Natural Science
Vol.06 No.09(2014), Article ID:45820,12 pages
10.4236/ns.2014.69062
Comparison of Protein Expression Profiles of Novel Halomonas smyrnensis AAD6T and Halomonas salina DSMZ 5928T
Aydan Salman Dilgimen1,2,3*, Kazim Yalcin Arga1*, Volker A. Erdmann2#, Brigitte Wittmann-Liebold4#, Aziz Akin Denizci5, Dilek Kazan1*†
1Bioengineering Department, Faculty of Engineering, Marmara University (Goztepe Campus), Istanbul, Turkey
2Institute for Chemistry/Biochemistry, Freie University, Berlin, Germany
3Present Address: Department of Biochemistry & Molecular Biology, University of Calgary, Calgary, Canada
4Wita GmbH, Berlin, Germany
5The Scientific and Technological Research Council of Turkey (TUBITAK), Marmara Research Center (MAM), Genetic Engineering and Biotechnology Institute, Gebze, Turkey
Email: †dkazan@marmara.edu.tr
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 5 January 2014; revised 5 February 2014; accepted 12 February 2014
ABSTRACT
In this work, the protein pattern of novel Halomonas smyrnensis AAD6T was compared to that of Halomonas salina DSMZ5928T, which is the closest species on the basis of 16S rRNA sequence, to understand how AAD6T differs from type strains. Using high resolution NEPHEGE technique, the whole cell protein composition patterns of both Halomonas salina DSMZ5928T and H. smyrnensis AAD6T were mapped. The expressed proteins of the two microorganisms were mostly located at the acidic side of the gels, at molecular weight values of 60 to 17 kDa, and at isoelectric points 3.8 to 6.0, where they share a significant number of common protein spots. Identification and characterization of protein spots via whole genome sequencing data indicated that these two microorganisms used similar pathways, especially TCA cycle, for their survival; in other words, for their energy requirements. On the other hand, the protein expression differences in AAD6T and H. salina DSMZ 5928T showed that they prefer different metabolic pathways for lipid biosynthesis and in adaptation to extreme environments. Thus, we suggested that phylogenetic dissimilarities between these microorganisms could be related to the protein expression differences; in other words, metabolic flux differences in AAD6T and H. salina DSMZ 5928T. This is the first study to explain the dissimilarities of phenotypic characters and DNA-DNA hybridization between type strain and novel strain AAD6T by using protein expression differences.
Keywords:
Proteomics, Halomonas salina DSMZ 5928T, Halomonas smyrnensis AAD6T, NEPHGE Technique, Genome

1. Introduction
All over the world, an important part of the diversity of life are microorganisms, especially extremophilic microorganisms that have added a new dimension to biodiversity and could represent the direct descending of ancestral forms of life [1] . Due to their ability to adapt to very hard environmental conditions like extreme temperature, pH, pressure and salinity, some of the extremophiles may be considered as “living fossils” [2] [3] . According to the requirements of the extreme parameters (temperature, pH, pressure and salinity) for survival; psychrophiles, thermophiles, acidophiles, alkaliphiles, barophiles and halophiles are the more common phenotypes. Within this group, moderately halophilic microorganisms have been in the center of industrial interest in the last decades owing to their growth in a wide range of salt concentrations.
Moderately halophilic novel bacterium, Halomonas smyrnensis AAD6T from Çamalti saltern, Turkey, produces levan, a fructose homopolymer with many potential uses in various industries [4] [5] . Halomonas smyrnensis is Gram-negative, facultatively aerobic, rod-shaped and exopolysaccharide (levan) producing bacterium. It is a moderate halophile that grows optimal at 10% NaCl but even tolerates NaCl concentration up to 25%. It requires magnesium ions for growth, but it doesn’t require 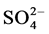 or K1+. 16S rRNA gene sequence similarities between strain AAD6T and type strains were 99.4% for Halomonas salina F8-11T, 99.4% for Halomonas halophila CCM 3662T, 98.1% for Halomonas maura S-31T, 98.0% Halomonas organivorans G-16.1T, 97.4% for Halomonas koreensis SS20T, 97.2% for Halomonas elongata DSM 2581T and 97.1% for Halomonas EX nitroreducens 11ST [4] [5] . Phylogenetic analysis of the draft genome of H. smyrnensis [6] showed that Halomonas salina (99.5%) and Halomonas halophila (99.5%) were found to be the closest species on the basis of 16S rRNA sequence comparison. However, according to DNA-DNA hybridization and phenotypic characteristics, strain AAD6T is distinguished from the type strains of closely related species as Halomonas salina, Halomonas halophila and other species of the genus Halomonas. For example, strain AAD6T differs from H. salina in the hydrolysis of tyrosine, in the absence of phenylalanine deaminase activity and for the reduction of nitrate. Additionally, strain AAD6T was oxidase-negative and able to hydrolyse starch, casein and Tween 80, while H. salina, was not. Also, strain AAD6T produced acid from glucose and mannose, while H. salina did not.
or K1+. 16S rRNA gene sequence similarities between strain AAD6T and type strains were 99.4% for Halomonas salina F8-11T, 99.4% for Halomonas halophila CCM 3662T, 98.1% for Halomonas maura S-31T, 98.0% Halomonas organivorans G-16.1T, 97.4% for Halomonas koreensis SS20T, 97.2% for Halomonas elongata DSM 2581T and 97.1% for Halomonas EX nitroreducens 11ST [4] [5] . Phylogenetic analysis of the draft genome of H. smyrnensis [6] showed that Halomonas salina (99.5%) and Halomonas halophila (99.5%) were found to be the closest species on the basis of 16S rRNA sequence comparison. However, according to DNA-DNA hybridization and phenotypic characteristics, strain AAD6T is distinguished from the type strains of closely related species as Halomonas salina, Halomonas halophila and other species of the genus Halomonas. For example, strain AAD6T differs from H. salina in the hydrolysis of tyrosine, in the absence of phenylalanine deaminase activity and for the reduction of nitrate. Additionally, strain AAD6T was oxidase-negative and able to hydrolyse starch, casein and Tween 80, while H. salina, was not. Also, strain AAD6T produced acid from glucose and mannose, while H. salina did not.
Proteomics are widely used to analyze the expressional changes of bacterial proteins at defined physiological conditions. Identification of differentially expressed proteins under given physiological conditions by proteomic analysis has gained fundamental importance for functional studies of cellular processes in recent years [7] . High-resolution two-dimensional electrophoresis (2-DE) is still one of the most powerful methods to separate thousands of proteins at once [8] . The availability of genome sequence information for a microorganism is crucial in proteomics studies, since the microbial genome sequence is the starting point for detailed analysis of identifying gene-protein associations.
In order to understand the dissimilarities between AAD6T and type strain, we compared the differential expression profiles of Halomonas smyrnensis AAD6T with closely related species Halomonas salina DSMZ 5928T on the basis of 16S rRNA sequence comparison. In this work, 2-DE by the NEPHGE technique [9] [10] followed by MALDI-TOF/MS, NANO-LC-ESI-Q TOF MS/MS, N-terminal sequencing and draft genome sequence of Halomonas smyrnensis AAD6T were used to compare the differential expression profiles of Halomonas salina DSMZ 5928T and Halomonas smyrnensis. This is the first study comparing the protein expression profiles of novel species and type strain to understand how closest species on the basis of 16S rRNA sequence differ from one another according to DNA-DNA hybridization and phenotypic characteristics.
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
Halomonas salina (DSMZ No 5928T) was supplied from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Germany. AAD6T was isolated from the soil samples of Çamalti saltern area near Izmir, Turkey [4] . Both microorganisms were cultured by shaking at 180 rpm at 37˚C in 250 ml flasks containing 100 ml of Brown medium containing 0.5% (w/v) yeast extract, 0.3% (w/v) trisodium citrate, 2% (w/v) magnesium sulphate heptahydrate, 0.2% (w/v) potassium chloride, 5% (w/v) sodium chloride at a pH of 7.0.
2.2. Sample Preparation and 2-DE Separation of Bacterial Proteins
Samples were harvested at their late exponential phase by centrifugation at 5000 rpm for 10 minutes at 4˚C and were washed two times with 50 mM Tris buffer (pH 7.8). The whole protein extractions of H. salina DSMZ 5928T and AAD6T were carried out by the SIGMA total protein extraction kit (SIGMA PROT-TOT) according to the manufacturer’s instructions.
Protein concentrations of the whole cell extracts were determined by employing the Bradford protein assay [11] , using BSA as a protein standard. Prepared samples were stored at −80˚C until their application to 2-DE. Bacterial proteins were separated by large gel 2-DE PAGE (30 × 23 × 0.15 cm) with the NEPHGE technique as described by Klose and Kobalz [12] . In the first dimension, bacterial samples were separated on capillary rods (for analytical gels, capillaries with inner diameter of 0.9 mm, and for preparative gels 1.5 mm inner diameter were used with lengths of 23 cm) using polyacrylamide gels containing 9 M urea, 3.5% acrylamide, 0.3% piperazine diacrylamide and 4% ampholyte mixture providing a pH range of 3 - 7.5. For the analytical and preparative gels, 80 µg and 150 µg of whole cell protein extracts were loaded respectively. The electrophoresis was run starting from 100 V for 60 minutes following 200 V for 60 minutes, 400 V 1050 minutes, 600 V 60 minutes and 1000 V for 45 minutes, respectively. At the end of the run, samples were incubated for 10 minutes at room temperature with gentle shaking in 125 mM Tris buffer containing 40% Glycerin (w/v), 3% SDS (w/v) and 64 mM DTT.
For the second dimensional, sodium dodecyl sulphate-PAGE was performed in 15% separating gels. SDS- PAGE electrophoresis was performed at 65 mA for 15 minutes followed by 100 mA for 400 minutes. The analytical gels were silver stained [13] and preparative gels were stained with coomassie G-250. The gels of each sample were dried between cellophanes under vacuum. Dried samples were scanned and analysed.
2.3. Mass Spectrometer Analyses for Protein Identification
Protein gel spots of interest were excised from the gels with a scalpel and transferred to clean 500 µl Eppendorf tubes. Trypsin solution at a concentration of 0.4 µg/µl was added to the gel pieces and incubated overnight at 37˚C to digest. Trypsin-digested proteins were eluted with a buffer containing 85% MeCN/0.1% TFA. Extracts were dried by a speed-vac concentrator and kept at −20˚C until further analysis by mass spectromety.
Two different MS techniques were applied to various selected protein samples. One of the MS method applied was MALDI-TOF/MS (Waters/Micromass) with a positive ion mode reflection system. The digested and dried protein samples were dissolved in elution buffer containing 85% acetonitrile and 0.1% TFA, mixed 1:1 (v/v) with a freshly prepared matrix solution and spotted onto the MALDI target. Peptides were analysed with MALDI-TOF/MS.
The other MS technique applied was NANO-LC-ESI-Q TOF MS/MS (Micromass). The dried and digested samples were resolved with 0.1% formic acid solution and injected into the sample loop of the MS system. Mass spectra of the samples were recorded in the Nano LC ESI-QTOF MS/MS mass spectrometer. The peptide fingerprint spectra of proteins were identified in the protein database using the MASCOT search engine, http://www.matrixscience.com.
2.4. N-Terminal Sequencing and Genome Sequence of H. smyrnensis AAD6T
Following the two-dimensional electrophoresis, gels of newly isolated Halomonas smyrnensis AAD6T were blotted on PVDF membranes with a semi dry blotter (Bio-Rad) using Towbin buffer. The blotting conditions were 300 mA for two hours in the cold room. Blotted membranes were stained using coomassie R-250, dried and kept in a clean environment until N-Terminal sequencing. CBB-R 250 stained protein spots on PVDF membranes were cut out very carefully with a scalpel and placed into the sequencer cartridge. After applying the membrane to the cartridge, Applied Biosystems Model 492A Procise sequencer (WITA GmbH, Teltow, Germany) was run automatically according to the manufacturer’s directions.
The genome sequence of H. smyrnensis AAD6T was recently published [6] and deposited at DDBJ/EMBL/ GenBank under the accession AJKS00000000. The sequence data used in the present study is the second version, with accession numbers AJKS02000001 to AJKS02000034. Based on the sequence data, gene prediction and genome annotation were carried out using the RAST auto-annotation server [14] . The gene function and classifications were based on the subsystem annotation of the RAST server. Information on enzyme encoding genes was taken from Kyoto Encyclopedia of Genes and Genomes (KEGG) [15] and Expasy databases [16] . Transport protein coding genes were annotated using the similarity searches against the Transfer Classification Database (TCDB) [17] .
3. Results and Discussion
During the last decade, the extensive studies on hypersaline environments carried out in many geographical areas have permitted the isolation and taxonomic characterization of a large number of moderately halophilic species. Although some Gram-negative species were considered members of different genera, phenotypic and phylogenetic data support their close relationship, and they are currently included in the family Halomonadaceae as members of two genera: Halomonas and Chromohalobacter [18] [19] . Halomonas smyrnensis strain AAD6T represents a novel species within the genus Halomonas according to physiological, biochemical and phylogenetic properties. Although, there is 99.4% 16S rRNA gene sequence similarity between strain AAD6T and Halomonas salina DSMZ 5928T [4] , there are many dissimilarities between phenotypic characters of type strain and strain AAD6T. Proteome analysis, comparative profiling of protein, is one of the powerful tools to analyse and manipulate the microorganisms, since most cellular metabolic activities are directly or indirectly mediated by proteins. So that, in order to elucidate the differences in protein level, we analysed the protein expression patterns of newly isolated moderately halophilic bacteria AAD6T and those of H. salina DSMZ 5928T. Because omics data alone is not enough to understand cellular physiology and regulatory mechanisms [20] , we combined proteomics data with genome data of Halomonas smyrnensis AAD6T.
In this work, the NEPHGE technique was used to carry out the first dimension of 2-DE gel electrophoresis. Cho and his group [21] studied 2-DE electrophoresis of Halobacterium salinarum by IPG strips and they obtained poor resolution with IPG strips. Shukla [22] studied Halobacterium NRC-I also with the 2-DE IPG strip technique and modified the 2-DE procedure in order to improve resolution and to minimize streaking of proteins in the acidic range. He also noted that the common unmodified 2-DE procedure resulted in horizontal and vertical streaking at the high MW range. In this work, by using the high resolution NEPHEGE technique, proteins were well resolved in the acidic range predominant for halophilic organisms and very few streaking effects were observed (Figure 1 and Figure 2).
Protein patterns for Halomonas salina DSMZ5928T and H. smyrnensis AAD6T are documented in Figure 1 and Figure 2. It can be seen from the gels that the whole cell protein composition patterns were mostly located at the acidic side of the gels. A number of studies have suggested that the halophilic adaptation correlates with an increase in acidic amino acids in the protein composition [23] . The fraction of acidic residues is extremely large in the surface composition of the halophilic proteins. The large number of acidic residues on the surface of halophilic proteins has been rationalized on the basis of their superior water-binding abilities in the charged forms [24] . An increase in salt concentration from 1 M to 5 M, increases the pKa value of aspartic acid from 4.0 to 4.9, and that of glutamic acid from 4.4 to 5.3 [25] and decreases the population of the charged forms of these acidic residues at the neutral pH.
At the studied conditions, the expressed proteins of the two microorganisms were mostly located at molecular weight values of 60 to 17 kDa and at isoelectric points 3.8 to 6.0 where they share a number of common protein spots on their 2-DE profiles. However there were some proteins that were not expressed by H. salina DSMZ 5928T while produced by AAD6T, or visa versa. This is not a surprise that phylogenetically related organisms have many proteins in common, while this number is lower for more distantly related species [23] .
As given in Table 1, 11 proteins as A1, A2, A3, A4, A5, A6, A7, A8, A9, A10 and A11 are common proteins expressed by both H. salina DSMZ 5928T and Halomonas smyrnensis AAD6T. From the common proteins identified, both from H. salina DSMZ 5928T and strain AAD6T, protein spot A1 was a pore forming outer membrane protein precursor which was known to take a role in porin formation for solute diffusion. This protein showed high similarities to the major outer membrane protein precursor from various microorganisms. A2, A3, A4, A5 and A7 were identified as aconitate hydratase, succinate semialdehyde dehydrogenase, Acetyl-CoA Acetyltrans- ferase, Malate dehydrogenase and Acetyl CoA synthase. These enzymes are responsible in the energy metabol- ism. Aconitate hydratase (A2) is an enzyme that catalyses the stereospecific isomerization of citrate to isocitrate

Figure 1. Silver stained 2-DE gel of H. Salina DSMZ5928T with the numbered spots showing the identified proteins.

Figure 2. Silver stained 2-DE gel of Halomonas sp. AAD6T with the numbered spots showing the identified proteins.

Table 1. Overview of H. salina DSMZ 5928T and H. smyrnensis AAD6T proteins.
Spot numbers coded with “A” identified from both microorganisms, *Protein spots belong to H. salina DSMZ5928T; **Protein spots belong to H. smyrnensis AAD6T; aWGS represent the whole genome sequence of H. smyrnensis AAD6 T. The genome identifiers in the form of peg.X are based on Sogutcu et al., 2012. [6] ; bScore is −10*Log(P), where P is the probability that the observed match is a random event. Protein scores greater than 50 are significant (p < 0.05) according to NCBI Nucleic acid database.
via cis-aconitate in the tricarboxylic acid (TCA) cycle. It serves as a protective buffer against oxidative stress [26] . Succinate semialdehyde dehydrogenase (A3) participates in the degradation of glutamate, while Acetyl- CoA Acetyltransferase participate in pyruvate metabolism. Acetyl-CoA C-Acetyltransferase belonging to the thiolase family which catalyzes the thiolysis of a linear fatty acid CoA Acetoacetyl-CoA thiolase (also called thiolase II) is specific for the thiolysis of acetoacetyl-CoA and involved in biosynthetic pathways such as poly β-hydroxybutyrate synthesis or steroid biogenesis. Its main function is the synthesis of acetoacyl-CoA from two molecules of acetyl-CoA, which shows its importance in several biosynthetic pathways.
Malate and oxaloacetate of the citric acid cycle were the key metabolites that served as the gateway to gluco- neogenesis. Succinic semialdehyde dehydrogenase catalyses the (NAD (P) +)-dependent catabolism reaction of succinic semialdehyde to succinate for metabolism by the TCA cycle. Spot A5 was identified as malate dehy- drogenase, which participates in the TCA cycle, and which belongs to the MDH (malate dehydrogenase) type 2 family of LDH/MDH (Lactate dehydrogenase/Malate dehydrogenase) superfamily and catalyzes the reversible oxidation of malate to oxaloacetate utilizing the NAD/NADH cofactor system.
Acetate-CoA ligase belonging to the ATP-dependent AMP-binding enzyme family (also known as acetate- CoA synthetase and acetyl-activating enzyme) is an ubiquitous enzyme, found in both prokaryotes and eukaryotes, which catalyses the formation of acetyl-CoA from acetate, coenzyme A (CoA) and ATP [27] . The activity of this enzyme is crucial for maintaining the required levels of acetyl-CoA, a key intermediate in many important biosynthetic and catabolic processes (some prokaryotic species can also activate acetate by either acetate kinase/phosphor-transacetylase or by ADP-forming acetyl-CoA synthase).
Spot A6 is a periplasmic phosphate binding protein, which is found in the periplasmic space of Gram-negative bacteria and serves as an initial high affinity precursor in the uptake of specific nutrient phosphate. Spot A8 is a dipeptide binding ABC transporter. These proteins work with the ABC transport system, in either direction and they can also function in the initiation of sensory transduction pathways.
Another common protein was putative cobalt-precorrin-6A synthase (spot A9) which may catalyze the methylation of C-1 in cobalt-precorrin-5 and the subsequent extrusion of acetic acid from the resulting intermediate to form cobalt-precorrin-6A, taking role mainly in cofactor biosynthesis. Protein A10 was found as electron transfer flavoprotein (ETFs) beta-subunit serving as specific electron acceptors for primary dehydrogenases, transferring the electrons to terminal respiratory systems. Group II ETFs produced by some prokaryotes under specific growth conditions; receive electrons only from the oxidation of specific substrates [28] . ETFs are heterodimeric proteins containing an FAD cofactor and AMP [29] - [31] . FAD is bound in a cleft between domains II and III, while domain III binds the AMP molecule. Interactions between domains I and III stabilize the protein, forming a shallow bowl where domain II resides. Spot A11 which was identified as a single-strand binding protein, is also known as the helix-destabilizing protein. It binds tightly, as a homotetramer, to single-stranded DNA (ss-DNA) and plays an important role in DNA replication, recombination and repair.
Common proteins expressed by H. salina DSMZ 5928T and H. smyrnensis AAD6T show that these two microorganisms use similar pathways, especially the TCA cycle, for their survival, in other words, for their energy requirements.
We also searched for proteins in Halomonas smyrnensis AAD6T, which were not specifically expressed by H.salina DSMZ 5928T. The protein spots 626 and 60011 were identified as enzymes taking a role in ectoine synthesis. Spot 626 was identified as aspartate semialdehyde dehydrogenase, which is found in the cytoplasm, it belongs to the oxidoreductase. Other known names of the enzyme are aspartic semialdehyde dehydrogenase, and L-aspartate-beta-semialdehyde dehydrogenase. This protein takes roles in lysine biosynthesis I, homoserine biosynthesis, and ectoine biosynthesis. The protein has also an oxidoreductase activity, acting on the aldehyde or oxo group of donors. Spot 60011 showed high similarity to L-ectoine synthase (fragment), which is present in various halophilic bacteria and archaea in blast search. This protein belongs to the ectoine synthase family, which plays a role in ectoine synthesis [32] [33] and which is activated by NaCl. It also takes a role in amine and polyamine biosynthesis catalyzing the circularization of gamma-N-actyl-alpha-gamma-diaminobutric acid to ectoine (1,4,5,6-tetrahydro-2-methyl-4-pyrimidine carboxylic acid), which is an excellent osmo-protectant.
The protein spots 62, 617, 629 and 60013 were identified as proteins taking a role in defense mechanisms, which act under stress conditions. Spot 62 showed similarity to alkyl hydroperoxide reductase (AhpC), which is responsible for directly reducing organic hyperoxides in their reduced dithiol form. Thiol specific antioxidant (TSA) is a physiologically important antioxidant, which constitutes an enzymatic defense against sulphur-con- taining radicals in blast search. It is known that it is induced by heat shock, salt stress, oxidative stress and glucose limitation. Spot 617 showed high similarity to universal stress protein A (USP A), which is a small cytoplasmic bacterial protein whose expression is enhanced when the cell is exposed to stress agents by blast search of N-terminal sequencing. UspA enhances the rate of cell survival during prolonged exposure to such conditions, and may provide a general “stress endurance” activity. Spot 629 was identified as Type I restriction-modifica- tion system, M subunit, which protects a bacterial cell against invasion of foreign DNA by endonucleolytic cleavage of DNA that lacks a site-specific modification. The R-M system is a complex containing three polyeptides as M, S and R [34] . The M and S subunits together form a methyltransferase that methylates two adenine residues in complementary strands of a DNA recognition sequence. When the target site is unmodified the DNA is cut, when semimethylated, the complex acts as a maintenance methyltransferase and both strands become methylated. Spot 60013 showed high similarity to superoxide dismutase, which destroys radicals normally produced within the cells and which are toxic to biological systems. SODs (Superoxide dismutases) catalyse the conversion of superoxide radicals to molecular oxygen. Fe/Mn SODs are ubiquitous enzymes that are responsible for the majority of SOD activity in prokaryotes.
Protein spot 623 was identified as dihydrolipoamide dehydrogenase, which belongs to the disulfide oxidoreductase family and also has several other names such as lipoamide dehydrogenase, lipoamide oxidoreductase, dehydrolipoate dehydrogenase. It is a cytoplasmic enzyme, which is the E3 component of dehydrogenase complexes for pyruvate, 2-oxoglutarate, 2-oxoisovalerate, and acetoine. It can also serve as the L protein of the glycine cleavage system. The protein takes part in isoleucine degradation I, valine degradation I, TCA cycle, glycine cleavage complex, and pyruvate dehydrogenase. Protein spot 624 was identified as translation elongation factor TU Tu which is a member of the G-protein superfamily clan, EF-Tu/EF-1A subfamily. Elongation factors belong to a family of proteins that promote the GTP-dependent binding of aminoacyl tRNA to the A site of ribosomes during protein biosynthesis, and catalyze the translocation of the synthesized protein chain from the A to the P site. This protein promotes the GTP-dependent binding of aminoacyl-tRNA to the A-site of ribosomes during protein biosynthesis. EF1A (or EF-Tu) is responsible for the selection and binding of the cognate aminoacyl-tRNA to the A-site (acceptor site) of the ribosome. EF2 (or EF-G) is responsible for the translocation of the peptidyl-tRNA from the A-site to the P-site (peptidyl-tRNA site) of the ribosome, thereby freeing the A-site so that the next aminoacyl-tRNA can bind to the A-site. Elongation factors are responsible for achieving accuracy of translation and both EF1A and EF2 are remarkably conserved throughout evolution. Spot 60021 showed high similarity to the succinyl-CoA ligase [ADP-forming] subunit alpha, which belongs to the succinate/malate CoA ligase alpha subunit family. This enzyme is a bacterial enzyme that during aerobic metabolism functions in the TCA cycle, coupling the hydrolysis of succinyl-CoA to the synthesis of ATP.
Spots 602 was chaperon protein DnaJ. Chaperone DnaJ, Hsp40 (heat shock protein 40 kD), is a molecular chaperone protein and plays a role in regulating the ATPase activity of Hsp70 heat-shock proteins. Spot 606 was identified as DNA mismatch repair protein, which is involved in the repair of mismatches in DNA and it has also a weak ATPase activity. Sports 608 was determined as pantothenate kinase type III. Pantothenate kinase, an essential enzyme in bacteria and eukaryotes, is involved in catalysing the first step of conversion of pantothenate to coenzyme A (CoA). Three isoforms (type I, II and III) of this enzyme have been reported from various organisms, which can be differentiated from each other on the basis of their biochemical and structural characteristics. Spot 609 was DNA polymerase III subunit gamma and tau. The tau and gamma subunits of DNA polymerase III holoenzyme are both products of the dnaX gene. Since tau and gamma are required as stoichiometric components of the replicative complex, a mechanism must exist for the cell to coordinate their synthesis and ensure that both subunits are present in an adequate quantity and ratio for assembly [35] .
Seven more proteins from Halomonas salina DSMZ 5928T spot 2821, 2822, 2823, 281, 284, 289 and 294 were not expressed by Halomonas smyrnensis AAD6T, although the genes responsible for these proteins are present in H. smyrnensis AAD6T. Spot 2823 was identified as nucleoside diphosphate kinase, which is required for the synthesis of nucleoside triphosphates (NTP) other than ATP. They provide NTPs for nucleic acid synthesis, CTP for lipid synthesis, UTP for polysaccharide synthesis and GTP for protein elongation, signal transduction and microtubule polymerization. Spot 2821 and 2822 are identified as hypothetical proteins, of which spot 2821 has a probable function in type IV secretion [36] , while 2822 has so far an unknown function. 281, 284, 289 and 294 were identified as malonate transporter, Enoyl-CoA hydratase, DNA ligase, cysteine desulfurase and cystdylate kinase. Enoyl-CoA hydratase is essential to metabolizing fatty acids to produce both acetyl CoA and energy. Cysteine desulfurases are applicable to the production of cofactors and the bioconversion of useful compounds. Cystdylate kinase has a role in pyrimidine metabolism.
The protein expression differences in AAD6T and H. salina DSMZ 5928T showed that these microorganisms prefer different metbolic pathways for lipid synthesis and adaptation to extreme environments. Consequently, although, there are 99.4% 16S rRNA gene sequence similarities between strain AAD6T and Halomonas salina DSMZ 5928T [4] , dissimilarities between phenotypic characters of both strains is a result of metabolic flux differences.
4. Conclusions
Comparing the protein expression of Halomonas smyrnensis AAD6T with that of Halomonas salina DSMZ 5928T and comparing that with the genome sequence of strain AAD6T helped us to understand how the microorganism differs from one another, although there were 16S rRNA gene sequence similarities between both strains. Experimental results indicated that as well as common proteins, different proteins were also expressed by AAD6T and H. salina DSMZ 5928T.
The protein expression differences in AAD6T and H. salina DSMZ 5928T were related to phylogenetic dissimilarities between these microorganisms. This is not a surprise that specific changes in the metabolism of the organism cause to change the protein expression pattern and the number of common proteins is lower for more distantly related species. To our knowledge, this is the first study to give an explanation for the dis- similarities of phenotypic characters and DNA-DNA hybridizatios between type strain and novel strain AAD6T by using protein expression differences.
Acknowledgements
This research was supported by The Scientific and Technical Research Council of Turkey, Engineering Group (TUBITAK-MAG) by the Project No. 104M236. The authors wish to thank Dr. Albrecht Otto (Max-Delbrück Centrum, Berlin-Buch), Dr. Christoph Radcke and Dr. Thomas Pohl (WITA GmbH, Teltow-Berlin) for their kind help in mass spectrometry and N-terminal analysis. The whole genome sequencing data of H. smyrnensis AAD6T was obtained under financial support by The Scientific and Technological Research Council of Turkey (TUBITAK) through Project No. 110M613.
References
- Winker, S. and Woese, C.R. (1991) A Definition of the Domains Archaea, Bacteria and Eucarya in Terms of Small Subunit Ribosomal RNA Characteristics. Systematic and Applied Microbiology, 14, 305-310. http://dx.doi.org/10.1016/S0723-2020(11)80303-6
- Madigan, M.T. and Oren, A. (1999) Thermophilic and Halophilic Extremophiles. Current Opinion in Microbiology, 2, 265-269. http://dx.doi.org/10.1016/S1369-5274(99)80046-0
- Ventosa, A., Nietro, J.J. and Oren, A. (1998) Biology of Moderately Halophilic Aerobic Bacteria. Microbiology and Molecular Biology Reviews, 62, 504-544.
- Poli, A., Nicolaus, B., Denizci, A.A., Yavuzturk, B. and Kazan, D. (2013) Halomonas smyrnensis sp. nov., a Moderately Halophilic, Exopolysaccharide-Producing Bacterium. IJSEM, 63, 10-18.
- Poli, A., Kazak, H., Gürleyendağ, B., Tommonaro, G., Pieretti, G., Oner, E.T. and Nicolaus, B. (2009) High Level Synthesis of Levan by a Novel Halomonas Species Growing on Defined Media. Carbohydrate Polymers, 78, 651-657. http://dx.doi.org/10.1016/j.carbpol.2009.05.031
- Sogutcu, E., Emrence, Z., Arikan, M., Cakiris, A., Abaci, N., Öner, E.T., Üstek, D. and Arga, K.Y. (2012) Draft Genome Sequence of Halomonas Smyrnensis AAD6T. Journal of Bacteriology, 194, 5690. http://dx.doi.org/10.1128/JB.00559-12
- Molloy, M.P., Herbert, B.R., Walsh, B.J., Tyler, M.I., Traini, M., Sanchez, J.C., Hochstrasser, D.F., Williams, K.L. and Gooley, A.A. (1998) Extraction of Membrane Proteins by Differential Solubilization for Separation Using Two-Di- mensional Gel Electrophoresis. Electrophoresis, 19, 837-844. http://dx.doi.org/10.1002/elps.1150190539
- Wittmann-Liebold, B., Graack, H.R. and Pohl, T. (2006) Two-Dimensional Gel Electrophoresis as Tool for Proteomics Studies in Combination with Protein Identification by Mass Spectrometry. Proteomics, 6, 4688-4703. http://dx.doi.org/10.1002/pmic.200500874
- Neuhoff, V., Stamm, R. and Eibl, H. (1985) Clear Background and Highly Sensitive Protein Staining with Coomassie Blue Dyes in Polyacrylamide Gels: A Systematic Analysis. Electrophoresis, 6, 427-448. http://dx.doi.org/10.1002/elps.1150060905
- Switzer, R.C., Merril, C.R. and Shifrin, S. (1979) A Highly Sensitive Silver Stain for Detecting Proteins and Peptides in Polyacrylamide Gels. Analytical Biochemistry, 98, 231-237. http://dx.doi.org/10.1016/0003-2697(79)90732-2
- Bradford, M.M. (1976) A Rapid and Sensitive for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analytical Biochemistry, 72, 248-254. http://dx.doi.org/10.1016/0003-2697(76)90527-3
- Klose, J. and Kobalz, U. (1995) Two-Dimensional Electrophoresis of Proteins: An Updated Protocol and Implications for a Functional Analysis of the Genome. Electrophoresis, 16, 1034-1059. http://dx.doi.org/10.1002/elps.11501601175
- Jungblut, P. and Seifer, R. (1990) Analysis by High Resolution Two-dimensional Electrophoresis of Differentiation-dependent Alterations in Cytosolic Protein Pattern of HL-60 Leukemic Cells. Journal of Biochemical and Bio- physical Methods, 21, 47-58. http://dx.doi.org/10.1016/0165-022X(90)90044-D
- Aziz, R.K., Bartels, D., Best, A.A., DeJongh, M., Disz, T., et al. (2008) The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics, 9, 75. http://dx.doi.org/10.1186/1471-2164-9-75
- Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. and Tanabe, M. (2012) KEGG for Integration and Interpretation of Large-Scale Molecular Datasets. Nucleic Acids Research, 40, 109-114. http://dx.doi.org/10.1093/nar/gkr988
- Artimo, P., Jonnalagedda, M., Arnold, K., Baratin, D., Csardi, G., de Castro, E., Duvaud, S., Flegel, V., Fortier, A., Gasteiger, E., Grosdidier, A., Hernandez, C., Ioannidis, V., Kuznetsov, D., Liechti, R., Moretti, S., Mostaguir, K., Redaschi, N., Rossier, G., Xenarios, I. and Stockinger, H. (2012) ExPASy: SIB Bioinformatics Resource Portal. Nucleic Acids Research, 40, 597-603. http://dx.doi.org/10.1093/nar/gks400
- Saier, M.H., Yen, M.R., Noto, K., Tamang, D.G. and Elkan, C. (2009) The Transporter Classification Database (TCDB): Recent Advances. Nucleic Acids Research, 37, 274-278. http://dx.doi.org/10.1093/nar/gkn862
- Dobson, S.J. and Franzmann, P.D. (1996) Unification of the Genera Deleya, Halomonas, and Halovibrio and the Species Paracoccus halodendrificans into a Single Genus, Halomonas, and Placement of the Genus Zymobacter in the Family Halomonadaceae. International Journal of Systematic Bacteriology, 46, 550-558. http://dx.doi.org/10.1099/00207713-46-2-550
- Franzmann, P.D., Wehmeyer, U. and Stackebrandt, E. (1988) Halomonadaceae fam. nov., A New Family of the Class Proteobacteria to Accommodate the Genera Halomonas and Deleya. Systematic and Applied Microbiology, 11, 16-19. http://dx.doi.org/10.1016/S0723-2020(88)80043-2
- Lee, J.C., Jeon, C.O., Lim, J.M., Lee, S.M., Lee, J.M., Song, S.M., Park, D.J., Li, W.J. and Kim, C.J. (2005) Halomonas taeanensis sp. nov., a Novel Moderately Halophilic Bacterium Isolated from a Solar Saltern in Korea. IJSEM, 55, 2027-2032.
- Cho, C.W., Lee, S.H., Choi, J., Park, S.J., Ha, D.J., Kim, H.J. and Kim, C.W. (2003) Improvement of the Two Dimensional Gel Electrophoresis Analyses for the Proteome Study of Halobacterium salinarum. Proteomics, 3, 2325- 2329. http://dx.doi.org/10.1002/pmic.200300525
- Shukla, H.D. (2006) Proteomic Analyses of Acidic Chaperons and Stress Proteins in Extreme Halophile Halo- bacterium NRC-I: A Comparative Proteomic Aproach to Steady Heat Shock Response. Proteome Science, 4, 6-14. http://dx.doi.org/10.1186/1477-5956-4-6
- Saum, S.H., Pfeiffer, F., Palm, P., Rampp, M., Schuster, S.C., Müller, V. and Oesterhelt, D. (2012) Chloride and Organic Osmolytes: A Hybrid Strategy to Cope with Elevated Salinities by the Moderately Halophilic, Chloride-De- pendent Bacterium Halobacillus halophilus. Environmental Microbiology, 15, 1619-1633. http://dx.doi.org/10.1111/j.1462-2920.2012.02770.x
- Kuntz, I.D. (1971) Hydration of Macromolecules: III. Hydration of Polypeptides. Journal of the American Chemical Society, 93, 514-516. http://dx.doi.org/10.1021/ja00731a036
- Elock, A.G. and McCommon, J.A. (1998) Electrostatic Contributions to the Stability of Halophilic Proteins. Journal of Molecular Biology, 280, 731-748. http://dx.doi.org/10.1006/jmbi.1998.1904
- Isarankura-Na-Ayudhya, P., Isarankura-Na-Ayudhya, C., Yainoy, S., Thippakorn, C., Singhagamol, W., Polprachum, W., Roytrakul, S. and Prachayasittikul, V. (2010) Proteomic Alterations of Escherichia coli by Paraquat. EXCLI Jour- nal, 9, 108-118.
- Starai, V.J. and Escalante-Semerena, J.C. (2004) Acetyl-Coenzyme A Synthetase (AMP forming). Cellular and Molecular Life Sciences, 61, 2020-2030. http://dx.doi.org/10.1007/s00018-004-3448-x
- Weidenhaupt, M., Rossi, P., Beck, C., Fischer, H.M. and Hennecke, H. (1996) Bradyrhizobium japonicum Possesses Two Discrete Sets of Electron Transfer Flavoprotein Genes: fixA, fixB and etfS, etfL. Archives of Microbiology, 165, 169-178.
- Roberts, D.L., Frerman, F.E. and Kim, J.J. (1996) Three-Dimensional Structure of Human Electron Transfer Flavoprotein to 2.1-A Resolution. Proceedings of the National Academy of Sciences, 93, 14355-14360. http://dx.doi.org/10.1073/pnas.93.25.14355
- Roberts, D.L., Salazar, D., Fulmer, J.P., Frerman, F.E. and Kim, J.J. (1999) Crystal Structure of Paracoccus denitrificans Electron Transfer Flavoprotein: Structural and Electrostatic Analysis of a Conserved Flavin Binding Domain. Biochemistry, 38, 1977-1989. http://dx.doi.org/10.1021/bi9820917
- Leys, D., Basran, J., Talfournier, F., Sutcliffe M.J. and Scrutton, N.S. (2003) Extensive Conformational Sampling in a Ternary Electron Transfer Complex. Nature Structural Biology, 10, 219-25. http://dx.doi.org/10.1038/nsb894
- Canovas, D., Vargas, C., Csonka, L.N., Ventosa, A. and Nieto, J.J. (1998) Synthesis of Glycine Betaine from Exogenous Choline in the Moderately Halophilic Bacterium Halomonas elongate. Applied and Environmental Microbiology, 64, 4095-4097.
- Canovas, D., Vargas, C., Calderon, M.I., Ventosa, A. and Nieto, J.J. (3043) Characterization of the Genes for the Biosynthesis of the Compatible Solute Ectoine in the Moderately Halophilic Bacterium Halomonas elongata DSM. Systematic and Applied Microbiology, 21, 487-497. http://dx.doi.org/10.1016/S0723-2020(98)80060-X
- Schouler, C., Clier, F., Lerayer, A.L., Ehrlich, S.D. and Chopin, M.C. (1998) A Type IC Restriction-Modification System in Lactococcus lactis. Journal of Bacteriology, 180, 407-411.
- Flower, A. and McHenry, C. (1991) Transcriptional Organization of the Escherichia coli dnaX Gene. Journal of Molecular Biology, 220, 649-658. http://dx.doi.org/10.1016/0022-2836(91)90107-H
- Bell, K.S., Sebaihia, M., Pritchard, L., Holden, M.T., Hyman, L.J., Holeva, M.C., Thomson, N.R., Bentley, S.D., Churcher, L.J., Mungall, K., Atkin, R., Bason, N., Brooks, K., Chillingworth, T., Clark, K., Doggett, J., Fraser, A., Hance, Z., Hauser, H., Jagels, K., Moule, S., Norbertczak, H., Ormond, D., Price, C., Quail, M.A., Sanders, M., Walker, D., Whitehead, S., Salmond, G.P., Birch, P.R., Parkhill, J. and Toth, I.K. (2004) Genome Sequence of the Enterobacterial Phytopathogen Erwinia carotovora subsp. Atroseptica and Characterization of Virulence Factors. Proceedings of the National Academy of Sciences, 101, 11105-11110. http://dx.doi.org/10.1073/pnas.0402424101
NOTES

*Addresses belong to the Institutions where the study was conducted. #Currently Prof. Wittmann and Prof. Erdmann retired.
†Corresponding author.