Health
Vol.5 No.7A5(2013), Article ID:34900,7 pages DOI:10.4236/health.2013.57A5002
Down-regulation of Rho-kinases induce tolerance in Ischemic preconditioning model after transient cerebral ischemia/reperfusion in rats
![]()
Department of Physiology, Faculty of Medicine and Heath Sciences, Alneelain University, Khartoum, Sudan; h_alsheekh82@yahoo.com
Copyright © 2013 Hiba A. Awooda. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 30 April 2013; revised 31 May 2013; accepted 17 June 2013
Keywords: Cerebral; Ischemic Preconditioning; ROCK; NO
ABSTRACT
Background: Ischemic preconditioning (IPC) is a brief episode of ischemia/reperfusion (I/R) that protects the brain from the damage induced by subsequent prolonged ischemia. Aim: To study the neuroprotective mechanism of IPC. Methods: 30 adult male Wistar rats (150 - 250 g) were divided into three groups 10 rats in each; the first group was sham-operated and served as a control, I/R group of rats subjected to 30 minutes of left common carotid artery occlusion (CCAO) followed by 24-hour of reperfusion, IPC group were treated with three episodes of 5- minutes of CCAO with 10 minutes of reperfusion in between, followed by 30 minutes of CCAO and then allowed for reperfusion for 24 hours. Neurobehavioral assessments were evaluated; Rhokinases (ROCK) and nitrite were measured in affected cerebral hemisphere. Results: Rats’ neurological deficits were significantly decreased in the I/R compared with the control group (P < 0.001) whereas rats treated by precondition stimuli showed significant improvement in neurological deficit compared to I/R group (P < 0.001). Nitrite level was significantly increased in the IPC rats compared to both control and I/R groups (P < 0.001). In contrast, the ROCK level was significantly higher in I/R group compared to control group and its level significantly decreased in IPC rats when compared to I/R group (P < 0.001). ROCK correlates negatively with the nitrite (CC = −0.695, P = 0.000). Conclusions: Downregulation of ROCK level following preconditioning stimuli with the potential involvement of Nitric oxide (NO) appear to be one of the neuroprotective mechanisms of IPC protection against a subsequent I/R challenge evidence by improvement in the neurological deficits.
1. INTRODUCTION
Cerebral ischemic preconditioning (IPC) is an incident of brief episodes of ischemia that protects the brain from subsequent, more severe ischemic insult [1,2]. Two phases of protection arise in IPC; rapid preconditioning occurs quickly after the preconditioning stimuli and lasts for up to 1 h, the second phase is more delayed with prolonged time window that usually detectable 24 h after preconditioning, peaks at 3 days and fades after 7 days [3]. Short episodes of IPC stimuli may imitate some of the characteristics of clinical transient ischemic attacks (TIAs). In which a TIA occurring before a stroke could lead to delayed tolerance [4]. Hence, understanding which proteins are involved during delayed tolerance can provide insight into the mechanisms of ischemic damage and afford therapeutic alternatives [5].
The molecular protective mechanisms of IPC are not fully understood, although several potentials have been implicated such as: alterations in heat shock proteins, lipid peroxidation, inflammation and mitochondrial metabolism [6,7]. Preconditioned brains actively suppressed genes that control metabolism, cell cycle regulation and ion channel activity indicating a complete reprogramming of the genomic response to ischemia. Thus, it results in shift the outcome from cell death to cell survival [8].
Nitric oxide (NO) is a cellular signaling molecule synthesized by the action of one nitric oxide synthase (NOS) isoforms [9]; endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS) [10]. NO has been implicated in several models of cerebral preconditioning [11]. Rho-kinases (also known as ROCK) are serine/threonine kinases that exists as two isomers Rhokinase α/ROCK2 and Rho-kinase β/ROCK1 [12]. ROCK implicated in the regulation of vascular smooth muscle cell migration, proliferation and differentiation [13,14]. It has been demonstrated that levels of Rho are up-regulated in the brains of patients who have died following focal cerebral infarction as well as mouse model of ischemia [15,16].
There is evidence that activation of ROCK could down-regulate eNOS expression and worsen the endothelial function [17]. Based on these observations, the aim of the present work was to study the neuroprotective mechanisms of IPC on Rho-kinase (ROCK II) and NO levels in the brain homogenate of rats and to explore the possible correlations between ROCK, NO and the subsequent neurological deficits in rats.
2. MATERIALS AND METHODS
2.1. Animals
The animals were handled in accordance with the ethical standards laid down in the US National Institutes of Health (NIH Publication No. 85 - 23) and its later revisions. 30 Male Wistar rats, weighing 150 - 250 g were selected and preserved at a constant temperature of 22˚C ± 2˚C with a fixed 12:12-h light-dark cycle. Nutritionally balanced pellets and water were freely available.
2.2. Experimental Design
Three randomly divided experimental groups were used, of 10 rats each: 1) sham operated: which included full surgical preparation without common carotid artery occlusion (CCAO) serve as control group; 2) ischemia/ reperfusion (I/R) group: Brain ischemia was maintained for 30 minutes of left CCAO followed by 24 hours reperfusion [18]; 3) IPC group were treated with three 5-minute episodes of CCAO with 10 minutes of reperfusion between stimuli, followed by thirty minutes of CCAO. Then the clips were removed to allow reperfusion of carotid artery for 24 hours [11].
2.3. Cerebral Ischemia Induction
The animals were fasted overnight prior to surgery with free access to tap water. Anesthesia was induced by ether inhalation and maintained by thiopental sodium (2.5 mg/kg) [19]. Body temperature was kept constant at 36.5˚C ± 0.5˚C using heating pad. A longitudinal cervical incision (2 cm) was made lateral to the midline and the CCA was carefully dissected. Ischemia was induced by placing non traumatic microvascular clip on left CCA just prior to its bifurcation [20]. During ischemia rats were monitored for body temperature and respiration pattern. The clips were removed to resume blood flow to the ischemic region for 24 hours [18]. Finally, the incisions were sutured, the animal was allowed to recover from anesthesia, and returned to a warm cage for recuperation during reperfusion period. The number of animals presented for each group is the number of rats that survived during 24-hour reperfusion period. The collected data of the animals that died during 24 hours reperfusion period were excluded.
2.4. Neurological and Behavioral Evaluation
Define Neurobehavioral tests of all experimental groups were assessed daily to determine the effect of ischemic injury on them. Neurobehavioral evaluations were performed three times: the day before surgery, the same day as surgery and just before killing the animals. Each rat was examined in the late afternoon hours so that rats that had been operated in the morning would fully recovered from the effects of anesthesia by the time of evaluation. The neurobehavioral study consisted of the following six tests: spontaneous activity, symmetry in the movement of the four limbs, forepaw outstretching, climbing, body proprioception and response to vibrissae touch. The score given to each rat at the end of the evaluation is the summation of all six individual test scores. The minimum neurological score was 3 and the maximum was 18 [21].
2.5. Laboratory Investigations
At the end of experimental period, the rats were sacrificed by decapitation. Brains were rapidly removed from the skull and washed with cold saline and stored at −20˚C for further analysis. A small part of each brain from the affected hemisphere was dissected to approximately 1 - 2 mm pieces and they were homogenized in 7 ml of ice-cold extraction buffer (1% Triton X-100, 10 mmol/l MgSO4, 1 mmol/l EDTA, 1 mmol/l dithiothreitol, 0.5 mol/l NaCl, 1% protease inhibitor cocktail, 20 mmol/l HEPES and pH of 7.5) [22]. The homogenate was centrifuged; the supernatant was taken and stored at −20˚C until being used. A modification of the method of Lowry was used for the determination of protein in the brain homogenate [23]. NO metabolite (nitrite) was measured colorimetrically [24] and Rho-kinase II was detected using enzyme-linked immunosorbent assay (ELISA) [25], purchased from Cell Biolabs, INC.
2.6. Data Analysis
Data were expressed as mean ± S.E.M. Statistical evaluation was performed using the Microsoft Office Excel (Microsoft Office Excel for windows; 2003) and SPSS
(SPSS for windows version 19). Screening studied rats’ groups for significant difference in the mean of ROCK and NO was performed using analysis of variance. P < 0.05 was considered significant.
3. RESULTS
Figure 1 reveals the significant decrease in the neurological deficit in rats subjected to ischemia/reperfusion (12.83 ± 0.7) compared to sham operated rats (17.60 ± 0.7, P < 0.001). While IPC rats showed a significant improvement in neurological deficit when compared to ischemia/reperfusion group (13.7 ± 0.25, P = 0.002).
As shown in Figure 2, the concentrations of brain ROCK in rats subjected to ischemia reperfusion (0.028 ± 0.00 ng/mg protein) was significantly higher compared to sham operated rats (0.02 ± 0. 005 ng/mg protein, P < 0.001). Rats subjected to IPC demonstrated a significant decrease in ROCK level (0.007 ± 0. 005 ng/mg protein, P < 0.001). Regarding brain nitrite level, IPC rats showed a significant increase in nitrite (84.65 ± 12.61 µmol/l) when compared to both control and ischemia/reperfusion groups (56.03 ± 11.38, 59.18 ± 10.86 µmol/l respectively, P < 0.001), while no significant difference between the shamoperated and ischemia/reperfusion rats (Figure 3). In addition, Table 1 demonstrated the correlation between ROCK, nitrite and the subsequent neurological deficits.
4. DISCUSSION
In the current study, three episodes of ischemic Preconditioning, each lasting 5 minutes, protect the brain against damage from subsequent 30 minutes of ischemia, evidenced by the improvement of the neurological score compared to the ischemia/reperfusion group. These findings provide support that IPC provokes tolerance to cerebral ischemia [2,5]. Several studies observed the protective mechanism of IPC in both global and focal ischemia [2,11].
The current data demonstrated a significant increased in ROCK level in rats subjected to ischemia/reperfusion, these findings were further supported by Yano et al. [26] and Li et al. [27], Yano et al. proved that Rho-kinase activity was increased in the ipsilateral cerebral hemisphere after 3 and 6 h but not 0.5 h compared with contralateral cerebral hemisphere in rats subjected to induced cerebral ischemia [26]. Li and his colleagues, observed an impaired motor activity and motor learning function in rats subjected to 2 h of middle cerebral artery occlusion (MCAO) followed by 24 h of reperfusion, with significant neuronal loss, nuclei condensation and inflammation responses in frontal cortex [27]. Furthermore, Feske et al. reported an increased leukocyte ROCK activity in patients after acute ischemic stroke [28]. These findings suggested the possibility that activation of Rho-kinase in neuronal cells may induce disturbance of neuronal networks and resulted in neurological deficits, and in the vascular cells it may induce contraction of blood vessels and reduction of cerebral blood flow, causing progression of infarction. These hypotheses are consistent with the improvement of outcome of cerebral infarction by Rho-kinase inhibitors [27].
Studies regarding ROCK in IPC are scarce. Inter-
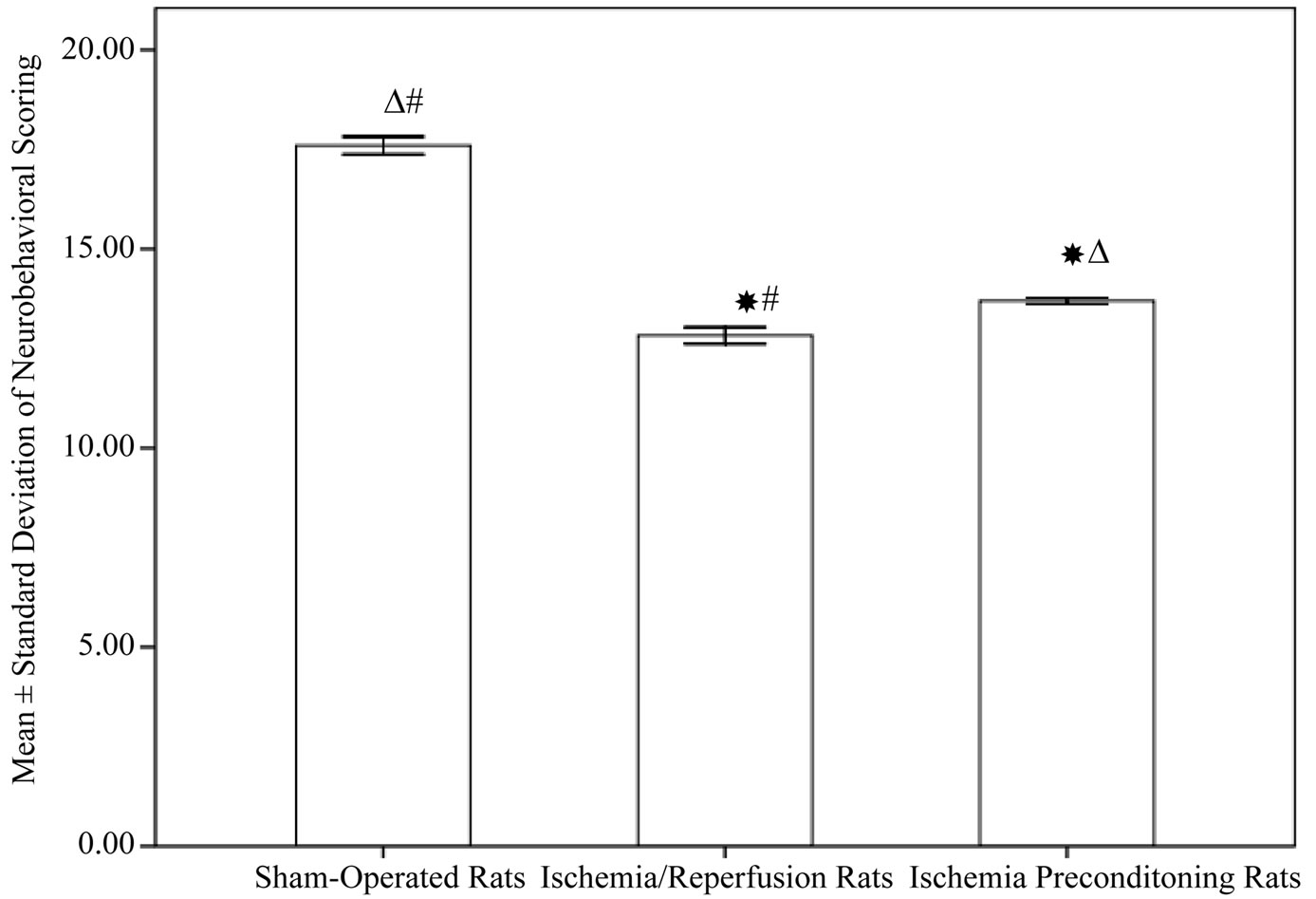
Figure 1. Neurological deficits among different experimental groups (10 rats in each group, data are expressed as mean ± SEM). Significant with controls; DSignificant with ischemia reperfusion; #Significant with ischemic preconditioning group.
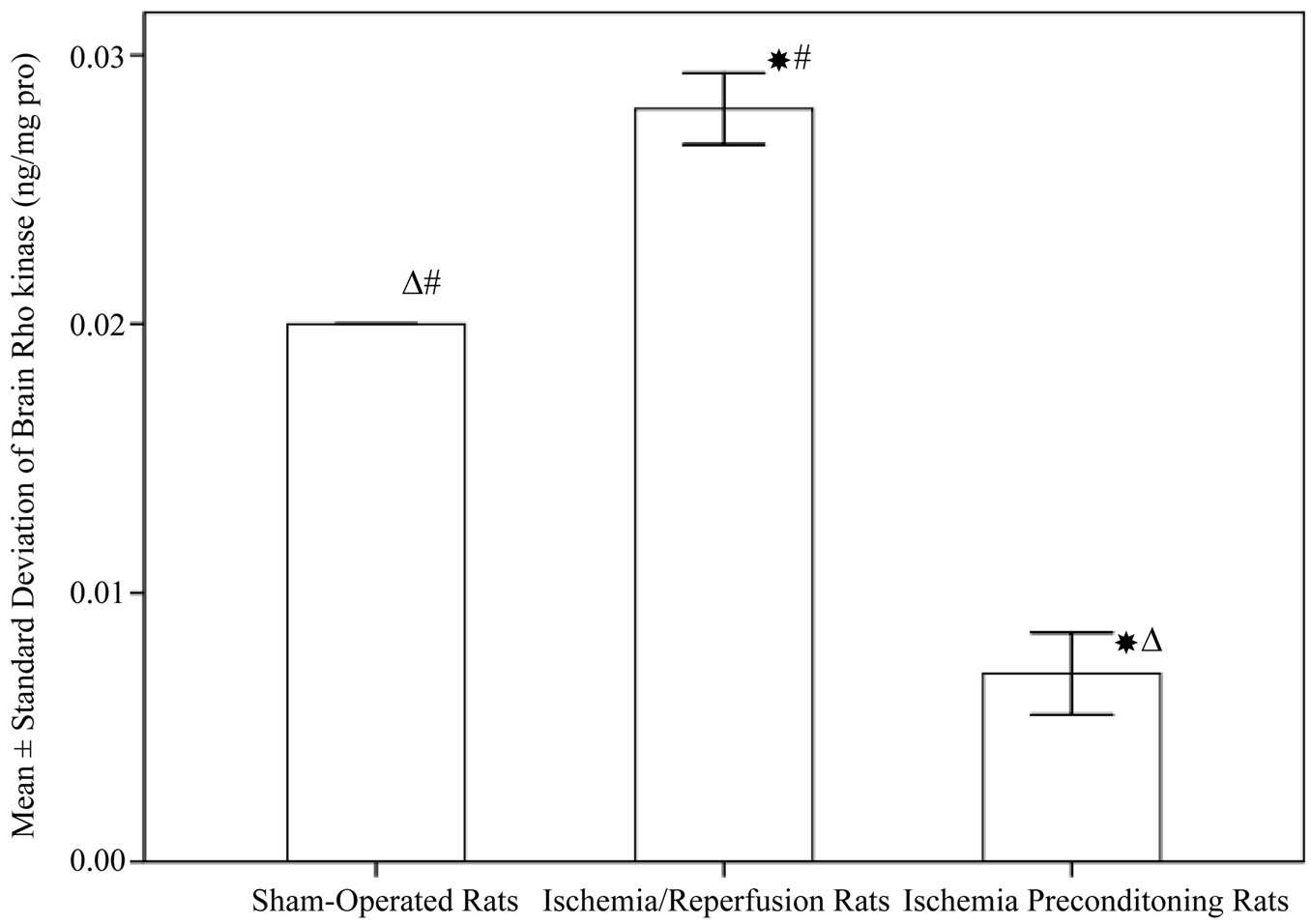
Figure 2. Rho-kinases in different experimental groups (10 rats in each group, data are expressed as mean ± SEM). Significant with controls; DSignificant with ischemia reperfusion; #Significant with ischemic preconditioning group.
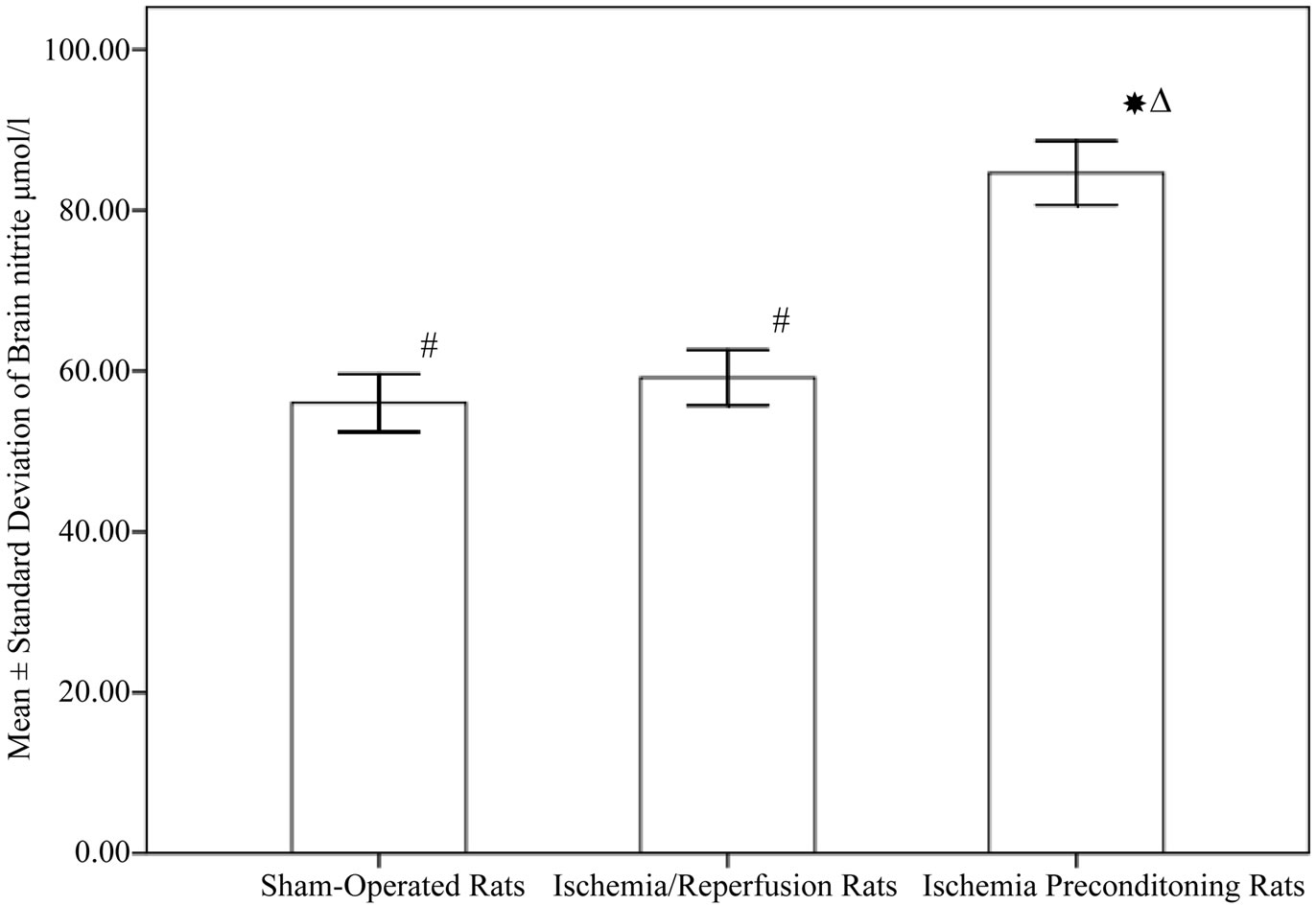
Figure 3. Nitrite level in different experimental groups (10 rats in each group, data are expressed as mean ± SEM). Significant with controls; DSignificant with ischemia reperfusion; #Significant with ischemic preconditioning group.
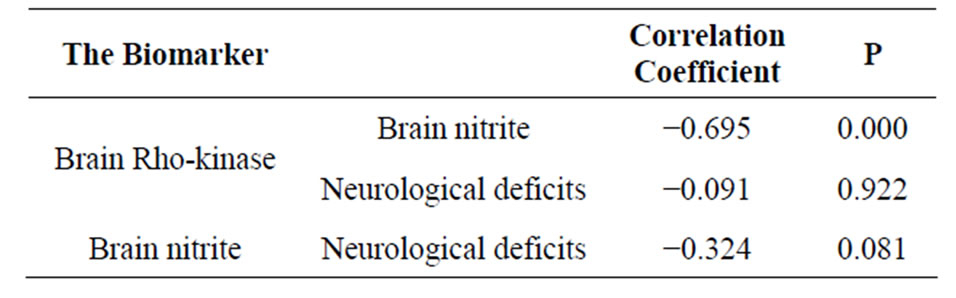
Table 1. Correlations analysis between studied biomarkers and neurological deficit.
estingly, we are the first to explore ROCK in IPC model and found that IPC rats demonstrated a significant decrease in ROCK level compared to I/R one.
Our IPC rats showed a significant increase in nitrite level. This consistence with the finding from Zhang, J. et al. [29], they applied a different model of IPC where rats subjected to repetitive 5 min of MCAO at 45-min intervals followed by 90-min MCAO were performed 24 to 96 hours later. They proved that two episodes of only 5-minute duration, which by themselves did not cause significant neuronal death, were effective in producing tolerance. This effect is associated with induction of iNOS expression, and this protection is blocked by the iNOS inhibitor aminoguanidine [30]. NO derived from eNOS is essential for eliciting early ischemic preconditioning (IP) [31], and these findings were suggesting that eNOS may mediate protection against ischemia. A shortcoming of this study is that it has not measured the enzymatic activity of different NOS isoforms.
The current study showed no significant increase in nitrite level in ischemia/reperfusion group, these findings are in accordance with the finding from Zhou et al. [32]. Although enhanced level of NO is a common, reproducible outcome of many previous works [33-36]. A possible explanation for this dispute is the type of NOS responseble for production of NO measured by different studies [32]. NO derived from eNOS has been suggested to have neuroprotective effects during cerebral ischemia [37] while NO produced by nNOS, iNOS can be neurotoxic [38].
The current study revealed a significant negative correlation between ROCK and nitrite, this may be explained by the work conducted by Fagan et al. [39], and Kataoka et al. [17], they suggested that hypoxia or ischemia may down-regulate eNOS activity via Rho-kinase activation and ROCK is a negative upstream regulator of eNOS via several mechanisms including changes in eNOS mRNA stability [40], subcellular translocation of eNOS because of reorganization of actin cytoskeleton, and eNOS phosphorylation at serine 1179 via phosphatidylinositol 3-kinase/Akt pathway [41]. Also Shin et al. [42], explored the relative contribution of these two mechanisms to the observed cerebral blood flow improvement in ischemic core and penumbra by testing hydroxyfasudil (ROCK inhibitor) in eNOS deficient mice, and showed that hydroxyfasudil did not augment cerebral blood flow in eNOS deficient mice. Thus they indicated that eNOS plays an obligatory role in this response, and that the direct vascular smooth muscle relaxing effect of Rho-kinase inhibitors does not augment cerebral blood flow in this model of focal ischemia.
In conclusion, the current study demonstrates that IPC affords protection against a subsequent ischemia/reperfusion challenge with possible contribution of NO and down regulation of ROCK in preconditioning neuroprotective mechanism. Further studies are needed to demonstrate the effect of selective NOS inhibitors and to measure each isoforms of NOS separately to evaluate the specific role of eNOS, iNOS and nNOS in different durations of cerebral ischemia.
5. ACKNOWLEDGEMENTS
To many colleagues in Alexandria University, Egypt, for whom I have great regard, and I wish to extend my sincere thanks to Professor Dr. Eman Shaat and Dr. Ali Kobil.
REFERENCES
- Smith, W.S. (2004) Pathophysiology of focal cerebral ischemia: A therapeutic perspective. Journal of Vascular and Interventional Radiology, 15, S3-S12. doi:10.1097/01.RVI.0000108687.75691.0C
- Miao, Y., Zhang, W., Lin, Y., Lu, X. and Qiu, Y. (2010) Neuroprotective effects of ischemic preconditioning on global brain ischemia through up-regulation of acidsensing ion channel 2a. International Journal of Molecular Sciences, 11, 140-153. doi:10.3390/ijms11010140
- Barone, F.C., White, R.F., Spera, P.A., Ellison, J., Currie, R.W., Wang, X. and Feuerstein, G.Z. (1998) Ischemic preconditioning and brain tolerance: Temporal histologycal and functional outcomes, protein synthesis requirement and interleukin-1 receptor antagonist and early gene expression. Stroke, 29, 1937-1950.
- Sitzer, M., Foerch, C., Neumann-Haefelin, T., Steinmetz, H., Misselwitz, B., Kugler, C. and Back, T. (2004) Transient ischaemic attack preceding anterior circulation infarction is independently associated with favourable outcome. Journal of Neurology, Neurosurgery & Psychiatry, 75, 659-660. doi:10.1136/jnnp.2003.015875
- Stenzel-Poore, M.P., Stevens, S.L., Xiong, Z., Lessov, N.S., Harrington, C.A., Mori, M., Meller, R., Rosenzweig, H.L., Tobar, E., Shaw, T.E., Chu, X. and Simon, R.P. (2003) Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: Similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet, 362, 1028-1037. doi:10.1016/S0140-6736(03)14412-1
- Dave, K.R., Saul, I., Busto, R., Ginsberg, M.D., Sick, T.J. and Perez-Pinzon, M.A. (2001) Ischemic preconditioning preserves mitochondrial function after global cerebral ischemia in rat hippocampus. Journal of Cerebral Blood Flow & Metabolism, 21, 1401-1410. doi:10.1097/00004647-200112000-00004
- Chimon, G.N. and Wong, P.T. (1998) Ischemic tolerance and lipid peroxidation in the brain. Neuroreport, 9, 2269- 2272. doi:10.1097/00001756-199807130-00023
- Stenzel-Poore, M.P., Stevens, S.L. and Simon, R.P. (2004) Genomics of preconditioning. Stroke, 35, 2683-2686. doi:10.1161/01.STR.0000143735.89281.bb
- Garthwaite, J. (2008) Concepts of neural nitric oxide-mediated transmission. European Journal of Neuroscience, 27, 2783-2802. doi:10.1111/j.1460-9568.2008.06285.x
- Ito, Y., Ohkubo, T., Asano, Y., Hattori, K., Shimazu, T., Yamazato, M., Nagoya, H., Kato, Y. and Araki, N. (2010) Nitric oxide production during cerebral ischemia and reperfusion in eNOSand nNOS-knockout mice. Current Neurovascular Research, 7, 23-31. doi:10.2174/156720210790820190
- Atochin, D.N., Clark, J., Demchenko, I.T., Moskowitz, M.A. and Huang, P.L. (2003) Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke, 34, 1299-1303. doi:10.1161/01.STR.0000066870.70976.57
- Offermanns, N.W.S. (2003) Rho/Rho-kinase mediated signaling in physiology and pathophysiology.
- Owens, G.K., Kumar, M.S. and Wamhoff, B.R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Reviews, 84, 767-801. doi:10.1152/physrev.00041.2003
- Kamiyama, M., Utsunomiya, K., Taniguchi, K., Yokota, T., Kurata, H., Tajima, N. and Kondo, K. (2003) Contribution of Rho A and Rho kinase to platelet-derived growth factor-BB-induced proliferation of vascular smooth muscle cells. Journal of Atherosclerosis and Thrombosis, 10, 117-123. doi:10.5551/jat.10.117
- Ginsberg, M.D. (2003) Adventures in the pathophysiology of brain ischemia: Penumbra, gene expression, neuroprotection: The 2002 Thomas Willis Lecture. Stroke, 34, 214-223. doi:10.1161/01.STR.0000048846.09677.62
- Heuschmann, P.U., Berger, K., Misselwitz, B., Hermanek, P., Leffmann, C., Adelmann, M., Buecker-Nott, H.J., Rother, J., Neundoerfer, B. and Kolominsky-Rabas, P.L. (2003) Frequency of thrombolytic therapy in patients with acute ischemic stroke and the risk of in-hospital mortality: The German Stroke Registers Study Group. Stroke, 34, 1106-1113. doi:10.1161/01.STR.0000065198.80347.C5
- Kataoka, C., Egashira, K., Inoue, S., Takemoto, M., Ni, W., Koyanagi, M., Kitamoto, S., Usui, M., Kaibuchi, K., Shimokawa, H. and Takeshita, A. (2002) Important role of Rho-kinase in the pathogenesis of cardiovascular inflammation and remodeling induced by long-term blockade of nitric oxide synthesis in rats. Hypertension, 39, 245-250. doi:10.1161/hy0202.103271
- Kuluz, J.W., Prado, R.J., Dietrich, W.D., Schleien, C.L. and Watson, B.D. (1993) The effect of nitric oxide synthase inhibition on infarct volume after reversible focal cerebral ischemia in conscious rats. Stroke, 24, 2023- 2029. doi:10.1161/01.STR.24.12.2023
- Keefer, L.K., Garland, W.A., Oldfield, N.F., Swagzdis, J.E. and Mico, B.A. (1985) Inhibition of N-nitrosodimethylamine metabolism in rats by ether anesthesia. Cancer Research, 45, 5457-5460.
- Renolleau, S., Aggoun-Zouaoui, D., Ben-Ari, Y. and Charriaut-Marlangue, C. (1998) A model of transient unilateral focal ischemia with reperfusion in the P7 neonatal rat: Morphological changes indicative of apoptosis. Stroke, 29, 1454-1460. doi:10.1161/01.STR.29.7.1454
- Furuya, K., Zhu, L., Kawahara, N., Abe, O. and Kirino, T. (2005) Differences in infarct evolution between lipopolysaccharide-induced tolerant and nontolerant conditions to focal cerebral ischemia. Journal of Neurosurgery, 103, 715-723. doi:10.3171/jns.2005.103.4.0715
- Star, R.A. (1998) Treatment of acute renal failure. Kidney International, 54, 1817-1831. doi:10.1046/j.1523-1755.1998.00210.x
- Lowry, O.H., Rosebrough, N.J., Farr, A.L. and Randall, R.J. (1951) Protein measurement with the Folin phenol reagent. The Journal of Biological Chemistry, 193, 265- 275.
- Montgomery, H.A.C. and Dymock, J. (1961) The determination of nitrite in water. Analyst, 86, 414-416.
- Riento, K. and Ridley, A.J. (2003) Rocks: Multifunctional kinases in cell behaviour. Nature Reviews Molecular Cell Biology, 4, 446-456. doi:10.1038/nrm1128
- Yano, K., Kawasaki, K., Hattori, T., Tawara, S., Toshima, Y., Ikegaki, I., Sasaki, Y., Satoh, S., Asano, T. and Seto, M. (2008) Demonstration of elevation and localization of Rho-kinase activity in the brain of a rat model of cerebral infarction. European Journal of Pharmacology, 594, 77- 83. doi:10.1016/j.ejphar.2008.07.045
- Li, Q., Huang, X.J., He, W., Ding, J., Jia, J.T., Fu, G., Wang, H.X. and Guo, L.J. (2009) Neuroprotective potential of fasudil mesylate in brain ischemia-reperfusion injury of rats. Cellular and Molecular Neurobiology, 29, 169-180. doi:10.1007/s10571-008-9308-8
- Feske, S.K., Sorond, F.A., Henderson, G.V., Seto, M., Hitomi, A., Kawasaki, K., Sasaki, Y., Asano, T. and Liao, J.K. (2009) Increased leukocyte ROCK activity in patients after acute ischemic stroke. Brain Research, 1257, 89-93. doi:10.1016/j.brainres.2008.12.045
- Zhang, J., Yang, Z.J., Klaus, J.A., Koehler, R.C. and Huang, J. (2008) Delayed tolerance with repetitive transient focal ischemic preconditioning in the mouse. Stroke, 39, 967-974. doi:10.1161/STROKEAHA.107.497412
- Kapinya, K., Penzel, R., Sommer, C. and Kiessling, M. (2000) Temporary changes of the AP-1 transcription factor binding activity in the gerbil hippocampus after transient global ischemia and ischemic tolerance induction. Brain Research, 872, 282-293. doi:10.1016/S0006-8993(00)02503-8
- Xuan, Y.T., Guo, Y., Zhu, Y., Wang, O.L., Rokosh, G. and Bolli, R. (2007) Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase C epsilon p44/42 mitogen-activated protein kinase pSer-signal transducers and activators of transcription1/3 pathway. Circulation, 116, 535-544. doi:10.1161/CIRCULATIONAHA.107.689471
- Zhou, L. and Zhu, D.Y. (2009) Neuronal nitric oxide synthase: Structure, subcellular localization, regulation and clinical implications. Nitric Oxide, 20, 223-230. doi:10.1016/j.niox.2009.03.001
- Hirabayashi, H., Takizawa, S., Fukuyama, N., Nakazawa, H. and Shinohara, Y. (2000) Nitrotyrosine generation via inducible nitric oxide synthase in vascular wall in focal ischemia-reperfusion. Brain Research, 852, 319-325. doi:10.1016/S0006-8993(99)02117-4
- Hata, R., Maeda, K., Hermann, D., Mies, G. and Hossmann, K.A. (2000) Evolution of brain infarction after transient focal cerebral ischemia in mice. Journal of Cerebral Blood Flow & Metabolism, 20, 937-946. doi:10.1097/00004647-200006000-00006
- Margaill, I., Allix, M., Charriaut-Marlangue, C., Boulu, R.G. and Plotkine, M. (1995) Loss of NADPH-diaphorase containing neurones after reversible focal ischaemia in rats delayed by L-NAME. British Journal of Pharmacology, 116, 2344-2345. doi:10.1111/j.1476-5381.1995.tb15076.x
- Kader, A., Frazzini, V.I., Solomon, R.A. and Trifiletti, R.R. (1993) Nitric oxide production during focal cerebral ischemia in rats. Stroke, 24, 1709-1716. doi:10.1161/01.STR.24.11.1709
- Cui, X., Chopp, M., Zacharek, A., Zhang, C., Roberts, C. and Chen, J. (2009) Role of endothelial nitric oxide synthetase in arteriogenesis after stroke in mice. Neuroscience, 159, 744-750. doi:10.1016/j.neuroscience.2008.12.055
- Zhao, X., Haensel, C., Araki, E., Ross, M.E. and Iadecola, C. (2000) Gene-dosing effect and persistence of reduction in ischemic brain injury in mice lacking inducible nitric oxide synthase. Brain Research, 872, 215-218. doi:10.1016/S0006-8993(00)02459-8
- Fagan, K.A., Oka, M., Bauer, N.R., Gebb, S.A., Ivy, D.D., Morris, K.G. and McMurtry, I.F. (2004) Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rhokinase. American Journal of Physiology—Lung Cellular and Molecular Physiology, 287, L656-L664. doi:10.1152/ajplung.00090.2003
- Laufs, U. and Liao, J.K. (1998) Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. The Journal of Biological Chemistry, 273, 24266-24271. doi:10.1074/jbc.273.37.24266
- Ming, X.F., Viswambharan, H., Barandier, C., Ruffieux, J., Kaibuchi, K., Rusconi, S. and Yang, Z. (2002) Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Molecular and Cellular Biology, 22, 8467-8477. doi:10.1128/MCB.22.24.8467-8477.2002
- Shin, H.K., Salomone, S., Potts, E.M., Lee, S.W., Millican, E., Noma, K., Huang, P.L., Boas, D.A., Liao, J.K., Moskowitz, M.A. and Ayata, C. (2007) Rho-kinase inhibition acutely augments blood flow in focal cerebral ischemia via endothelial mechanisms. Journal of Cerebral Blood Flow & Metabolism, 27, 998-1009.
ABBREVIATIONS
Ischemic preconditioning (IPC);
Ischemia/reperfusion (I/R);
Left common carotid artery occlusion (CCAO);
Rho-kinase (ROCK);
Nitric oxide (NO);
Transient ischemic attacks (TIAs);
Endothelial NOS (eNOS);
Neuronal NOS (nNOS);
Inducible NOS (iNOS);
Middle cerebral artery occlusion (MCAO).