Journal of Cancer Therapy
Vol. 4 No. 4 (2013) , Article ID: 32384 , 10 pages DOI:10.4236/jct.2013.44093
Induction of Apoptosis and Anoikis by Bit1 in Pancreatic Cancer Cells
![]()
1Department of Biology, Xavier University of Louisiana, New Orleans, USA; 2Sanford-Burnham Medical Research Institute, La Jolla, USA.
Email: *hbiliran@xula.edu
Copyright © 2013 Kelly Leleux et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 6th, 2013; revised May 5th, 2013; accepted May 12th, 2013
Keywords: Anoikis; Apoptosis; Integrin; Pancreatic Cancer
ABSTRACT
Pancreatic cancer is a highly aggressive disease with a very high mortality rate among all human cancers. The poor prognosis is in part due to intrinsic resistance to the apoptosis-inducing effects of radioand chemotherapy. To find alternative cell death pathways that can bypass the apoptotic resistance of pancreatic cancer cells, we examined the role of the novel anoikis effector Bit1 (Bcl-2 inhibitor of transcription) in the survival and apoptotic resistance of pancreatic cancer cells. Bit1 is a mitochondrial protein that induces a caspase-independent apoptosis upon its release into the cytosol following loss of integrin-mediated attachment to extracellular matrix (anoikis). In this report, we observed that ectopic expression of Bit1 in the cytosol reduced viability and induced caspase-independent apoptosis in human pancreatic cancer cell lines, Miapaca-2 and PANC-1. While increased expression of mitochondrial Bit1 in these cells did not induce apoptosis under attached conditions, detachment significantly induced higher level of apoptosis in mitochondrial Bit1-transfected cells than in control transfected cells. Conversely, downregulation of endogenous Bit1 in PANC-1 cells further enhanced their anoikis resistance. Furthermore, exogenous expression of mitochondrial Bit1 in Miapaca-2 cells inhibited their anchorage-independent growth and enhanced their sensitivity to etoposide-mediated apoptosis. Mechanistically, we found that the Bit1 apoptosis function is in part dependent on the groucho related Amino-terminal Enhancer of Split (AES) expression and is abrogated by the transcriptional corepressor TLE1 protein. Consistent with our in vitro findings that Bit1 is an effector of apoptosis in pancreatic tumor cells, we find that Bit1 is significantly downregulated in a fraction of advanced stages of human pancreatic carcinoma tissues. Taken together, these findings indicate that the Bit1-apoptotic pathway can be targeted to trigger cell death in pancreatic cancer cells and implicate Bit1 as a novel therapeutic agent in attenuating pancreatic chemoresistance.
1. Introduction
Pancreatic cancer is a highly aggressive disease which has a dismal prognosis and currently has no viable mode of treatment. The severity of this disease can be appreciated by the fact that most new patients diagnosed with this cancer eventually die of the disease [1,2]. The poor prognosis associated with this malignancy is in part due to the aggressiveness and resistance of pancreatic cancer cells to conventional chemotherapeutic agents. Considerable evidence indicates that resistance to apoptosis by pancreatic cancer cells represent as the major obstacle to pancreatic cancer treatment.
Key survival mechanisms that allow pancreatic cancer cells to evade cell death include up regulation of antiapoptotic heat shock protein 70 (HSP 70) and activation of the NF-kB survival pathway [2-5]. Importantly, intrinsic apoptotic resistance to current therapy in pancreatic cancer cells has also been associated with inhibition of the caspase-dependent apoptotic pathway [1,6]. To circumvent the apoptotic resistance of pancreatic cancer cells, it is imperative to find alternative functional cell death pathways in these cells. Indeed, several studies have documented the apoptotic activity of several agents via a caspase-independent mechanism. Of noteworthy, Bobel and HDAC inhibitors, two novel apoptotic agents, induce caspase-independent cell death in several pancreatic cancer cell lines at least in part via the function of Apoptosis Inducing Factor (AIF) [7,8]. These studies indicate the functionality of caspase-independent cell death program regardless of apoptotic resistance in pancreatic cancer cells.
This study examines the role of the novel anoikis effector Bcl2-inhibitor of transcription (Bit1) in the apoptosis resistance and survival of pancreatic cancer cells. Bit1 is a mitochondrial protein that is released to the cytosol following loss of cell attachment, and subsequently induces a caspase-independent apoptosis (anoikis) [9,10]. The independence of Bit1 apoptosis from caspases is based on the findings that caspase inhibitors cannot prevent Bit1-induced cell death and by the lack of PARP cleavage in Bit1 transfected cells [9]. In addition to its caspase-independence, it is noteworthy that several antiapoptotic factors such as bcl2, bclx-l, and Pi3k/Akt are ineffective in inhibiting cytoplasmic Bit1 apoptosis function. On the other hand, the integrin-mediated cell attachment seems to be the only upstream factor that can prevent cytoplasmic Bit1 apoptosis, and hence Bit1 has been coined as “the guardian of anchorage-dependence”. Jan et al. 2004 previously demonstrated that the α5β1 integrin (the main fibronectin receptor) blocks Bit1 apoptosis in part by preventing the complex formation of cytoplasmic localized Bit1 with the groucho related Aminoterminal Enhancer (AES) protein.
In light of the recent findings demonstrating the utility of the caspase-independent cell death machinery in pancreatic cancer, we investigated the role of the Bit1 apoptotic pathway as a molecular apoptotic effector in pancreatic cancer cells. Considering that pancreatic cancer cells are highly resistant to apoptosis and such apoptosis resistance may contribute to pancreatic tumor aggressiveness, we first tested whether Bit1 apoptotic pathway is bypassed during panreatic cancer development. Here, we show that Bit1 expression is selectively downregulated in a subset of advanced human pancreatic tumors. Targeting Bit1 expression in the cytosol in human pancreatic cancer cell lines, Miapaca-2 and PANC-1, induced caspase-independent apoptosis while exogenous expression of mitochondrial-localized Bit1 in these cells attenuated their anoikis resistance and enhanced their sensitivity to etoposide-induced apoptosis. The role of Bit1 as an anoikis effector in pancreatic cancer cells was further evident in the observed increased anoikis resistance following specific downregulation of endogenous Bit1 expression. Mechanistically, we show that induction of the Bit1 apoptotic pathway is dependent on AES expression and is inhibited by the nuclear corepressor TLE1 protein. Taken together, these findings implicate Bit1 as a potential novel therapeutic target in rendering pancreatic cancer cells more prone to apoptotic stimuli such as following loss of attachment and upon exposure to chemotherapeutic agents.
2. Materials and Methods
2.1. Cell Culture and Transfection Assays
PANC-1 and Miapaca-2 cells from American Type Culture Collection (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with glutamine containing 10% fetal bovine serum, penicillin, and streptomycin. Transfections were carried out with lipofectamine 2000 (Invitrogen) in OPTI-MEM (Invitrogen) according to the manufacturer’s protocol with cells plated 18 hr before transfection. The total amount of plasmid used per transfection was normalized with the corresponding empty vector constructs. For siRNA transfection, cells were transfected with AES specific siRNAs or control siRNAs (obtained from Ambion) with the Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s recommendation [11]. To generate stable Bit1 knockdown and control pools of clones, PANC-1 cells infected with controlor Bit1-shRNA lentiviral particles (Santa Cruz Biotechnology) in a six-well plate format in the presence of polybrene (5 ug/ml) [11,12]. 24 h postinfection, cells were treated with 2 ug/ml puromycin to select for stable control and Bit1 knockdown clones. Several puromycin resistant control and Bit1 knockdown clones were harvested by ring-cloning, and the level of Bit1 knockdown was confirmed by immunoblotting against a specific Bit1 antibody. Two controlshRNA clones and three positive Bit1shRNA knockdown clones were pooled together to generate the controlshRNA pool and Bit1shRNA pool, respectively. The resulting controlshRNA and Bit1shRNA pools were then again subjected to immunoblotting using a specific antibody to Bit1 to confirm the downregulation of Bit1 expression.
2.2. Chemical Reagents, Antibodies, and Plasmids
Poly(2-hydroxyethyl methacrylate) (Polyhema) and the mouse monoclonal anti-β-actin antibody were obtained from Sigma Chemical Co. (St. Louis, Mo). The mouse monoclonal anti-FLAG, anti-AES, and anti-B-actin antibodies were purchased from Sigma (St. Louis, MO). The caspase inhibitor zVad-fmk and etoposide were purchased from Calbiochem (La Jolla, CA). The anti-c-myc antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The specific antibody to Bit1 used for immunoblotting was generated as described previously [9], and the affinity purified rabbit anti-Bit1 antibody (HPA012897, Sigma) was used for immunohistochemistry studies [12]. The mammalian expression vector encoding for mitochondrial Bit1 was generated as described previously [9]. The DDK-TLE1 plasmids were obtained from Origene (Rockville, MD).
2.3. Analysis of Apoptosis, Anoikis, Cell Viability, and Anchorage-Independent Growth
Apoptosis was assessed by determining the level of cytosolic nucleosomal fragments with the use of Cell Death Detection ELISA kit (Roche Molecular Biochemicals), according to the manufacturer’s instructions [11]. To assess for anoikis cell death, cells were plated onto Polyhema coated 96 well plates in complete growth medium containing 0.5% methylcellulose at a density of 1.0 × 104/well at various time points as previously described [11,12]. Detached cells were then collected and subjected to the Cell Death ELISA apoptosis assay. In some experiments, cell viability was quantified by clonogenic assay [13] or with the use of MTT (Thiazolyl Blue Tetrazolium Bromide, Sigma). Briefly, fifteen µl of MTT solution (5 mg/ml in PBS) was added and further incubated for 4 h. The resulting MTT formazan was solubilized by addition of 100 µl of SDS solution (20% in 10 mM HCL), and the absorbance was measured 24 h at 550 nm and a reference wavelength of 690 nm using a microplate reader. The anchorage-independent growth of cells was measured using the 96-well plate format [11]. Briefly, 5000 cells in 0.3% agar solution was plated onto wells precoated with 0.6% agar in culture medium. The anchorage-independent growth of cells was quantified by alarmar blue staining (Invitrogen) and fluorescence reading at 530 - 560 nm excitation wavelength and 590 nm emission wavelength.
2.4. Protein Preparation, Western Blotting, and Co-Immunoprecipitation Assays
Protein preparation and western blotting were performed as described previously [11,12]. Briefly, cells were harvested 24 - 48 hr after transfection with various constructs or siRNAs by adding ice-cold NP-40 lysis buffer (1% NP-40; 20 mM Tris-HCL [pH 7.4]; 150 mM NaCl; 10% glycerol, 2 mM sodium vanadate; 1 mM henylmethylsulfonyl fluoride; 10 μg/ml leupeptine; and 5 μg/ml aprotinin) and incubating at 4˚C for 20 min. For immunoblot analysis, equal amounts of proteins were resolved on 4% - 20% gradient Tris-glycine gels (Invitrogen) and electrophoretically transferred to nitrocellulose membrane. The membranes were incubated with primary antibodies overnight at 4˚C followed by secondary antibodies conjugated with horseradish peroxidase. Membranes were developed using the ECL detection system. For co-immunoprecipitation assays, transfected cells were harvested in ice-cold Nonidet P-40 lysis buffer, and cell debris was removed by centrifugation. Flag-tagged Bit1 was immunoprecipitated with anti-Flag-agarose conjugate (Sigma) and thoroughly washed with lysis buffer. Bound proteins were resolved by SDS-PAGE, and western blotting was performed using anti-Flag or anti-Myc antibodies [10].
2.5. Human Pancreatic Tumor Array Analysis
Pancreatic tumor tissue array slides were obtained from US Biomax, Inc. (Rockville, MD). The immunohistochemistry procedure was performed by Biomax Inc. on two tissue microarray slides. As described previously [11,12], tissue array slides were deparaffinised, hydrated and subjected to antigen retrieval. The slides were then incubated in 2.5% normal horse serum for 30 min at room temperature followed by incubation with the primary antibody (1:100 dilution) for 1 h at room temperature. Rabbit normal serum was used as negative control antibody to replace the primary antibody on control slide with 1 hr incubation. Tissue array slides were then washed and incubated with ImmPRESS reagent (Vector Laboratories) followed by treatment with peroxidise substrate DAB solution (DAKO Cytomation). Each of the experimental and control slide was scored for average staining intensity by two investigators with no knowledge of the pathologic status of the samples. These investigators scored staining intensity as 0, no staining; 1 low staining; 2 medium staining; or 3 high staining.
2.6. Statistical Analysis
Data are presented as means (±S.E.). For western blots and anoikis assays, experiments were performed at least three times with duplicates. Statistical differences between groups were established at a P value < 0.05 using the two-tailed Student’s t test. For pancreatic tumor tissue array analysis, a one-way ANOVA with subsequent post hoc testing using the Tukey-Kramer multiple comparison test was used to compare the average staining intensity of each case type [11,12]. All calculations were done using the NCSS statistical software (NCSS, Kasville, UT).
3. Results
3.1. Bit1 Protein Expression Is Downregulated in Advanced Pancreatic Adenocarcinoma Tumors
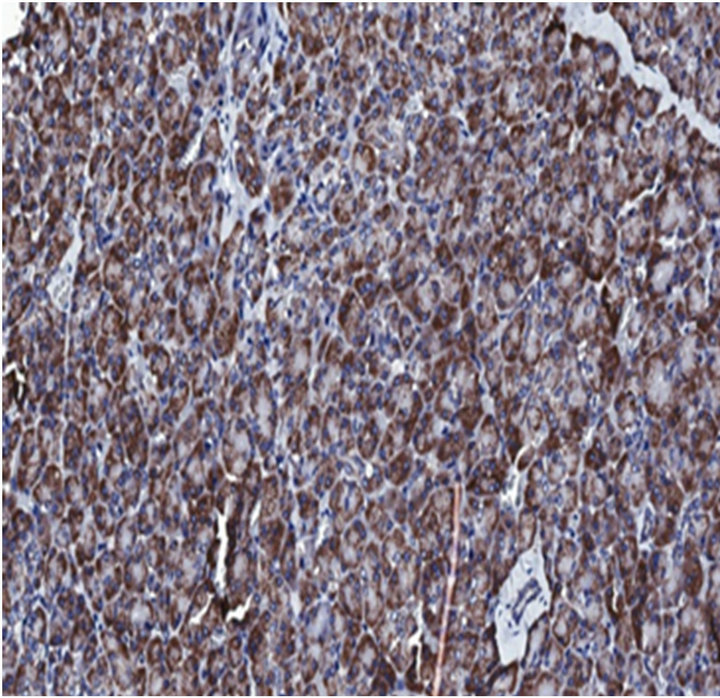
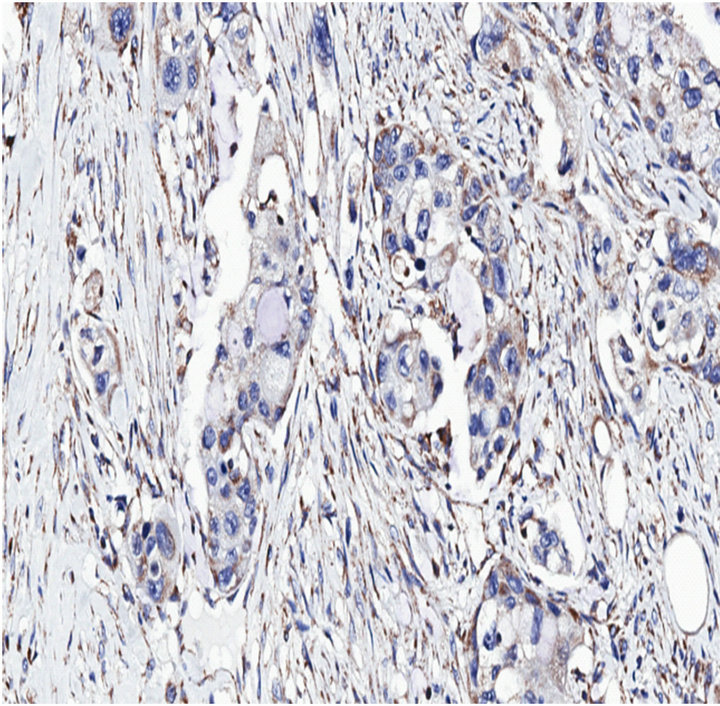
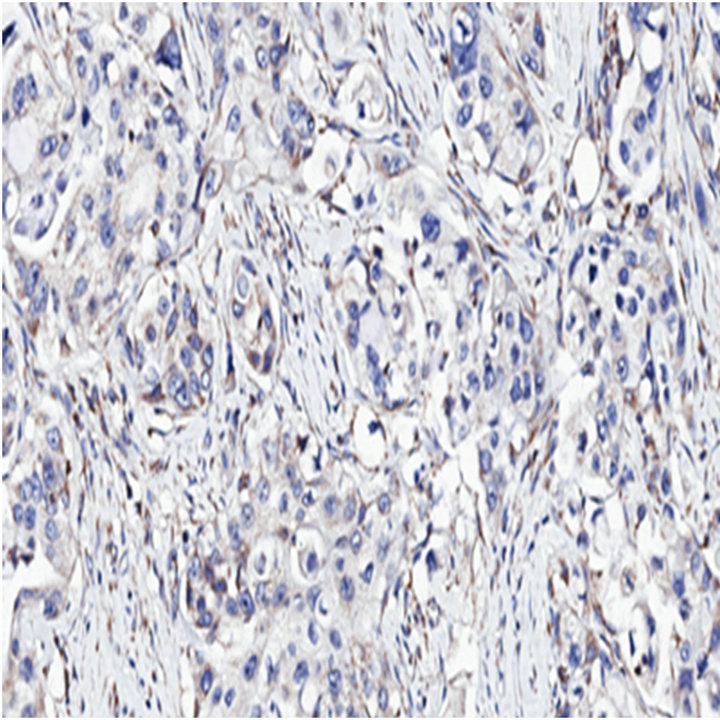
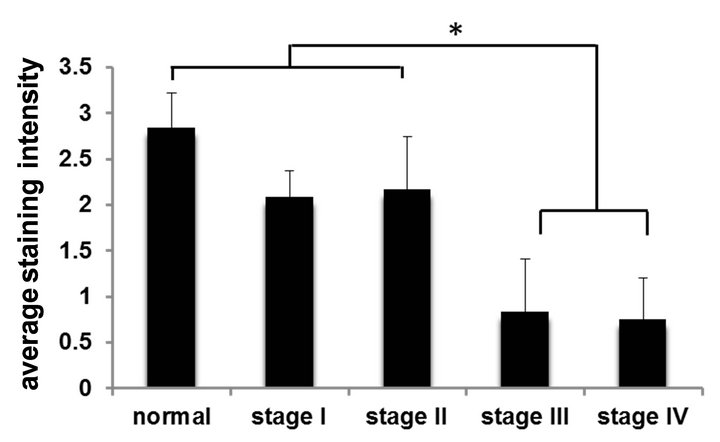
Since anoikis resistance is a primary determinant of tumorigenesis and/or metastasis [14], we hypothesized that the anoikis effector Bit1 is downregulated or dysfunctional in advanced tumors. We performed immunohistochemistry to examine Bit expression in a human pancreatic tumor tissue microarray with the use of an affinity purified polyclonal antihuman Bit1 antibody (Sigma). This polyclonal anti-Bit1 antibody was developed and its specificity validated by the Human Proteome Resource (HPR) project [11,12]. Immunoreactivity was graded as 0 (none), 1 (slight), 2 (moderate), or 3 (strong). In normal pancreatic tissue, both the exocrine acinar cells and the islet cells showed strong cytoplasmic staining for Bit1 (Figure 1(a(i))). The strong Bit1 staining was particularly homogenous within the pancreatic acini. As compared to the strong immunoreactivity of Bit1 in the normal pancreatic tissue, a subset of advanced pancreatic tumors (20 of 42 (47%) of stage III (Figure 1(a(ii)) and (22 of 39 (56%) of stage IV (Figure 1(a(iii)) exhibited significantly reduced cytoplasmic Bit1 immunoreactivity. Quantification of the average Bit1 staining (Figure 1(b)) confirmed the reduction of Bit1 immunoreactivity of advanced pancreatic tumor tissues relative to its normal counterpart. These findings indicate that Bit1 expression is lost in advanced pancreatic cancer, suggesting that downregulation of Bit1 expression may be an important event in the progression of pancreatic cancer.
3.2. Ectopic Expression of Cytoplasmic Bit1 Induces Caspase-Independent Apoptosis in Pancreatic Cancer Cells
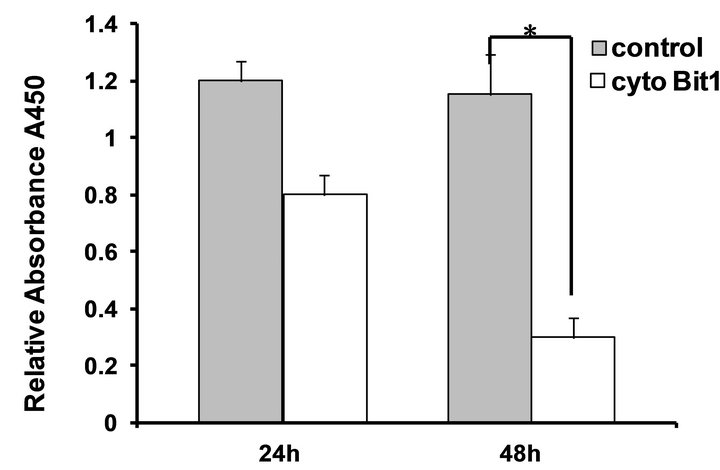
To test the functionality of the Bit1 apoptotic pathway in pancreatic cancer cells, we ectopically overexpressed Bit1 in the cytoplasm of human pancreatic cancer cell line Miapaca-2 via transfection with either a vector control or N-terminally myc-tagged Bit1 construct [10]. Placement of tag at the N-terminal region of the Bit1 protein results in its cytoplasmic localization [9]. The cytoplasmic Bit1 transfected cells showed a dramatic decrease in cell viability 48 hr post transfection, as assessed by MTT (Figure 2(a)) and clonogenic (Figure 2(b)) assays. The reduction in cell viability in cytoplasmic Bit1 transfected cells was associated with increased levels of DNA-histone fragments (Figure 2(c)), indicating the induction of apoptotic form of cell death by Bit1. Similar induction of apoptosis was also observed upon targeting Bit1 expression in the cytoplasm of PANC-1 cells (Figure 2(d)). Consistent with previous findings [9], the induction of Bit1-induced cell death was not abrogated by the pan-caspase inhibitor zVAD-fmk, indicating the caspase-independence mode of Bit1-mediated apoptosis (Figure 2(e)).
3.3. Alterations in Mitochondrial Bit1 Expression Modulate Anoikis Sensitivity of Pancreatic Cancer Cells
We evaluated the effects of overexpressing the mito-
(i) (ii) (iii)
(a)
(b)
Figure 1. Bit1 expression is downregulated in advanced stage pancreatic tumors. (a) Pancreatic tumor tissue array slides were stained with affinity purified anti-Bit1 (Sigma). Images are representative of each respective case type: normal pancreatic tissue 10× (i), pancreatic adenocarcinoma (stage III) 10× (ii), and pancreatic adenocarcinoma (stage IV) 10× (iii); (b) The average staining intensity of each subgroup was determined. While no significant difference was found between normal and stage I and stage II pancreatic tumors, the normal pancreatic tissue was statistically significant from the stage III and stage IV pancreatic tumors using the ANOVA and subsequent Tukey post-hoc analysis (see Materials and Methods) (*, P < 0.05).
 (a)
(a)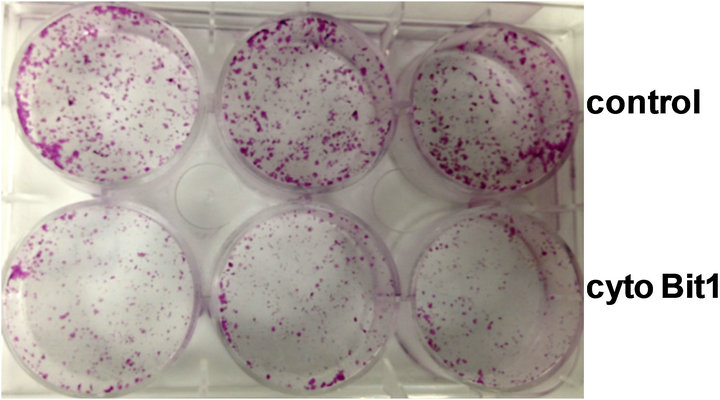 (b)
(b)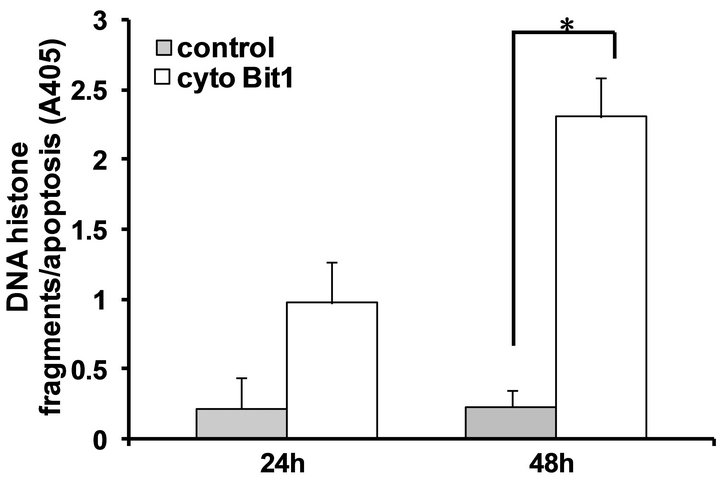 (c)
(c)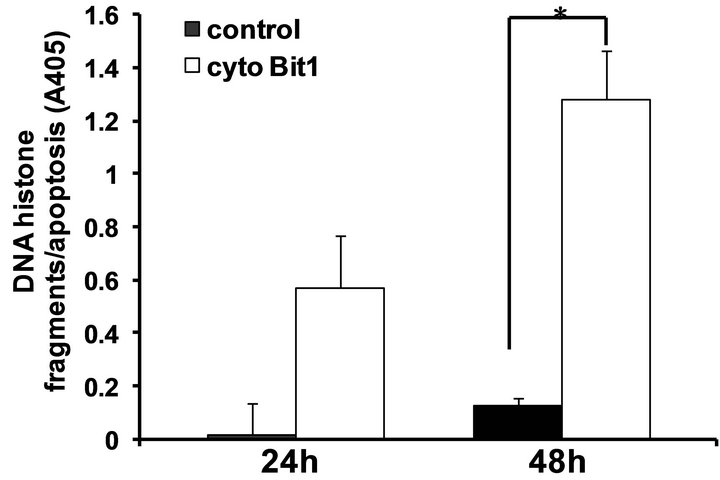 (d)
(d)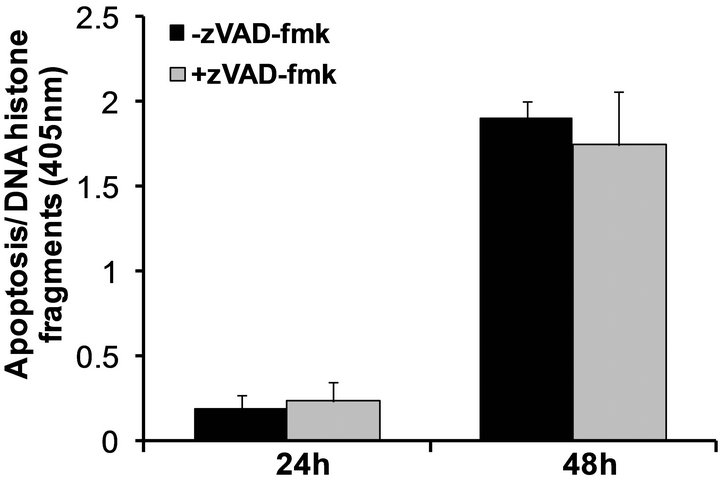 (e)
(e)
Figure 2. Targeted expression of Bit1 in the cytoplasm reduces cell viability and and induces caspase-independent apoptosis. (a) Miapaca-2 cells were transfected with empty vector (control) or N-terminally myc tagged cytoplasmic localized Bit1 (cyto Bit1), and 24 and 48 h post transfection, cells were subjected to MTT assay; (b) Control or N-terminally myc tagged Bit1 (cyto Bit1) transfected Miapaca-2 cells were subjected to clonogenic assay 48 hr post-transfection; (c) Control or N-terminally myc tagged Bit1 transfected Miapaca-2 cells were also subjected to Cell Death ELISA 24 h and 48 h post-transfection to determine the level of apoptosis; (d) PANC-1 cells were transfected with empty vector or N-terminally myc tagged Bit1, and 24 and 48 h post transfection, cells were subjected to Cell Death ELISA assay; (e) Miapaca-2 cells transfected with empty vector or N-terminally myc tagged Bit1 were cultured in the presence of z-Vad-fmk (50 uM) or DMSO for 24 h. Cells were then harvested and subjected to Cell Death ELISA. In (a), (c), (d), and (e), three independent experiments were performed in triplicates, *indicates P < 0.05 by Student’s t test.
chondrial localized Bit1 on the anoikis sensitivity of pancreatic cancer cells. Mitochondrial Bit1 was overexpressed in Miapaca-2 cells via transfection of C-terminally myc tagged Bit1 or vector construct [9,10]. Exogenous expression of mitochondrial Bit1 induced significantly higher level of apoptosis following loss of cell attachment as compared with control cells (Figure 3(a)). In contrast, both control and mitochondrial Bit1-transfected cells showed similar levels of basal apoptosis in attached conditions (Figure 3(a)). To complement the results from exogenous mitochondrial Bit1 studies, we also examined the effect of downregulating Bit1 expression in PANC-1 cells, which exhibit moderate levels of endogenous Bit1. To this end, we generated stable Bit1 knockdown clones by infecting PANC-1 cells with lentiviral expression vectors for Bit1 specific shRNAs or control shRNAs [12]. As shown in Figure 3(b), the Bit1 expression was reduced by 70% in a pool of Bit1 shRNA clones as compared with control shRNA pool of clones. The control shRNA and Bit1 shRNA pools exhibited
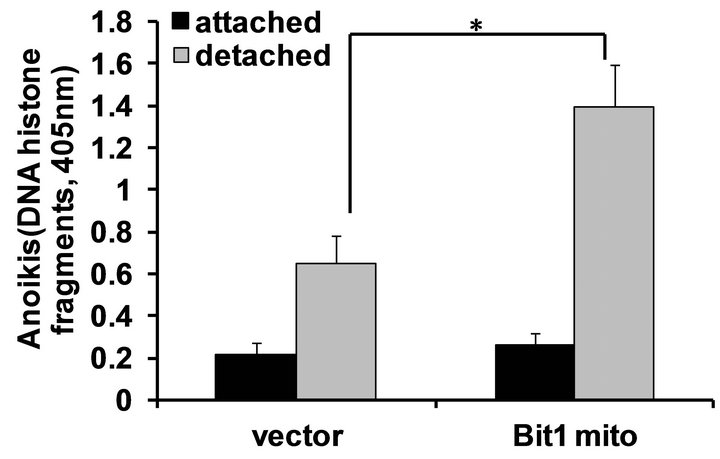
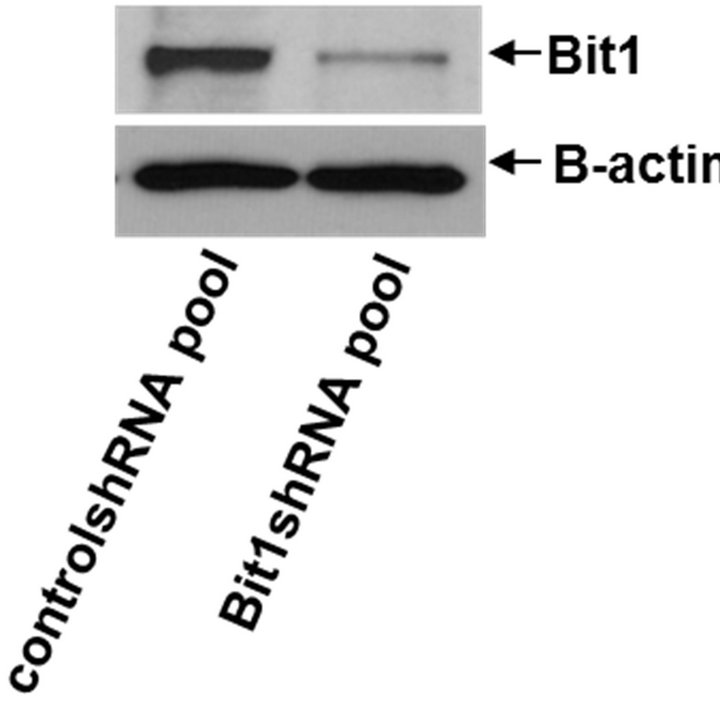
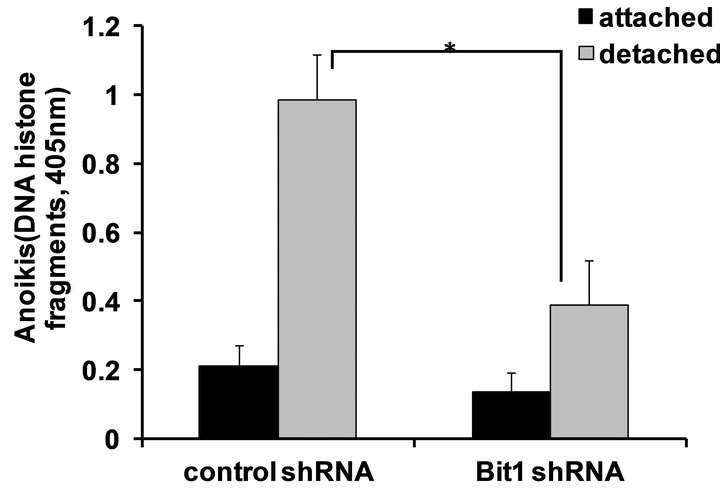
Figure 3. Mitochondrial localized Bit1 regulates anoikis sensitivity of pancreatic cancer cells. (a) Miapaca-2 cells were transfected with C-terminally tagged myc-tagged mitochondrial localized Bit1 (Bit1 mito) or vector construct, and 24 h post-transfection, cells were plated onto polyhema coated or uncoated tissue culture plates. Following 48 h in culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa); (b) Stable PANC-1 control shRNA and Bit1 shRNA knockdown pools were generated as described in materials and methods, and the total cell lysates derived from control shRNA and Bit1shRNA knockdown pools were subjected to immunobloting using specific antibodies to Bit1 and B-actin; (c) Stable PANC-1 derived Bit1shRNA knockdown and control shRNA pools were plated onto polyhema coated tissue culture plates. Following 48 hr in suspension culture, cells were harvested and analyzed for apoptosis by measuring the amount of DNA histone fragments (Cell Death Elisa). In (a) and (c), three independent experiments were performed in triplicates, *indicates P < 0.05 by Student’s t test.
similar levels of basal apoptosis (Figure 3(c)) in monolayer culture. In contrast, when cultured without anchorage, the Bit1 shRNA pool displayed a significantly decreased apoptosis than the control shRNA pool. Taken together, these results indicate that Bit1 is an anoikis effector in pancreatic adenocarcinoma cells.
3.4. Overexpression of Mitochondrial Bit1 Attenuates Anchorage-Independent Growth and Enhances Sensitivity to Etoposide-Induced Apoptosis in Pancreatic Cancer Cells
Since anoikis resistance is a requirement of anchorageindependent growth, a hallmark of transformation [14, 15], we then examined the effect of exogenous mitochondrial Bit1 expression on the growth of Miapaca-2 cells in a soft agar assay. Consistent with their enhanced anoikis sensitivity (Figure 3(a)), mitochondrial Bit1 transfected cells showed significant reduction in anchorage-independent growth relative to control transfected cells (Figure 4(a)). The Bit1 mitochondrial-transfected cells resulted in smaller and fewer colonies than the control cells (Figures 4(b) and (c)).
Since chemoresistance is an important determinant of pancreatic tumor aggressiveness, we examined whether induction of the Bit1 apoptotic pathway may enhance the chemosensitivity of the highly chemoresistant Miapaca-2 cells. We overexpressed the mitochondrial localized Bit1 in Miapaca-2 cells and examined their responsiveness to the chemotherapeutic agent, etoposide. While vector transfected cells showed no or little response to low concentrations of etoposide, the Bit1 transfected cells exhibited significant apoptosis at these conditions (Figure 4(d)). At much higher doses of etoposide, the induction of cell death became evident in control cells albeit at a significantly lower level relative to mitochondrial Bit1 transfected cells. These findings indicate that mitochondrial Bit1 enhances the sensitivity of Miapaca-2 cells to etoposide-induced apoptosis.
3.5. Bit1 Apoptotic Function Is Dependent on AES Expression and Is Blocked by the Corepressor TLE1 Protein
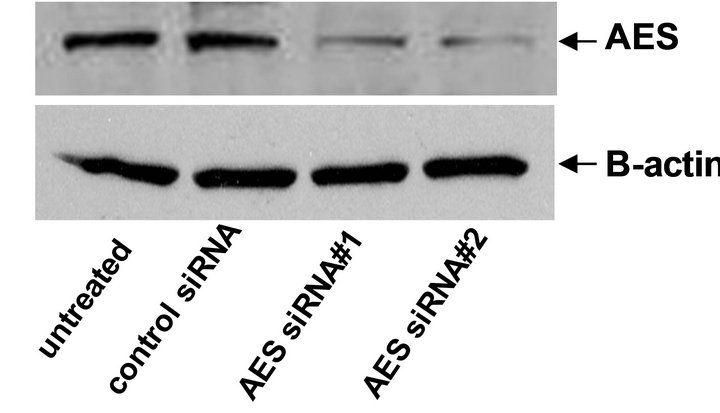
We have previously shown that Bit1, upon its release to or following its ectopic expression in the cytoplasm, associates with the groucho related AES protein to induce apoptosis [9,10]. To address the functional requirement of AES in Bit1 apoptosis function in pancreatic cancer cells, we evaluated the effects of alteration of AES expression on the sensitivity of cells to Bit1 apoptosis. Specific downregulation of AES expression in Miapaca- 2 cells attenuated cytoplasmic Bit1 apoptosis (Figures 5(a) and (b)). Conversely, exogenous AES expression further enhanced the induction of apoptosis by Bit1 (Figure 5(c)). We then evaluated the association of Bit1 and AES protein via co-immunoprecipitation studies. Miapaca-2 cells were transfected with N-terminally flagtagged Bit1 (cyto Bit1) and myc-tagged AES. Immunoprecipitation with anti-flag and blotting of the precipitate with anti-myc antibodies revealed Bit1/AES complexes
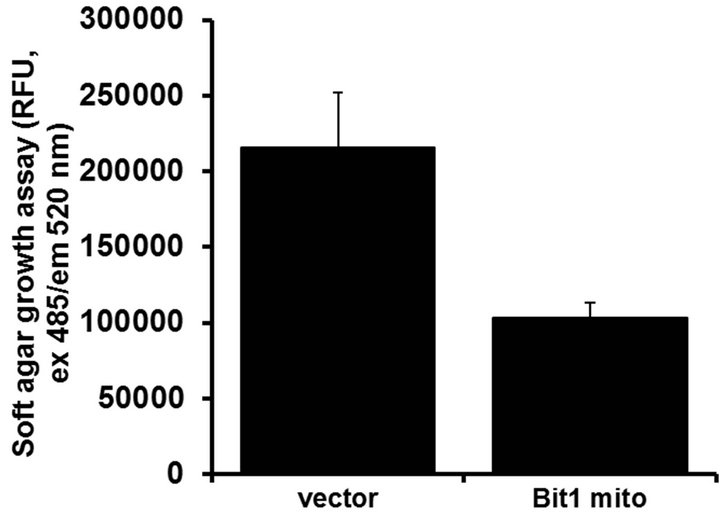
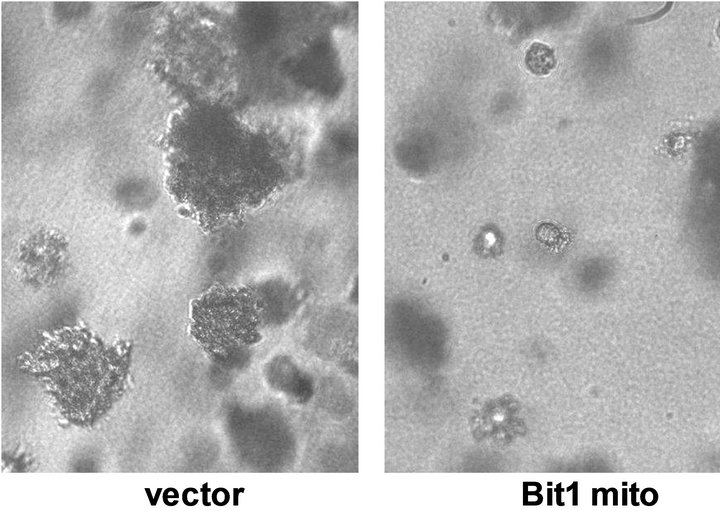
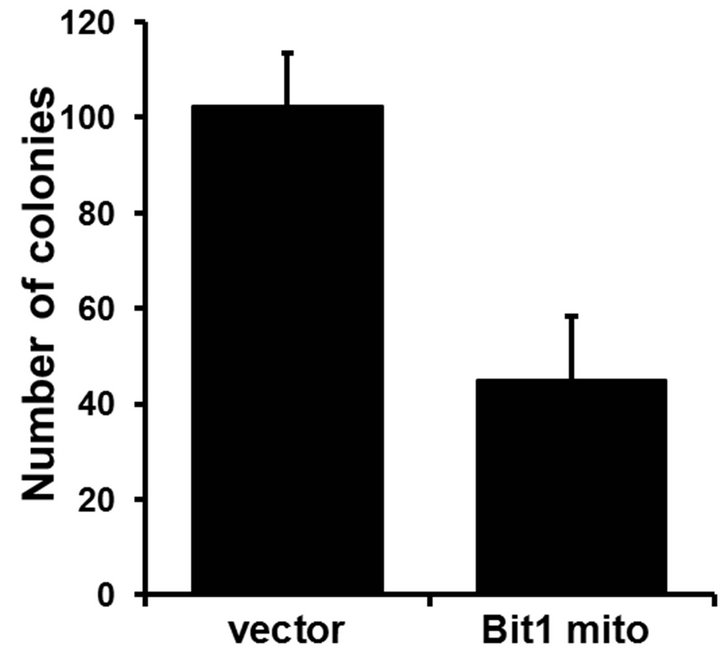
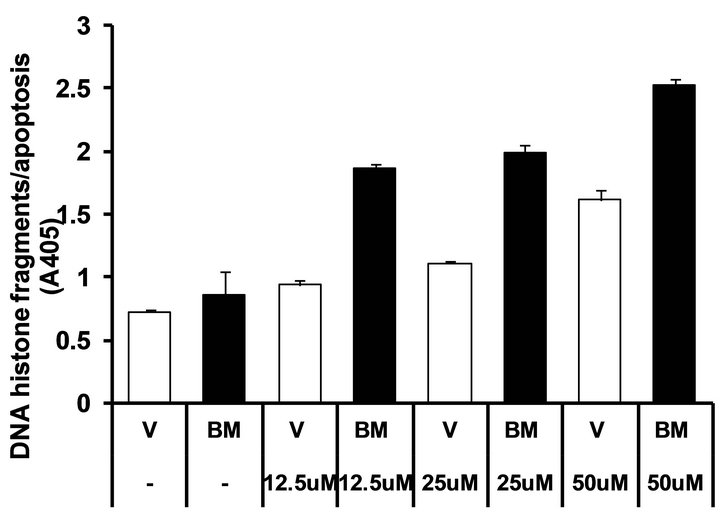
Figure 4. Exogenous mitochondrial Bit1 attenuates the anchorage-independent growth and chemoresistance of pancreatic cancer cells. (a) Miapaca-2 cells were transfected with C-terminally tagged mitochondrial localized Bit1 (Bit1 mito) or vector construct, and 24 h post-transfection, cells were subjected to soft agar assay, and the level of anchorage-independent growth was quantified by alamar blue staining and subsequent fluorescent reading; (b) The representative colonies of vector and Bit1 mito transfected cells were visualized microscopically; (c) The extent of anchorage-independent growth of vector or Bit1 mito transfected cells was also measured by counting the number of visible colonies (with a diameter greater than 30 uM); (d) Vector (V) or Bit1 mito (BM) transfected cells were treated with various dosage of etoposide, and 24 following drug treatment, cells were subjected to Cell Death Elisa Kit to determine the level of drug-induced apoptosis. In (a), (c) and (d), three independent experiments were performed in triplicates, *P < 0.05 as compared with the vector transfected cells (Student’s t test).
in the transfected cells (Figure 5(d)). The groucho related nuclear TLE1 corepressor blocks the Bit1-AES complex formation (Figure 5(d)) and abrogates Bit1 apoptosis (Figure 5(e)). Taken together, these findings indicate that Bit1 associates and cooperates with AES to induce apoptosis in pancreatic cancer cells.
4. Discussion
The poor prognosis associated with pancreatic cancer is in part due to its propensity to disseminate to distant organs and its high resistance to conventional therapy. The aggressive nature of this cancer undoubtedly depends on its survival mechanisms and unresponsiveness to apoptotic stimuli. The major challenge therefore in combating this deadly disease is to find new modalities or molecular regulators of alternative cell death programmes that may remain functional in pancreatic cancer. This study examines the role of the novel anoikis effector named Bcl2- inhibitor of transcription 1 (Bit1) in the apoptosis resistance and survival of pancreatic cancer cells. Here, we show that ectopic expression of Bit1 in the cytosol results in reduced viability and induces significant apoptosis in two aggressive, highly chemoresistant pancreatic cancer cell lines, Miapaca-2 and PANC-1. Moreover, genetic alteration of mitochondrial Bit1 expression in these cells impacts their ability to undergo cell death following loss of cell attachment (anoikis). Mechanistically, we observed that Bit1 apoptotic function in pancreatic cancer cells involves association with the groucho AES protein.
Due to defects in signalling pathways leading to the activation of caspases in a variety of tumors, the use of the caspase-independent cell death program as an alternative strategy in triggering tumor cell death has recently gained interest [7,8]. This is particularly true in pancreatic cancer where inhibition of caspase-dependent apoptotic pathways has been strongly associated with intrinsic resistance to chemotherapy. In this report, we show that ectopic overexpression of cytoplasmic-localized Bit1 can significantly induce a caspase-independent form apoptosis in two pancreatic cancer cell lines, Miapaca-2 and PANC-1. As described previously, the Bit1-induced apoptosis is not associated with significant activation of the caspase-dependent pathway. In addition to its cas-
 (a)
(a)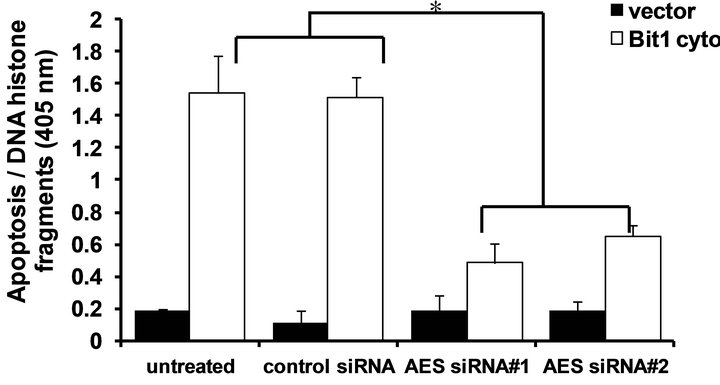 (b)
(b)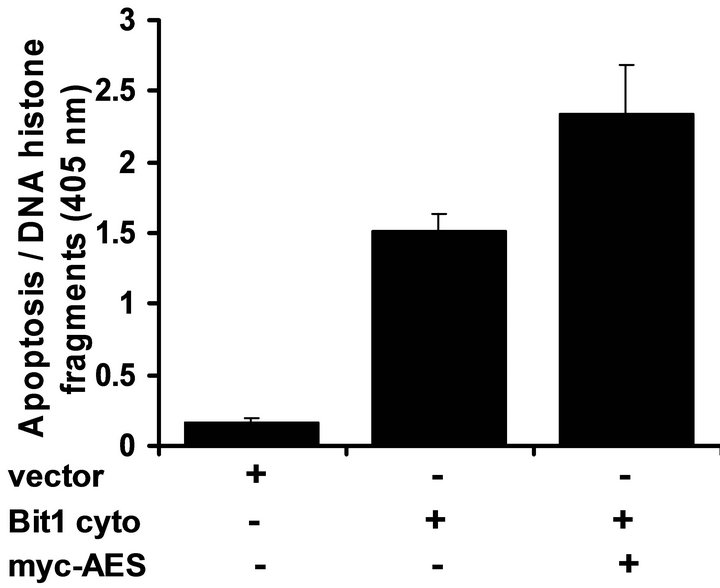 (c)
(c)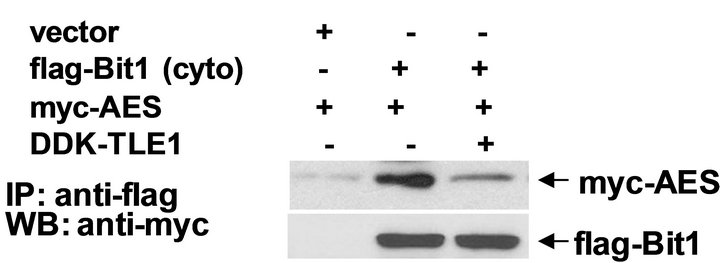 (d)
(d)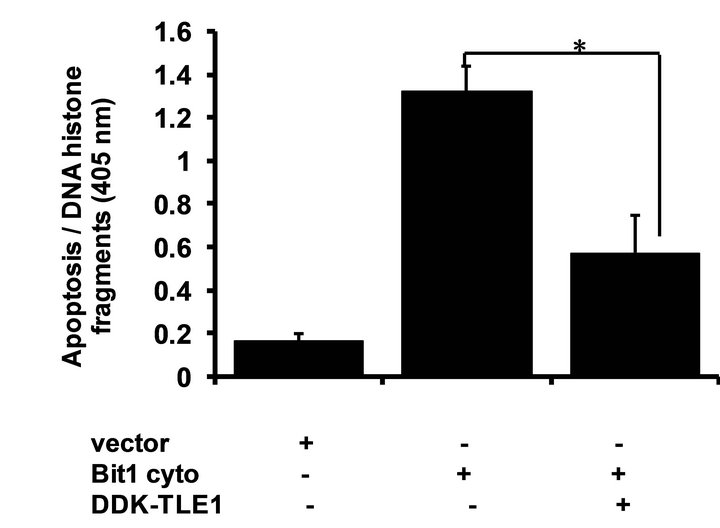 (e)
(e)
Figure 5. Bit1 apoptosis is dependent on AES expression and is antagonized by TLE1. (a) Miapaca-2 cells were transfected with controlor AES specific siRNAs, and 48 h post-transfection, cells were harvested and subjected to immunoblotting (a) with antibodies against AES and B-actin; In (b) controland AES siRNA treated cells were transfected with the N-terminally flag-tagged Bit1 (cyto Bit1) or vector, and 48 h later cells were harvested and subjected to Cell Death ELISA assay to measure the amount of DNA histone fragments; (c) Miapaca-2 cells were transfected with the cyto Bit1 together with myc-AES construct. The amount of plasmid transfected into cells was normalized via the vector construct. 48 h post transfection, cells were harvested and subjected to Cell Death ELISA assay; (d) Miapaca-2 cells were transfected with the respective construct as indicated. 24 h post-transfection, cells were harvested and cell extracts were prepared, immunoprecipitated with agarose-immobilized anti-flag, and immunoblotted with anti-myc antibody; (e) Miapaca-2 cells were transfected with the cyto Bit1 together with DDK-TLE1 construct. 48 h post transfection, cells were then harvested and subjected to Cell Death ELISA assay. In (b), (c), and (e), three independent experiments were performed in triplicates, *indicates P < 0.05 by Student’s t test.
pase-independent nature, Bit1 apoptotic function is uniquely regulated by integrin-mediated cell attachment and remains unresponsive to various anti-apoptotic treatments including Akt, Bcl-xl, and Bcl-2 [9]. These unique features indicate the usefulness of Bit1 as a potential therapeutic target in triggering cell death in the highly apoptotic resistant pancreatic cancer. Recent reports demonstrated that histone deacetylase (HDAC) inhibitors [7] and a series of cyclooxygenase and 5-lypoxygenase inhibitors (Bobel-24) inhibitors [8] can induce caspaseindependent cell death in a variety of pancreatic cancer cell lines, at least in part through the apoptotic function of apoptosis inducting factor (AIF). It will be interesting to determine whether the induction of caspase-independent apoptosis by HDAC and Bobel-24 may also involve Bit1. Our findings together with these previous reports indicate that the caspase-independent mechanism may represent as viable target to induce cell death in pancreatic cancer cells regardless of caspase-dependent apoptotic blockade.
The poor prognosis of pancreatic cancer is also in part due to the highly invasive and metastatic nature of this malignancy even in its early stage [1,16]. At the time of diagnosis, most pancreatic cancer present with metastatic or locally advanced disease. It is therefore imperative to search for factors that can inhibit the aggressive biology of pancreatic cancer. Since anoikis resistance is an essential step in acquiring malignant transformation and metastatic potential of tumor cells [14,15], re-establishing anoikis sensitivity may serve as an attractive therapeutic strategy to combat the aggressiveness pancreatic cancer cells. Our results indicate that activation of the Bit1 anoikis pathway significantly enhances the anoikis sensitivity and chemosensitivity of pancreatic cancer cells. As the sole caspase-independent anoikis effector known to date, Bit1 may act as a potent suppressor of anoikis resistance and aggressiveness of pancreatic cancer cells. Indeed, our immunohistochemistry studies on multiple pancreatic tumor tissue arrays indicate that Bit1 is suppressed in advanced stages of pancreatic cancer, suggesting that abrogation of the Bit1 apoptotic pathway may contribute to the progression and development of pancreatic carcinogenesis. We are currently investigating the potential impact of Bit1 in the tumorigenic growth and metastatic progression of pancreatic cancer in vivo.
Resistance to chemotherapy by tumor cells has been associated with dysregulation of apoptotic machinery that allows them to evade drug-induced cell death [1, 6,16,17]. In particular, defects in the caspase-dependent apoptotic pathway may render cells resistant to antitumor chemotherapy. Clearly, new modes of cell death programmes have to be discovered in order to bypass the apoptotic resistance of some tumors including pancreatic carcinomas. Recent studies have indicated the existence of the caspase-independent mechanisms by which chemotherapeutic drugs induce cell death in cancer cells [7,8]. In particular, the caspase-independent apoptotic effector AIF has been shown to be critical in inducing cell death by several anticancer agents. It is noteworthy that components of the caspase-independent cell death machinery have not been exhaustively identified, and the therapeutic value of this cell death pathway in circumventing tumor chemoresistance will be highly dependent on deciphering the key caspase-independent effectors.
Our data indicate that the Bit1 apoptotic pathway may be specifically targeted to enhance impact pancreatic cancer cell sensitivity to chemotherapy. Considering the intricate interaction and interdependency between caspaseand caspase-independent pathways, we will not exclude the possibility that the caspase-independent Bit1 apoptotic function following drug treatment may activate the caspase-dependent cell death pathway and their synergistic actions may effectively induce apoptosis in the highly chemoresistant pancreatic cancer cells.
To date, the molecular mechanism underlying Bit1 apoptotic function remains to be fully elucidated. Consistent with our previous studies [9-12], the groucho AES protein is a critical component of the Bit1 apoptoic pathway. Ablation of AES attenuates Bit1 apoptosis while exogenous AES accelerated Bit1 apoptosis in pancreatic cancer cells. As evidenced in our previous studies [9,10], the AES protein acts as pro-apoptotic partner of Bit1 in mediating apoptosis. The Bit1/AES complex is a critical component of the Bit1 apoptotic pathway since inhibiting the Bit1/AES complex formation, such as through integrin-mediated cell attachment or via nuclear TLE1 corepressor expression [9], attenuates Bit1 apoptosis. The ability of TLE1 to block Bit1/AES complex formation is in part due to the ability of TLE1 to compete with Bit1 for AES binding and to sequester AES in the nucleus [11].
In summary, we have shown that the caspase-independent Bit1 apoptotic pathway is a viable alternative cell death pathway that can be reactivated to induce cell death in pancreatic cancer cells. Induction of apoptosis by cytoplasmic Bit1 involves biological interaction with AES protein. Based on the potent apoptotic activity of cytoplasmic Bit1 in pancreatic cancer cells, it is not surprising that advanced pancreatic tumors exhibit low levels of Bit1 expression. Selective targeting of Bit1 in primary and metastatic pancreatic tumors may represent a unique therapeutic strategy in conjunction with chemotherapy to potently effect apoptosis in pancreatic cancer cells.
5. Acknowledgements
We thank Dr. Shubha Kale Ireland for comments on the manuscript. This work was supported by Louisiana Cancer Research Consortium (LCRC) Start up Grant (HB), NIH RCMI G12RR026250-03 Grant (to Xavier University of Losuiana), NIH SC2CA153382 grant (HB), and Research Competitiveness Subprogram (Louisiana Board of Regents) (HB).
REFERENCES
- G. Schneider, J. T. Siveke, F. Eckel and R. M. Schmid, “Pancreatic Cancer Basic and Clinical Aspects,” Gastroenterology, Vol. 128, No. 6, 2005, pp. 1606-1625. doi:10.1053/j.gastro.2005.04.001
- A. Aghdassi, “Heat Shock Protein Increases Tumorigenicity and Inhibits Apoptosis in Pancreatic Adenocarcinoma,” Cancer Research, Vol. 67, 2007, pp. 616-625. doi:10.1158/0008-5472.CAN-06-1567
- P. Phillips, V. Dudeja, J. McCarroll, D. Borja-Cacho, R. Dawra, W. Grizzle, S. Vickers and A. Saluja, “Triptolide Induces Pancreatic Cancer Cell Death via Inhibition of Heat Shock Protein 70,” Cancer Research, Vol. 67, No. 19, 2007, pp. 9409-9416. doi:10.1158/0008-5472.CAN-07-1077
- A. Arlt, J. Vorndamm, M. Breitenbroich, U. Folsh, H. Kalthoff, W. Schmidt and H. Schafer, “Inhibition of NFkB Sensitizes Human Pancreatic Carcinoma Cells to Apoptosis Induced by Etoposide (VP16) or Doxorubicin,” Oncogene, Vol. 20, No. 7, 2001, pp. 859-868. doi:10.1038/sj.onc.1204168
- A. Arlt, A. Gehrz, S. Muerkoster, J. Vorndamm, M. Kruse, U. Folsch and H. Schafer, “Role of NF-kB and Akt/P13K in the Resistance of Pancreatic Carcinoma Cell Lines against Gemcitabine-Induced Cell Death,” Oncogene, Vol. 22, 2003, pp. 3243-3251. doi:10.1038/sj.onc.1206390
- S. Westphal and H. Kalthoff., “Apoptosis: Targets in Pancreatic Cancer,” Molecular Cancer, Vol. 2, 2003, p. 6. doi:10.1186/1476-4598-2-6
- P. Garcia-Morales, A. Gomez-Martinez, A. Carrato, I, Martinez-Lacaci, V. Barbea, J. Soto, E. Carrasco-Garcia, E., M. Menendez-Guterrez, M. Castro-Galache, J. Ferragut and M. Saceda, “Histone Deacetylase Inhibitors Induced Caspase-Independent Apoptosis in Human Pancreatic Adenocarcinoma Cell Lines,” Molecular Cancer Therapeutics, Vol. 4, No. 8, 2005, pp. 1222-1230. doi:10.1158/1535-7163.MCT-04-0186
- M. Parreno, I. Casanova, M. Cespedes, J. Vaque, M. Pavon, J. Leon and R. Mangues, “Bobel-24 and Derivatives Induce Caspase-Independent Death in Pancreatic Cancer Regardless of Apoptotic Resistance,” Cancer Research, Vol. 68, No. 15, 2008, pp. 6313-6323. doi:10.1158/0008-5472.CAN-08-1054
- Y. Jan, M. Matter, J. T. Pai, Y. L. Chen, J. Pilch, M. Komatsu, E. Ong, M. Fukuda and E. Ruoslahti, “A Mitochondrial Protein, Bit1, Mediates Apoptosis Regulared by Integrins and Groucho/TLE Corepressors,” Cell, Vol. 116, No. 5, 2004, pp. 751-762. doi:10.1016/S0092-8674(04)00204-1
- H. Biliran, Y. Jan, R. Chen, E. B. Pasquale and E. Ruoslahti, “Protein Kinase D is a Positive Regulator of Bit1 Apoptotic Function,” Journal of Biological Chemistry, Vol. 283, No. 42, 2008, pp. 28029-28037. doi:10.1074/jbc.M803139200
- C. Brunquell, H. Biliran, H. Tram, S. K. Ireland and E. Ruoslahti, “TLE1 Is an Anoikis Regulator and Is Downregulated by Bit1 in Breast Cancer Cells,” Molecular Cancer Research, Vol. 10, No. 11, 2012, pp. 1482-1495. doi:10.1158/1541-7786.MCR-12-0144
- P. P. Karmali, C. Brunquell, H. Tram, S. K. Ireland, E. Rouslahti and H. Biliran, “Metastasis of Tumor Cells Is Enhanced by Downregulation of Bit1,” PLoS One, Vol. 6, No. 8, 2011, Article ID: e23840. doi:10.1371/journal.pone.0023840
- H. Biliran, Y. Wang, S. Banerjee, H. Xu, H. Heng, A. Thakur, A. Bollig, F. H. Sarkar and J. D. Liao, “Overexpression of Cyclin D1 Promotes Tumor Cell Growth and Confers Resistance to Cisplatin-Mediated Apoptosis in an Elastase-Myc Transgene-Expressing Pancreatic Tumor Cell Line,” Clinical Cancer Research, Vol. 11, No. 16, 2005, pp. 6075-6086. doi:10.1158/1078-0432.CCR-04-2419
- S. M. Frisch and H. Frances, “Disruption of Epithelial Cell-Matrix Interactions Induce Apoptosis,” Journal of Cell Biology, Vol. 124, No. 4, 1994, pp. 619-626. doi:10.1083/jcb.124.4.619
- S. M. Frisch and E. Ruoslahti, “Integrins and Anoikis,” Current Opinion in Cell Biology, Vol. 9, No. 5, 1997, pp. 701-706. doi:10.1016/S0955-0674(97)80124-X
- C. J. Yeo, R. A. Abrams, L. B, Grochow, T. A. Sohn, S. E. Ord, R. H. Hruban, M. L. Zahurak, W. C. Dooley, J. Coleman, P. K. Sauter, H. A. Pitt, K. D. Lillemoe and J. L. Cameron, “Pancreaticoduodenectomy for Pancreatic Adenocarcinoma: Postoperative Adjuvant CHemoradiation Improves Survival: A Prospective, Single-Institution Experience,” Annals of Surgery, Vol. 225, No. 5, 1997, pp. 621-636. doi:10.1097/00000658-199705000-00018
- M. Donadelli, C. Costanzo, S. Beghelli, et al., “Synergistic Inhibition of Pancreatic Adenocarcinoma Cell Growth by Trichostatin A and Gemcitabine,” Biochimica et Biophysica Acta, Vol. 1773, No. 7, 2007, pp. 1095-1106. doi:10.1016/j.bbamcr.2007.05.002
Abbreviations
Bit1, Bcl-2 inhibitor of transcription
NOTES
*Corresponding author.