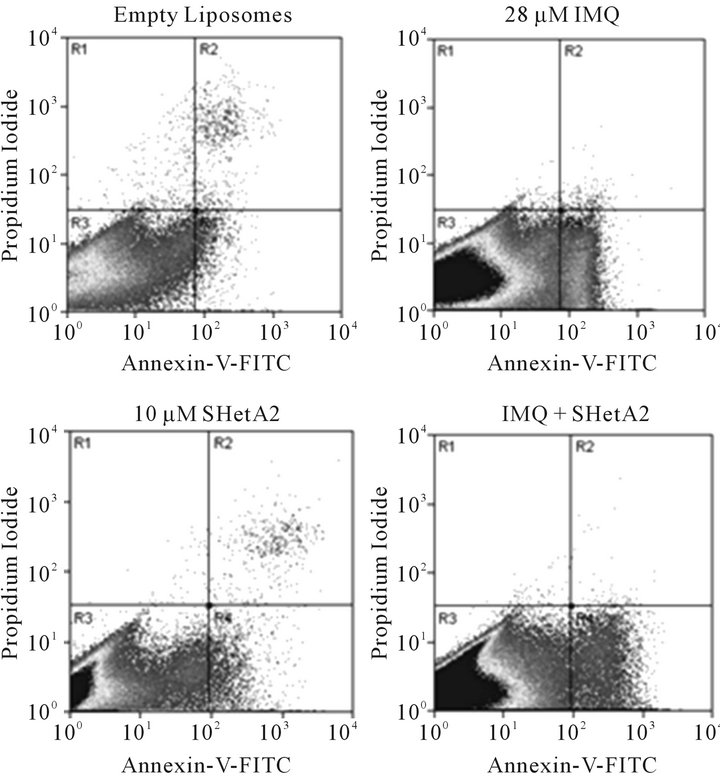
Figure 2. Induction of apoptosis in A2780 ovarian cancer cells by imiquimod and SHetA2. A2780 cultures were incubated in the presence of empty liposomes, 11 μg/ml imiquimod (IMQ), 10 μM SHetA2, or the same concentrations of drugs combined. After 30 hours of treatment, the cells were trypsinized, pelleted, and resuspended in Annexin-V-FITC propidium iodide (PI) and analyzed by dual channel flow cytometric analysis. The x-axis represents Annexin-V-FITC staining of apoptosis and the Y axis represents propidium iodide staining for dead. The lower left quadrant (R3) represents live cells, the lower right quadrant (R4) represents apoptosing cells and the upper right quadrant represents necrosis and late apoptosis.
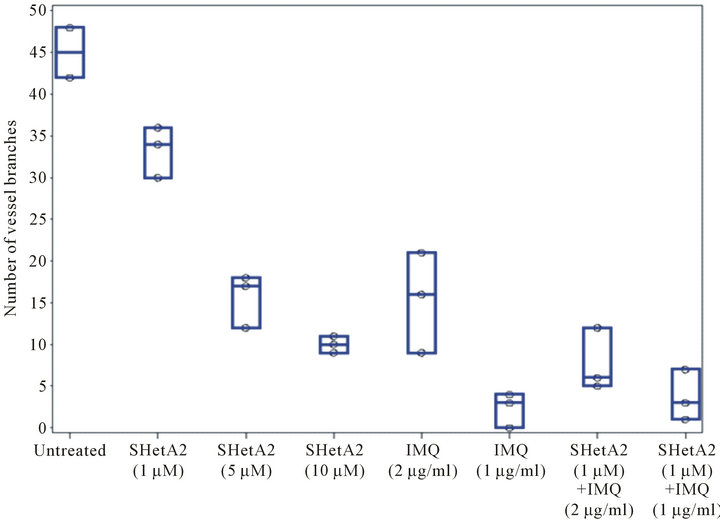
Figure 3. Inhibition of endothelial tube formation. EAhy.926 endothelial cells were plated in triplicate on matrigel in various concentration of SHerA2 or IMQ or combinations of the two drugs. Twenty hours later an investigator, who was blind to treatment administration, took representative photomicrographs (A) and counted vessel branches in each well (B). This experiment was repeated 4 times with similar results.
 (a)
(a)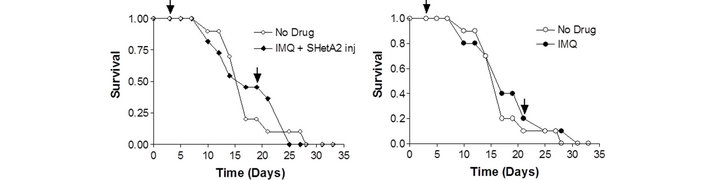 (b)
(b)
Figure 4. Survival of mice bearing B16 syngeneic melanoma xenografts. C57 BJ/6 immunocompetent mice were injected with B16 melanoma cells. Mice were divided into groups of 10 animals for each treatment group. Half of the untreated controls were gavaged with PEG400 solvent and the other half were injected with solvent. Survival of the untreated control was compared with the groups receiving 60 mg/kg oral SHerA2 (a); 9.25 mg/kg intratumoral injected SHetA2 (b); imiquimond (IMQ) and oral SHetA2 (c); intratumorally-injected SHetA2 and IMQ (d); and IMQ (e). In all groups, IMQ was injected intratumorally. The arrows indicate the initiation and termination of treatments.
One lived 3 years and one month after tumor injection and the other lived beyond that time. Autopsies of the deceased mice by an expert veterinary pathologist found no evidence of melanoma or other diseases. These two mice had tumors that regressed with treatment in spite of their aggressive growth.
Exploratory modeling of the distribution of survival times suggested that the two surviving mice (one in the oral SHetA2 treatment group and one in the intratumoral SHetA2 injected group) represent relative rarities. The probability that an individual would survive 36 days in a population of mice modeled from this experiment is 0.023. We arrived at this conclusion by using Kolmogorov-Smirnov tests to check the similarity of distributions of survival times among six groups of mice that were treated but not cured, then combining the groups’ survival times and modeling them as a Weibull distribution (illustrated in the superimposed curve in Figure 5) with specific parameters (threshold = 12.45; scale = 14.66; shape = 2.8). These parameters were used to calculate the probability of specific durations of survival.
3.5. B16 Xenograft Tumor Histology
Tumor sections were stained with H&E to evaluate the histology in the six different treatment groups (Figure 6). In the untreated control group, the tumors were amorphous, necrotic and discohesive (tended to fall apart when handled) and therefore could not be accurately measured or weighed. The tumors sampled from mice who received either oral or injected SHetA2 were similar, and exhibited more differentiated architecture with cells arranged into large nests. The most impressive feature was that the cells were reasonably uniform with more cytoplasm. In the tumors from animals treated with imiquimod, massive necrosis was seen in most areas of the tumor. In tumors from the animals treated with both SHetA2 and the immune stimulant, massive necrosis was also frequent. Also noted in the tumors of animals treated with both agents was a greater tendency of the tumor cells to form nests and a loss of pleomorphism compared with tumors in untreated controls. CD34 staining of endothelial cells revealed a decrease in microvessel density in all treatment groups (Data not shown). Detailed evaluation of angiogenesis in the tumors could not be performed due to the extensive necrosis. The tumors in each group of treated mice demonstrated lower mitotic indices (measured by Ki-67 staining) than those in untreated mice (Figure 7). Among the treatment groups, the mitotic indices of tumors from mice that received imiquimod alone
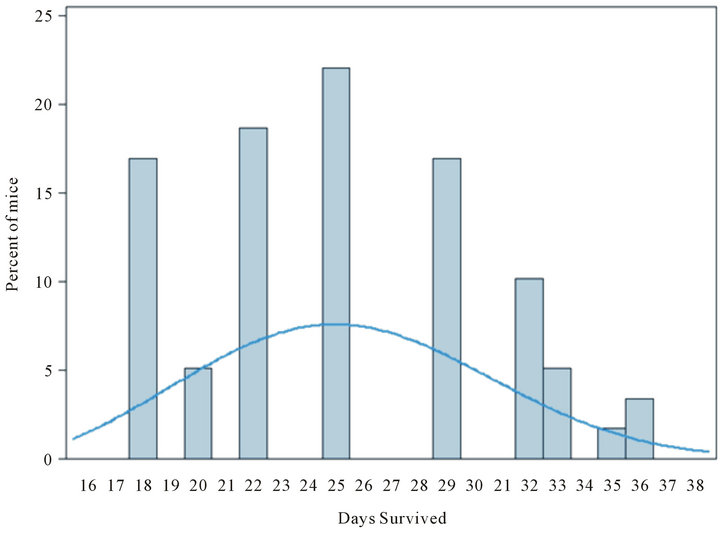
Figure 5. Exploratory modeling of distribution of survival times for treatment groups receiving oral SHetA2 or intratumoral injection of SHetA2. On the basis of a Weibull distribution (illustrated by superimposed smooth curve) with hypothetical parameters (threshold = 12.45; scale = 14.66; shape = 2.8), the probability that an individual mouse would survive 36 days is 0.023.
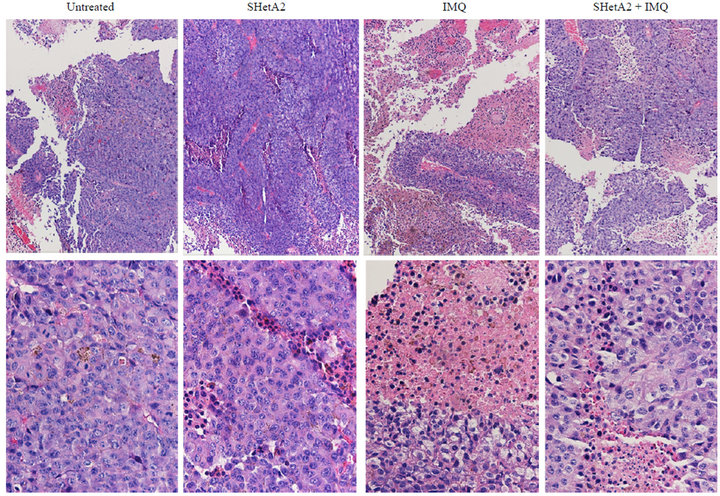
Figure 6. Representative Hi&E statined sections of B16 xenograft tumors. The upper panel is 10× and the lower panel is 40×.
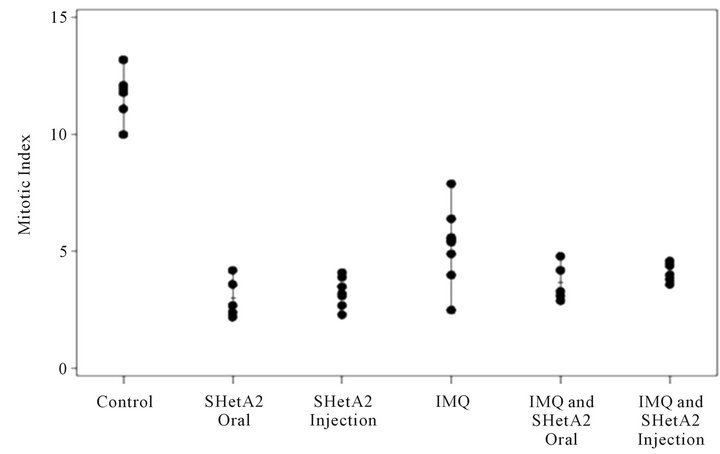
Figure 7. Inhibition of mitotic indices. After animal euthanasia, tumors were removed, fixed in formalin and embedded in paraffin. Tumor sections were stained with Ki67 and the mean number of K67-positive cells per 10 high powered fields were counted and recorded for each tumor.
did not significantly differ from that of the combined imiquimod and SHetA2 injection, but exhibited a larger range than that of the other three treatment groups.
6. Discussion
The results of this study demonstrate the efficacy of imiquimod and SHetA2 alone and in combination against ovarian cancer and melanoma cells. The interaction of these two agents for inhibition of cancer cell growth was either additive or synergistic, depending on the cell line. Both agents were also active in inhibiting endothelial tube formation as single agents and exerted an additive effect when used in combination. The different mechanisms of action of imiquimod and SHetA2 are likely responsible for their interaction. Imiquimod induces both apoptosis and necrosis in association with development of autophagic vesicles and induction of toll like receptors [69], while SHetA2 induces apoptosis through direct effects on the mitochondria in association with G1 cell cycle arrest and development of autophagic vesicles and ER stress [53,54,70].
Angiogenesis is an important target for drug development because the development of blood vessels is required to support tumor growth and metastasis [71]. Inhibition of angiogenesis can prevent progression of premalignant lesions and is considered a rationale target for secondary chemoprevention [72]. The ability of imiquimod and SHetA2 to inhibit endothelial tube formation increases the potential of these compounds to be effective against cancer through their alterations of the tumor microenvironment. The mechanism of SHetA2 inhibition of angiogenesis involves regulation of both cancer and endothelial cells [73]. The direct effects on cancer cells involve inhibition of angiogenic cytokine secretion, while the direct effects on endothelial cells involve inhibition of cell cycle progression.
Post-surgical platinum-based chemotherapy is the standard of care for ovarian cancer and cancer recurrences are treated with platinum-based therapy until the patient’s cancer becomes platininum resistant [74]. The activities of imiquimod and SHetA2 against both cisplatin-sensitive and -resistant ovarian cancer cell lines support the use of these agents in both first and second line treatment. There is also significant toxicity associated with platinum-based chemotherapy [75]. Thus, low-toxicity compounds, such as imiquimod and SHetA2 might provide an alternative or supplement to the highly toxic practice of repeated platinum-based therapies for ovarian cancer primary therapy and recurrence. Oral administration without toxicity is the optimal situation for a chemoprevention agent. SHetA2 is oral bioavailability and did not cause evidence of toxicity in a 28-day dog toxicity study resulting in a no observed adverse effect level (NOAEL) at a dose (2000 mg/kg/day) 33 fold above the dose used in this study (60 mg/kg/day), and caused a lowest observed adverse effect level (LOAEL) in a 28-day rat toxicity study at a dose (1500 mg/kg/day) 25 fold above that used in this study [63].
The in vivo anti-cancer efficacy of SHetA2 is indicated by complete tumor regression and long-term melanomafree survival of two mice in the syngeneic B16 melanoma model. This cure of one out of ten mice in each of the SHetA2-treated groups in the B16 syngeneic animal model is a rare event. Although statistical analysis found no significant differences in overall survival between the various groups, probability analysis suggests that the complete regression of the tumors and the long-term survival of the two animals is significant. The cure of one mouse in each of the two SHetA2 treated groups regardless of the route of administration, oral or direct intratumoral injection, strengthens the significance of this finding. If this result translated to humans, cure of one out of ten melanoma patients would be a major breakthrough in the treatment of melanoma. The lack of effect on overall survival by single agent imiquimod or SHetA2 alone or in combination may be due to the highly aggressive nature of the syngeneic B16 melanoma model. The promising results of two out of twenty cures, justify studying SHetA2 in additional animal models including ovarian cancer. Previous studies demonstrated single agent activity of SHetA2 against aggressively growing OVCAR- 3 xenograft tumors [51].
In conclusion, imiquimod and SHetA2 demonstrate in vitro activity against cancers that cannot easily be treated with conventional chemotherapeutic agents. Their mechanisms and toxicities are different from traditional agents, indicating that they may be effective as primary therapy and as second line approaches to overcome resistance to traditional chemotherapy. The cure of melanoma in two out of twenty SHetA2-treated mice regardless of the route of administration, and without toxicity, justifies further study of SHetA2 for treatment of melanoma.
7. Acknowledgements
We thank the following individuals for working on the animal model: Sara E. Cook (University of Oklahoma Health Sciences Center, Department of Obstetrics and Gynecology), Taimur L. Chaudhry, (University of Oklahoma Health Sciences Center, Department of Obstetrics and Gynecology), Shylet Chengedza, (University of Oklahoma Health Sciences Center, Department of Biochemistry and Molecular Biology), Daynelle M. Dedmond (University of Oklahoma Health Sciences Center, Department of Obstetrics and Gynecology), Lance W. Gill (Southwestern Oklahoma State University Department of Chemistry), Margaret P. O’Connell, (University of Oklahoma Health Sciences Center, College of Medicine), Jordan D. Harrison (Oklahoma Medical Research Foundation Department of Arthritis and Immunology), Thuan H. Nguyen (Oklahoma Medical Research Foundation Department of Arthritis and Immunology), Chasity J. Jackson (Langston University, Department of Biology), Anna C. Nelson (Southwestern Oklahoma State University Department of Chemistry), Bethany N. Hannafon (University of Oklahoma Health Sciences Center, Department of Obstetrics and Gynecology). Sara Cook also performed the cytotoxicity assays and Bethany Hannafon performed the apoptosis assays.
REFERENCES
- M. A. Cheever, J. Schlom, L. M. Weiner, H. K. Lyerly, M. L. Disis, A. Greenwood, O. Grad, W. G. Nelson and Translational Research Working, “Translational Research Working Group Developmental Pathway for Immune Response Modifiers,” Clinical Cancer Research, Vol. 14, No. 18, 2008, pp. 25692-25699. doi:10.1158/1078-0432.CCR-08-1266
- S. A. Rosenberg and M. E. Dudley, “Adoptive Cell Therapy for the Treatment of Patients with Metastatic Melanoma,” Current Opinion in Immunology, Vol. 21, No. 2, 2009, pp. 233-240. doi:10.1016/j.coi.2009.03.002
- L. E. Kandalaft, D. J. Powell, N. Singh and G. Coukos, “Immunotherapy for Ovarian Cancer: What’s Next?” Journal of Clinical Oncology, Vol. 29, No. 7, 2011, pp. 925- 933. doi:10.1200/JCO.2009.27.2369
- M. B. Lens and M. Dawes, “Global Perspectives of Contemporary Epidemiological Trends of Cutaneous Malignant Melanoma,” British Journal of Dermatology, Vol. 150, No. 2, 2004, pp. 179-185. doi:10.1111/j.1365-2133.2004.05708.x
- M. S. Evans, S. V. Madhunapantula, G. P. Robertson and J. J. Drabick, “Current and Future Trials of Targeted Therapies in Cutaneous Melanoma,” Advances in Experimental Medicine & Biology, Vol. 779, 2013, pp. 223-255. doi:10.1007/978-1-4614-6176-0_10
- G. Bollag, P. Hirth, J. Tsai, J. Zhang, P. N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhang, G. Habets, E. A. Burton, B. Wong, G. Tsang, B. L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K. Y. J. Zhang, D. R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. D/’Andrea, A. Koehler, M. Stumm, P. S. Lin, R. J. Lee, J. Grippo, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. B. Chapman, K. T. Flaherty, X. Xu, K. L. Nathanson and K. Nolop, “Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAF-Mutant Melanoma,” Nature, Vol. 467, No. 7315, 2010, pp. 596- 599. doi:10.1038/nature09454
- S. L. Topalian, F. S. Hodi, J. R. Brahmer, S. N. Gettinger, D. C. Smith, D. F. McDermott, J. D. Powderly, R. D. Carvajal, J. A. Sosman, M. B. Atkins, P. D. Leming, D. R. Spigel, S. J. Antonia, L. Horn, C. G. Drake, D. M. Pardoll, L. Chen, W. H. Sharfman, R. A. Anders, J. M. Taube, T. L. McMiller, H. Xu, A. J. Korman, M. Jure-Kunkel, S. Agrawal, D. McDonald, G. D. Kollia, A. Gupta, J. M. Wigginton and M. Sznol, “Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer,” New England Journal of Medicine, Vol. 366, No. 26, 2012, pp. 2443- 2454. doi:10.1056/NEJMoa1200690
- F. S. Hodi, S. J. O’Day, D. F. McDermott, R. W. Weber, J. A. Sosman, J. B. Haanen, R. Gonzalez, C. Robert, D. Schadendorf, J. C. Hassel, W. Akerley, A. J. M. van den Eertwegh, J. Lutzky, P. Lorigan, J. M. Vaubel, G. P. Linette, D. Hogg, C. H. Ottensmeier, C. Lebbé, C. Peschel, I. Quirt, J. I. Clark, J. D. Wolchok, J. S. Weber, J. Tian, M. J. Yellin, G. M. Nichol, A. Hoos and W. J. Urba, “Improved Survival with Ipilimumab in Patients with Metastatic Melanoma,” New England Journal of Medicine, Vol. 363, No. 8, 2010, pp. 711-723. doi:10.1056/NEJMoa1003466
- I. Romero and R. C. Bast Jr., “Minireview: Human Ovarian Cancer: Biology, Current Management, and Paths to Personalizing Therapy,” Endocrinology, Vol. 153, No. 4, 2012, pp. 1593-1602. doi:10.1210/en.2011-2123
- M. A. Bookman, M. F. Brady, W. P. McGuire, P. G. Harper, D. S. Alberts, M. Friedlander, N. Colombo, J. M. Fowler, P. A. Argenta, K. DeGeest, D. G. Mutch, R. A. Burger, A. M. Swart, E. L. Trimble, C. Accario-Winslow and L. M. Roth, “Evaluation of New Platinum-Based Treatment Regimens in Advanced-Stage Ovarian Cancer: A Phase III Trial of the Gynecologic Cancer Intergroup,” Journal of Clinical Oncology, Vol. 27, No. 9, 2009, pp. 1355-1358. doi:10.1200/JCO.2008.19.1684
- C. Hesling, M. D’Incan, S. Mansard, F. Franck, A. Corbin-Duval, C. Chevenet, P. Dechelotte, J. C. Madelmont, A. Veyre, P. Souteyrand and Y. J. Bignon, “In Vivo and in Situ Modulation of the Expression of Genes Involved in Metastasis and Angiogenesis in a Patient Treated with Topical Imiquimod for Melanoma Skin Metastases,” British Journal of Dermatology, Vol. 150, No. 4, 2004, pp. 761-767. doi:10.1111/j.0007-0963.2004.05898.x
- A. Steinmann, J. O. Funk, G. Schuler and P. von den Driesch, “Topical Imiquimod Treatment of a Cutaneous Melanoma Metastasis,” Journal of the American Academy of Dermatology, Vol. 43, No. 3, 2000, pp. 555-556. doi:10.1046/j.1365-2133.2002.488811.x
- I. H. Wolf, J. Smolle, B. Binder, L. Cerroni, E. Richtig and H. Kerl, “Topical Imiquimod in the Treatment of Metastatic Melanoma to Skin,” Archives in Dermatology, Vol. 139, No. 3, 2003, pp. 273-276. doi:10.1001/archderm.139.3.273
- A. B. Bong, B. Bonnekoh, I. Franke, M. P. Schön, J. Ulrich and H. Gollnick, “Imiquimod, a Topical Immune Response Modifier, in the Treatment of Cutaneous Metastases of Malignant Melanoma,” Dermatology, Vol. 205, No. 2, 2002, pp. 135-138. doi:10.1159/000063904
- S. Ugurel, A. Wagner, C. Pfohler, W. Tilgen and U. Reinhold, “Topical Imiquimod Eradicates Skin Metastases of Malignant Melanoma But Fails to Prevent Rapid Lymphogenous Metastatic Spread,” British Journal of Dermatology, Vol. 147, No. 3, 2002, pp. 621-624. doi:10.1046/j.1365-2133.2002.488811.x
- P. Vereecken, A. Mathieu, M. Laporte, M. Petein, T. Velu, A. Awada and M. Heenen, “Management of Cutaneous Locoregional Recurrences of Melanoma: A New Therapeutic Perspective with Imiquimod,” Dermatology, Vol. 206, No. 3, 2003, pp. 279-280. doi:10.1159/000068901
- C. Loquai, D. Nashan, D. Metze, U. Beiteke, K. W. Ruping, T. A. Luger and S. Grabbe, “Imiquimod, Pegylated Interferon-Alpha-2b and Interleukin-2 in the Treatment of Cutaneous Melanoma Metastases,” Hautarzt, Vol. 55, No. 2, 2004, pp. 176-181. doi:10.1007/s00105-003-0625-z
- B. Berman, V. N. Poochareon and A. M. Villa, “Novel Dermatologic Uses of the Immune Response Modifier Imiquimod 5% Cream,” Skin Therapy Letters, Vol. 7, No. 9, 2002, pp. 1-6. http://www.skintherapyletter.com/2002/7.9/1.html
- R. Sidbury, “What’s New in Pediatric Dermatology: Update for the Pediatrician,” Current Opinion in Pediatrics, Vol. 16, No. 4, 2004, pp. 410-414. doi:10.1097/01.mop.0000133081.24760.08
- M. I. Martinez, I. Sanchez-Carpintero, P. E. North and M. C. Mihm Jr., “Infantile Hemangioma: Clinical Resolution with 5% Imiquimod Cream,” Archives in Dermatology, Vol. 138, No. 7, 2002, pp. 881-884. doi:10.1001/archderm.138.7.881
- R. Sidbury, N. Neuschler, E. Neuschler, P. Sun, X. Q. Wang, R. Miller, M. Tomai, E. Puscasiu, S. Gugneja and A. S. Paller, “Topically Applied Imiquimod Inhibits Vascular Tumor Growth in Vivo,” Journal of Investigative Dermatology, Vol. 121, No. 5, 2003, pp. 1205-1209. doi:10.1046/j.1523-1747.2003.12521.x
- S. Majewski, M. Marczak, B. Mlynarczyk, B. Benninghoff and S. Jablonska, “Imiquimod Is a Strong Inhibitor of Tumor Cell-Induced Angiogenesis,” International Journal of Dermatology, Vol. 44, No. 1, 2005, pp. 14-19. doi:10.1111/j.1365-4632.2004.02318.x
- P. Savage, V. Horton, J. Moore, M. Owens, P. Witt and M. E. Gore, “A Phase I Clinical Trial of Imiquimod, an Oral Interferon Inducer, Administered Daily,” British Journal of Cancer, Vol. 74, No. 9, 1996, pp. 1482-1486. doi:10.1038/bjc.1996.569
- M. Naylor, “Verbal Communication with 3M Pharmaceuticals.”
- D. K. Armstrong, B. Bundy, L. Wenzel, H. Q. Huang, R. Baergen, S. Lele, L. J. Copeland, J. L. Walker, R. A. Burger and Gynecologic Oncology Group, “Intraperitoneal Cisplatin and Paclitaxel in Ovarian Cancer,” New England Journal of Medicine, Vol. 354, No. 1, 2006, pp. 34- 43. doi:10.1056/NEJMoa052985
- C. Ingves and G. B. Jemec, “Combined Imiquimod and Acitretin for Non-Surgical Treatment of Basal Cell Carcinoma,” Scandinavian Journal of Plastic Reconstructive Surgery and Hand Surgery, Vol. 37, No. 5, 2003, pp. 293-295. doi:10.1080/02844310310001724
- B. Giannotti, L. Vanzi, E. M. Difonzo and N. Pimpinelli, “The Treatment of Basal Cell Carcinomas in a Patient with Xeroderma Pigmentosum with a Combination of Imiquimod 5% Cream and Oral Acitretin,” Clinical and Experimental Dermatology, Vol. 28, No. 1, 2003, pp. 33-35. doi:10.1046/j.1365-2230.28.s1.11.x
- M. Gaboli, D. Gandini, L. Delva, Z. G. Wang and P. P. Pandolfi, “Acute Promyelocytic Leukemia as a Model for Cross-Talk between Interferon and Retinoic Acid Pathways: From Molecular Biology to Clinical Applications,” Leukemia & Lymphoma, Vol. 30, No. 1-2, 1998, pp. 11- 22.
- G. Fierlbeck, T. Schreiner and G. Rassner, “Combination of Highly Purified Human Leukocyte Interferon and 13- cis-Retinoic Acid for the Treatment of Metastatic Melanoma,” Cancer Immunology and Immunotherapy, Vol. 40, No. 3, 1995, pp. 157-164. doi:10.1007/BF01517347
- A. Haque, A. Das, L. M. Hajiaghamohseni, A. Younger, N. L. Banik and S. K. Ray, “Induction of Apoptosis and Immune Response by All-Trans Retinoic Acid Plus Interferon-Gamma in Human Malignant Glioblastoma T98G and U87MG Cells,” Cancer Immunology and Immunotherapy, Vol. 56, No. 5, 2007, pp. 615-625. doi:10.1007/s00262-006-0219-6
- S. Vertuani, E. Dubrovska, V. Levitsky, M. J. Jager, R. Kiessling and J. Levitskaya, “Retinoic Acid Elicits Cytostatic, Cytotoxic and Immunodulatory Effects in Uveal Melanoma Cells,” Cancer Immunology and Immunotherapy, Vol. 56, No. 2, 2007, pp. 193-204. doi:10.1007/s00262-006-0185-z
- E. Garattini, L. Mologni, I. Ponzanelli and M. Terao, “Cross- Talk between Retinoic Acid and Interferons: Molecular Mechanisms of Interaction in Acute Promyelocytic Leukemia Cells,” Leukemia & Lymphoma, Vol. 30, No. 5-6, 1998, pp. 467-475.
- D. V. Kalvakolanu, “The GRIMs: A New Interface between Cell Death Regulation and Interferon/Retinoid Induced Growth Suppression,” Cytokine & Growth Factor Reviews, Vol. 15, No. 2, 2004, pp. 169-194. doi:10.1016/j.cytogfr.2004.01.002
- F. Recchia, G. Saggio, A. Cesta, G. Candeloro, A. Di Blasio, G. Amiconi, M. Lombardo, A. Nuzzo, A. Lalli, E. Alesse, S. Nceozione and S. Rea, “Phase II Study of Interleukin-2 and 13-cis-Retinoic Acid as Mainenance Therapy in Metastatic Colorectal Cancer,” Cancer Immunology and Immunotherarpy, Vol. 56, No. 5, 2006, pp. 699- 708.
- D. S. Alberts, O. M. Colvin, A. H. Conney, V. L. Ernster, J. E. Garber, P. Greenwald, L. Gudas, K. W. Hong, G. J. Kelloff, R. A. Kramer, C. E. Lerman, D. J. Mangelsdorf, A. Matter, J. D. Minna, W. G. Nelson, J. M. Pezzuto, F. Prendergast, V. W. Rusch, M. B. Sporn, L. W. Wattenberg and I. B. Weinstein, “Prevention of Cancer in the Next Millennium: Report of the Chemoprevention Working Group to the American Association for Cancer Research,” Cancer Research, Vol. 59, No. 19, 1999, pp. 4743-4748. http://cancerres.aacrjournals.org/content/59/19/4743.long
- F. R. Khuri, S. M. Lippman, M. R. Spitz, R. Lotan and W. K. Hong, “Molecular Epidemiology and Retinoid Chemoprevention of Head and Neck Cancer,” Journal of the National Cancer Institute, Vol. 89, No. 3, 1997, pp. 199- 211. doi:10.1093/jnci/89.3.199
- G. de Palo, L. Mariani, T. Camerini, E. Marubini, F. Formelli, B. Pasini, A. Decensi and U. Veronesi, “Effect of Fenretinide on Ovarian Carcinoma Occurrence,” Gynecologic Oncology, Vol. 86, No. 1, 2002, pp. 24-27. doi:10.1006/gyno.2002.6663
- R. D. Alvarez, M. G. Conner, H. Weiss, P. M. Klug, S. Niwas, U. Manne, J. Bacus, V. Kagan, K. C. Sexton, C. J. Grubbs, I. E. Eltoum and W. E. Grizzle, “The Efficacy of 9-Cis-Retinoic Acid (Aliretinoin) as a Chemopreventive Agent for Cervical Dysplasia: Results of a Randomized Double-Blind Clinical Trial,” Cancer Epidemiology, Biomarkers and Prevention, Vol. 12, No. 2, 2003, pp. 114- 119. http://cebp.aacrjournals.org/content/12/2/114.full.pdf+html
- M. Follen, E. N. Atkinson, D. Schottenfeld, A. Malpica, L. West, S. Lippman, C. Zou, W. N. Hittelman, R. Lotan and W. K. Hong, “A Randomized Clinical Trial of 4-Hydroxyphenylretinamide for High-Grade Squamous Intraepithelial Lesions of the Cervix,” Clinical Cancer Research, Vol. 7, No. 11, 2001, pp. 3356-3365. http://clincancerres.aacrjournals.org/content/7/11/3356.long
- R. Masetti, C. Biagi, D. Zama, F. Vendemini, A. Martoni, W. Morello, P. Gasperini and A. Pession, “Retinoids in Pediatric Onco-Hematology: The Model of Acute Promyelocytic Leukemia and Neuroblastoma,” Advances in Therapy, Vol. 29, No. 9, 2012, pp. 747-762. doi:10.1007/s12325-012-0047-3
- V. Duong and C. Rochette-Egly, “The Molecular Physiology of Nuclear Retinoic acid Receptors. From Health to Disease,” Biochimica et Biophysica Acta, Vol. 1812, No. 8, 2011, pp. 1023-1031. doi:10.1016/j.bbadis.2010.10.007
- D. M. Benbrook, “Refining Retinoids with Heteroatoms,” Minireviews in Medicinal Chemistry, Vol. 2, No. 3, 2002, pp. 271-277. doi:10.2174/1389557023406160
- M. I. Dawson, P. D. Hobbs, K. Derdzinski, R. L.-S. Chan, J. Gruber, W.-R. Chao, S. Smith, R. W. Thies and L. J. Schiff, “Conformationally Restricted Retinoids,” Journal of Medicinal Chemistry, Vol. 27, No. 11, 1984, pp. 1516- 1531. doi:10.1021/jm00377a022
- C. Lindamood III, F. O. Cope, D. L. Dillehay, M. P. Everson, H. D. Giles, E. W. Lamon, D. J. McCarthy, J. L. Sartin and D. L. Hill, “Pharmacological and Toxicological Properties of Arotinoids SMR-2 and SMR-6 in Mice,” Fundamental and Applied Toxicology, Vol. 14, No. 1, 1990, pp. 15-29. doi:10.1093/toxsci/14.1.15
- C. I. Lindamood, D. L. Dillehay, E. W. Lamon, H. D. Giles, Y. F. Shealy, B. P. Sani and D. L. Hill, “Toxicologic and Immunologic Evaluations of N-(All-trans-Retinoyl)-DL-Leucine and N-(all-trans-Retinoyl)glycine,” Toxicolog and Applied Pharamcolology, Vol. 96, No. 2, 1988, pp. 279-295. doi:10.1016/0041-008X(88)90087-7
- D. M. Benbrook, M. M. Madler, L. W. Spruce, P. J. Birckbichler, E. C. Nelson, S. Subramanian, G. M. Weerasekare, J. B. Gale, M. K. Patterson Jr., B. Wang, W. Wang, S. Lu, T. C. Rowland, P. DiSilvestro, C. Lindamood III, D. L. Hill and K. D. Berlin, “Biologically Active Heteroarotinoids Exhibit Anticancer Activity and Decreased Toxicity,” Journal of Medicinal Chemistry, Vol. 40, No. 22, 1997, pp. 3567-3583. doi:10.1021/jm970196m
- K. M. Waugh, K. D. Berlin, W. T. Ford, E. M. Holt, J. P. Carroll, P. R. Schomber and L. J. Schiff, “Synthesis and Characterization of Selected Heteroarotinoids. Pharmacological Activity as Assessed in Vitamin a Deficient Hamster Tracheal Organ Cultures. Single Crystal X-Ray Diffraction Analysis of 4,4-Dimethylthiochroman-6-yl methyl ketone 1,1-Dioxide and Ethyl (E)-p-[2-(4,4-Dimethylthiochroman-6-yl)Benzoate,” Journal of Medicinal Chemistry, Vol. 28, No. 1, 1985, pp. 116-124. doi:10.1021/jm00379a021
- R. A. S. Chandraranta, “Tazarotene-First of a New Generation of Receptor-Selective Retinoids,” British Journal of Dermatology, Vol. 135, No. S49, 1996, pp. 18-25.
- S. Guruswamy, S. Lightfoot, M. Gold, R. Hassan, K. D. Berlin, R. T. Ivey and D. M. Benbrook, “Effects of Retinoids on Cancerous Phenotype and Apoptosis in Organotypic Culture of Ovarian Carcinoma,” Journal of the National Cancer Institute, Vol. 93, No. 7, 2001, pp. 516- 525. doi:10.1093/jnci/93.7.516
- S. Liu, C. W. Brown, K. D. Berlin, A. Dhar, S. B. Guruswamy, D. Brown, G. J. Gardner, M. J. Birrer and D. M. Benbrook, “Synthesis of Flexible Sulfur-Containing Heteroarotinoids that Induce Apoptosis and Reactive Oxygen Species with Discrimination between Malignant and Benign Cells,” Journal of Medicinal Chemistry, Vol. 47, No. 4, 2004, pp. 999-1007. doi:10.1021/jm030346v
- D. M. Benbrook, S. A. Kamelle, S. B. Guruswamy, S. A. Lightfoot, B. Hannafon, T. L. Rutledge, N. S. Gould, S. T. Dunn and K. D. Berlin, “Flexible Heteroarotinoids (Flex- Hets) Exhibit Improved Therapeutic Ratios as Anti-Cancer Agents over Retinoic Acid Receptor Antagonists,” Investigational New Drugs, Vol. 23, No. 5, 2005, pp. 417- 428. doi:10.1007/s10637-005-2901-5
- T. C. Le, K. D. Berlin, S. D. Benson, M. A. Eastman, G. Bell-Eunice, A. C. Nelson and D. M. Benbrook, “Heteroarotinoids with Anti-Cancer Activity against Ovarian Cancer Cells,” Open Medicinal Chemistry Journal, Vol. 1, 2007, pp. 11-23. doi:10.2174/1874104500701010011
- T.-Z. Liu, B. Hannafon, L. Gill, B. Kelly and D. M. Benbrook, “Flex-Hets Differentially Induce Apoptosis in Cancer over Normal Cells by Directly Targeting Mitochondria,” Molecular Cancer Therapy, Vol. 6, 2007, pp. 1814-1822. doi:10.1158/1535-7163.MCT-06-0279
- P. Masamha and D. Benbrook, “Cyclin D1 Degradation Is Sufficient to Induce G1 Cell Cycle Arrest despite Constitutive Expression of Cyclin E2 in SHetA2-Treated Ovarian Cancer Cells,” Cancer Research, Vol. 69, 2009, pp. 6565-6572. doi:10.1158/0008-5472.CAN-09-0913
- T. Liu, C. P. Masamha, S. Chengedza, K. D. Berlin, S. Lightfoot, F. He and D. M. Benbrook, “Development of Flexible-Heteroarotinoids for Kidney Cancer,” Molecular Cancer Therapeutics, Vol. 8, No. 5, 2009, pp. 1227-1238. doi:10.1158/1535-7163.MCT-08-1069
- K. C. Moxley, S. Chengedza and D. Mangiaracina, “Induction of Death Receptor Ligand-Mediated Apoptosis in Epithelial Ovarian Carcinoma: The Search for Sensitizing Agents,” Gynecologic Oncology, Vol. 115, No. 3, 2009, pp. 438-442. doi:10.1016/j.ygyno.2009.09.007
- F. A. Mic, A. Molotkov, D. M. Benbrook and G. Duester, “Retinoid Activation of RAR but Not RXR Is Sufficient for Mouse Embryonic Development,” Proceedings of the National Academy of Science, Vol. 100, No. 12, 2003, pp. 7135-7140. doi:10.1073/pnas.1231422100
- K.-H. Chun, D. M. Benbrook, K. D. Berlin, W. K. Hong and R. Lotan, “Induction of Apoptosis in Head and Neck Squamous Cell Carcinoma (HNSCC) Cell Lines by Heteroarotinoids through a Mitochondrial Dependent Pathway,” Cancer Resesearch, Vol. 63, 2003, pp. 3826-3832. http://cancerres.aacrjournals.org/content/63/13/3826.long
- E. Garattini, M. Gianni and M. Terao, “Retinoid Related Molecules an Emerging Class of Apoptotic Agents with Promising Therapeutic Potential in Oncology: Pharmacological Activity and Mechanisms of Action,” Current Pharmaceutical Design, Vol. 10, No. 4, pp. 433-448, 2004. doi:10.2174/1381612043453351
- N. Colombo, F. Formelli, M. G. Cantu, G. Parma, M. Gasco, A. Argusti, A. Santinelli, R. Montironi, E. Cavadini, L. Baglietto, A. Guerrieri-Gonzaga, G. Viale and A. Decensi, “A Phase I-II Preoperative Biomarker Trial of Fenretinide in Ascitic Ovarian Cancer,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 15, No. 10, 2006, pp. 1914-1919. doi:10.1158/1055-9965.EPI-06-0183
- M. Cazzaniga, C. Varricchio, C. Montefrancesco, I. Feroce and A. Guerrieri-Gonzaga, “Fenretinide (4-HPR): A Preventive Chance for Women at Genetic and Familial Risk?” Journal of Biomedicine & Biotechnology, Vol. 2012, No. 2012, 2012, Article ID: 172897. doi:10.1155/2012/172897
- Y. Zhang, Y. Hua, D. M. Benbrook, J. M. Covey, G. Dai, Z. Liu and K. K. Chan, “High Performance Liquid Chromatographic Analysis and Preclinical Pharma-cokinetics of the Heteroarotinoid Antitumor Agent, SHetA2,” Cancer Chemotherapy & Pharmacology, Vol. 58, No. 5, 2006, pp. 561-569. doi:10.1007/s00280-006-0211-z
- R. S. Doppalapudi, E. S. Riccio, Z. Davis, S. Menda, A. Wang, N. Du, C. Green, L. Kopelovich, C. V. Rao, D. M. Benbrook and I. M. Kapetanovic, “Genotoxicity of the Cancer Chemopreventive Drug Candidates CP-31398, SHetA2, and Phospho-Ibuprofen,” Mutation Research, Vol. 746, No. 1, 2012, pp. 78-88. doi:10.1016/j.mrgentox.2012.03.009
- K. K. Kabirov, I. M. Kapetanovic, D. M. Benbrook, N. Dinger, I. Mankovskaya, A. Zakharov, C. Detrisac, M. Pereira, T. Martín-Jiménez, E. Onua, A. Banerjee, R. B. van Breemen, D. Nikolić, L. Chen and A. V. Lyubimov, “Oral Toxicity and Pharmacokinetic Studies of SHetA2, a New Chemopreventive Agent, in Rats and Dogs,” Drug and Chemical Toxicology, Vol. 36, No. 3, 2012, pp. 284- 295. doi:10.3109/01480545.2012.710632
- Z. Liu, Y. Zhang, Y. F. Hua, J. M. Covey, D. M. Benbrook and K. K. Chan, “Metabolism of a Sulfur-Containing Heteroarotionoid Antitumor Agent, SHetA2, Using Liquid Chromatography/Tandem Mass Spectrometry,” Rapid Communications in Mass Spectrometry, Vol. 22, No. 21, 2008, pp. 3371-3381. doi:10.1002/rcm.3744
- J. J. Emeiss and C. J. Edgell, “Fibrinolytic Properties of a Human Endothelial Hybrid Cell Line (Ea.hy 926),” Blood, Vol. 71, 1988, pp. 1669-1675. http://bloodjournal.hematologylibrary.org/content/71/6/1669.long
- D. S. Grant, P. I. Lelkes, K. Fukuda and H. K. Kleinman, “Intracellular Mechanisms Involved in Basement Membrane Induced Blood Vessel Differentiation in Vitro,” In Vitro Cellular and Developmental Bioliology, Vol. 27, No. 4, 1991, pp. 327-336. doi:10.1007/BF02630910
- C. J. Kuo, K. R. J. LaMontagne, G. Garcia-Cardena, B. D. Ackley, D. Kalman, S. Park, R. Christofferson, J. Kamihara, Y.-H. Ding, K.-M. Lo, S. Gillies, J. Folkman, R. C. Mulligan and K. Javaherian, “Oligomerization-Dependent Regulation of Motility and Morphogenesis by the Collagen XVIII NC1/Endostatin Domain,” The Journal of Cell Biology, Vol. 152, No. 6, 2001, pp. 1233-1246. doi:10.1083/jcb.152.6.1233
- M. Y. Ahn, S. M. Kwon, H. H. Cheong, J. H. Park, J. Lee, S. K. Min, S. G. Ahn and J. H. Yoon, “Toll-Like Receptor 7 Agonist, Imiquimod, Inhibits Oral Squamous Carcinoma Cells through Apoptosis and Necrosis,” Journal of Oral Pathology & Medicine, Vol. 41, No. 7, 2012, pp. 540-546. doi:10.1111/j.1600-0714.2012.01158.x
- D. M. Benbrook and A. Long, “Integration of Autophagy, Proteasomal Degradation, Unfolded Protein Response and Apoptosis,” Experimental Oncology, Vol. 34, No. 3, 2012, pp. 286-297. http://exp-oncology.com.ua/article/3596
- J. Folkman, “Angiogenesis in Cancer, Vascular, Rheumatoid and Other Disease,” Nature Medicine, Vol. 1, No. 1, 1995, pp. 27-31.
- A. Albini, F. Tosetti, V. W. Li, D. M. Noonan and W. W. Li, “Cancer Prevention by Targeting Angiogenesis,” Nature Reviews Clinical Oncology, Vol. 9, No. 9, 2012, pp. 498-509.
- T. Myers, S. Chengedza, S. Lightfoot, Y. Pan, D. Dedmond, L. Cole, Y. Tang and D. M. Benbrook, “Flexible Heteroarotinoid (Flex-Het) SHetA2 Inhibits Angiogenesis in Vitro and in Vivo,” Investigational New Drugs, Vol. 27, 2008, pp. 304-318. doi:10.1007/s10637-008-9175-7
- S. Bhoola and W. J. Hoskins, “Diagnosis and Management of Epithelial Ovarian Cancer,” Obstetrics & Gynecology, Vol. 107, No. 6, 2006, pp. 1399-1410. doi:10.1097/01.AOG.0000220516.34053.48
- J. T. Hartmann and H.-P. Lipp, “Toxicity of Platinum Compounds,” Expert Opinion on Pharmacotherapy, Vol. 4, No. 6, 2003, pp. 889-901. doi:10.1517/14656566.4.6.889
NOTES
*Corresponding author.