Journal of Modern Physics
Vol.4 No.12C(2013), Article ID:41491,11 pages DOI:10.4236/jmp.2013.412A3002
Coordination Interaction between Rare Earth and/or Transition Metal Centers and Thiophene Series Oligomer Derivatives in Ultrathin Langmuir-Blodgett Films
1Physics Department, Belarusian State University, Minsk, Belarus
2Belarusian State University of Informatics and Radioelectronics, Minsk, Belarus
Email: grushevskaja@bsu.by
Copyright © 2013 H. V. Grushevskaya et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received July 1, 2013; revised August 5, 2013; accepted September 3, 2013
Keywords: Langmuir Monolayer; Self-Organization; Metal-Containing Ultrathin Langmuir-Blodgett Film
ABSTRACT
It has been shown by thermodynamic and electro-chemical methods that coordination interactions are involved in the process of incorporation of rare-earth elements and/or Fe into Langmuir monolayers of oligomer with alkyl fragment. A coordination mechanism, which is responsible for self-organization of octahedral Fe(II)-complexes of nano-cyclic ligands obtained from oligomer of thiophene pyrrole series derivatives in compressed Langmuir monolayers has been proposed. It has been established that a coordinational-polymeric hexagonal network of nano-cyclic ligands with metal centers is formed at the phase transition when compressing the Langmuir monolayers of thiophene pyrrole series oligomer with alkyl fragment on surface of subphase with rare-earth (Sm, Er, Ce) and/or Fe ions.
1. Introduction
Metal-polymer Langmuir-Blodgett (LB) films with rareearth and transition elements as quantum spin-polarized conducting molecular layers (monolayers) are of great interest for optoand nano-electronics as well as for sensor technology. For example, Langmuir-Blodgett conducting films are utilized as a detecting element in the sensor for selective retention of ions and small molecules [1,2].
Functional area of nano-electronic device is multilayered one. Such methods as sputtering, centrifugation or deposition of metal-organic compounds allow to form only defective monolayer network. Alternative way of formation is a catalyst of metal-polymer networks in self-organized thin-film systems [3]. The advantage of self-organization is the formation of highly ordered thiophene polymer in the plane of stable two-dimensional (2D) low-defect systems. One of the promising directions to design low-dimensional films of metal-polymer compounds is their self-organized formation from metal complexes of macrocyclic coordination compounds. In [4] it was described a cooperative formation of polymetallic spirals from helicates having from two up to five metal centers.
The Langmuir-Blodgett technique is a method to produce monolayers self-assembling on liquid subphase surface. The monolayers are formed from ions of amphiphilic molecules and counter ions of the subphase by phase transition from two-dimensional gas to a crystalline state [5]. The LB technique is difficult to utilize because it is necessary to optimize the ratio of mutually exclusive properties of semiconductor hydrophilic and insulator lipophilic sublayers in the amphiphilic monolayer. Thus, for example, after polymerization in the presence of FeCl3, the LB film of hydrophobic conductive thiophene oligomers with attached hydrophilic groups acquires lateral conductivity [1,6]. However, high conductivity values are achievable only in the plane of highly ordered crystalline LB monolayer film.
To arrange condensed monolayer, instead of oligomer one should take a mixture consisting of thiophene polymer with additional hydrophilic polar groups and amphiphilic compound. Among the variety of such mixtures, coordination interactions occur in a monolayer formed from thiophene and iron-containing complex of pyrrole derivatives, such as, iron phthalocyanine [6]. The fact and an increasing conductivity by several orders of magnitude for the LB film, consisting of pyrrole and thiophene polymers and doped by NOPF6, [7] allow us to suggest that LB films of thiophene pyrrole series oligomer will have improved electro-physical characteristics. A condition for the existence of the metal-polymer is the resonance energy transfer of quasiparticle excitation on a metal (Fe) sub-system with excitation of a mixed state which is formed by localized d-electron orbitals of valent band and delocalized s(p)-electron orbitals of conduction band, or to be more accurate not a mixed but a generalized coherent electron state [8]. Since the transfer of an electron into conduction band changes the electronic configuration from 3d6 to 3d5, then the valence of iron is increased by one from Fe2+ to Fe3+. Therefore, such coordination systems are called systems with mixed valence [9,10].
An existence of the resonance between localized felectron states and delocalized d-, s(p)-electrons in the conduction band is known for inorganic compounds of rare-earth metals (Ln) [11]. In contrast to transition metal compounds with mixed valence, a mean or “fractional” valence of rare earth compounds is observed in experiments. Therefore, their valent state, rather than mixed, received the name of intermediate valence [11]. Though the term “intermediate valence” is mainly used in respect to compounds of rare earths and actinides, it is sometimes used in the description of transition metal atom states [12]. Except Ce, the transition of rare earth metal atom from a “normal” integer valent state into the intermediate valent state is stipulated by excitation of an electron from internal 4f-shell on external 5d-, 6s(p)- levels in conduction band. For Ce, delocalized electrons retain the behaviour of f-electrons and the transition 4f ® 5d is absent [13-15]. The intermediate valence state in Ce is considered as a resonance between slightly different in energy states 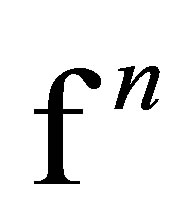 and
and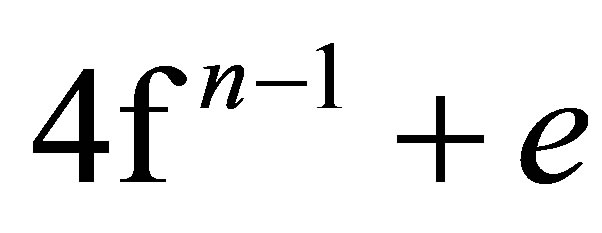 , being initial and final states of the electron.
, being initial and final states of the electron.
In solids with rare earth atoms, valent electrons usually either form chemical bonds or pass into conduction band. Electrons of partially filled 4f-shell remain localized in atomic core. Typical value of rare earth element valence is +3. This means that three electrons (5d, 6s(p)) are in conduction band. Some rare earths are mostly observed in valence states +4 (Се, Рr), +2 (Sm, Eu, Tm, Yb). The mixed valent state in SmS manifests as valence change from +2 to +3 in a process of phase transition taking place at pressure increase [16]. Similar effect can be achieved by varying Sm concentration in compounds doped with other lanthanides [17]. In papers [18,19] for samarium compounds Sm1−xRxS (R = Ce, Gd, Y) it has been shown that at low concentrations of samarium [Sm], 4f - 5d-, 6s (p)-hybridization of the orbitals in the same atom occurs instead of hybridization of 4f-orbitals of Sm atom with 5d-, 6s (p)-orbitals of the neighboring Sm atom. This leads to the valence tending to value +2 at low concentrations [Sm]®0 [18,19]. Ce atoms, along with a valence +3, at which the 4f-shell of the atom contains one electron, have a valence +4, when the 4fshell is empty. For example, valence resonance state for Ce in intermetallide Ce2Fe17 possesses intermediate valence in range between +3 and +4 [20]. Silicides of Ce, alloys Ce-Th, Ce-Yb are also referred as substances with metal atoms in the mixed valence state [12]. It is known [21-25] that the exchange interactions in rare earths sublattice and between sublattices of rare earth atoms and transition metal atoms (indirect exchange) are weak.
In this paper we show that, due to the weakness of the above-mentioned exchange interactions, the presence of rare-earth atoms in the metal-polymer compounds containing iron atoms in a mixed valence, leads to an increase in spin relaxation time and thus slows down the reorientation of the magnetic moments. The latter leads to stabilization of magnetic ordering of transition metal atoms impurity levels, and respectively to a prolonged coexistence of domains of semiconductor and metal phases with a valences Fe2+ and Fe3+, respectively. We shall show by thermodynamic and electro-chemical methods that coordination interactions are involved in the process of incorporation of rare-earth elements and/or Fe into monolayers of thiophene pyrrole series oligomer with alkyl fragment. We will also demonstrate that after first-order phase transition, the Langmuir monolayer represents itself a polymetallic organic complex in an intermediate valence state.
The goal of the paper is to establish a coordination mechanism, which provides self-organization of Fe(II)- complexes of nano-cyclic ligands formed from thiophene-pyrrole series oligomer in compressed Langmuir monolayers.
2. Materials and Methods
2.1. Reagents
Salts Fe(NO3)3∙9Н2О, almost colorless Ce2(SO4)3, Sm2(SO4)3∙8Н2О, and slightly colored in pink Er(NO3)3∙5Н2О (“Sigma-Aldric”, USA), hydrochloric acid, deionized water were used to preparate subphase.
Monolayers and Langmuir-Blodgett films was formed from amphiphilic oligomer of thiophene derivatives: 3-hexadecyl-2,5-di(thiophen-2-yl)-1H‑pyrrole (H-dithionilepyrrole, H-dithiopyrrole) which chemical structure is represented in Figure 1. H-dithionilepyrrole was synthesized by a method proposed in [26].
NMR Spectra (NMR spectrometer “Bruker Avance 400”, Germany) of H-dithionilepyrrole are: NMR Spectrum 1Н (δ, ppm) (400 MHz, CDCl3): 8.11 s. (1H), 7.34 - 6.93 m (6H), 2.64 s. (1H), 1.71 - 1.58 m (2H), 1.46 - 1.21 m (28H), 0.88 t (J = 6.7 Hz, 3H); NMR Spectrum 13С (δ,
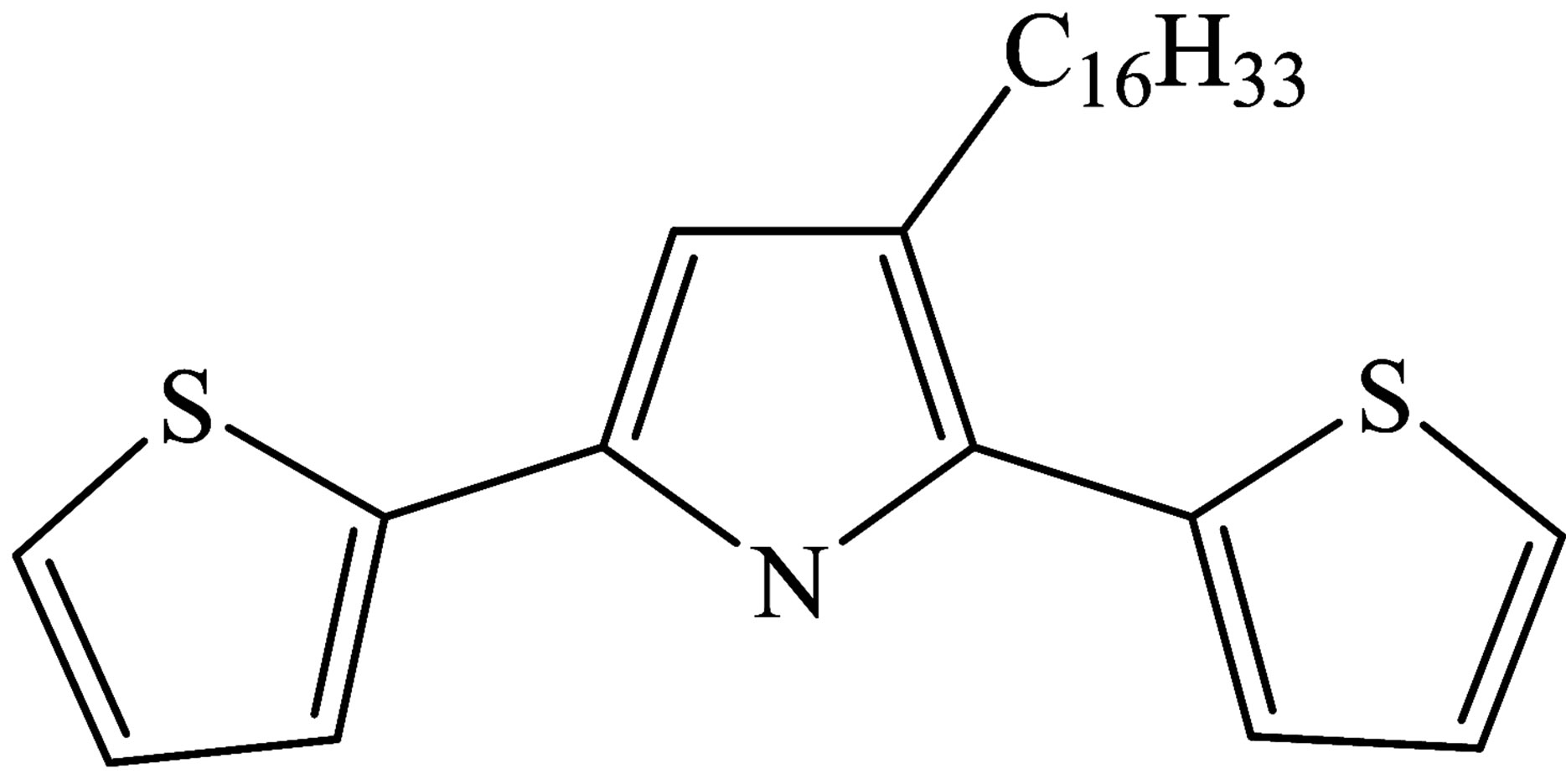
Figure 1. Chemical structure of H-dithiopyrrole molecule.
ppm) (100 MHz, CDCl3): 127.86, 127.64, 124.65, 123.50, 122.87, 122.74, 120.99, 109.21, 32.08, 30.90, 29.86, 29.70, 29.52, 26.79, 22.85, 14.29. An infrared spectrum (ν, cm–1) (KBr): 3411, 2917, 2849, 1503, 1471, 1416, 844, 799, 711, 686 of H-dithionilepyrrole was recorded by IR spectrometer VERTEX-70 (Bruker Austria GmbH). Working solution of H-dithionilepyrrole, 1.0 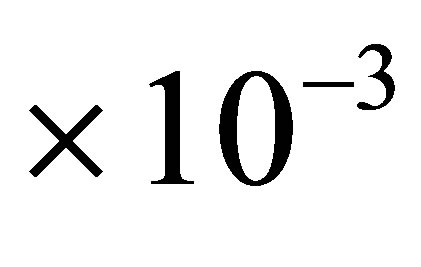 M was prepared by dissolving precisely weighed substances in hexane.
M was prepared by dissolving precisely weighed substances in hexane.
Experiments were performed in deionized water. All used materials belong to class of analytical pure reagents.
2.2. Formation of Langmuir Monolayers
Langmuir-Blodgett monolayer formation was carried out on an automated hand-made Langmuir trough with controlled deposition on a substrate, and with computer user interface working under Microsoft Windows operational system. Control of the surface tension has been performed by a highly sensitive resonant inductive sensor.
Preliminary, the H-dithionilepyrrole solution was dropped with help of a micropipette on a surface the liquid subphase. The subphase represents itself a water solution of a salt or salts with addition of hydrochloric acid. We have deposited crystalline monomolecular layers at surface pressures  equal to 24 mN/m. Amorphous monolayers formed before appearance of plateau on a
equal to 24 mN/m. Amorphous monolayers formed before appearance of plateau on a 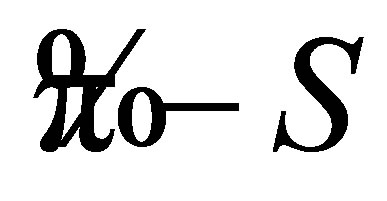 isotherm of first-order phase transition were deposited at
isotherm of first-order phase transition were deposited at , equal to 19 mN/m. The Y-type transposition of five monolayers was performed. The temperature range for forming Langmuir monolayer was equal to 17˚C ± 0.2˚C, a compression speed of 4 mm/min, pH of subphase 1.65 ± 0.1.
, equal to 19 mN/m. The Y-type transposition of five monolayers was performed. The temperature range for forming Langmuir monolayer was equal to 17˚C ± 0.2˚C, a compression speed of 4 mm/min, pH of subphase 1.65 ± 0.1.
The valence state of metals in fabricated 5-monolayer LB films was estimated by means of the ration of electrical capacitance of LB film deposited before the phase transition to the capacitance of LB film deposited after phase transition.
2.3. Impedance Analysis
For electrochemical studies, we use a planar capacitive sensor of interdigital-type on pyroceramics support [27]. Aluminum electrodes of the sensor are arranged in an Archimedes-type spiral configuration. Dielectric coating of the electrodes represents itself nanoporous anodic alumina layer (AOA) with a pore diameter of 10 nm on which LB-film is deposited.
To excite harmonic auto-oscillations of electric current (charge-discharge processes in capacitor), the sensor was connected as the capacitance C into the RC relaxation auto-generator [28]. Operation of such an autogenerator is based on the principle of self-excitation of an amplifier with a positive feedback on the quasi-resonance frequency [29]. The capacitance C of the sensor entered in measuring RC-oscillating circuit has been calculated by the formula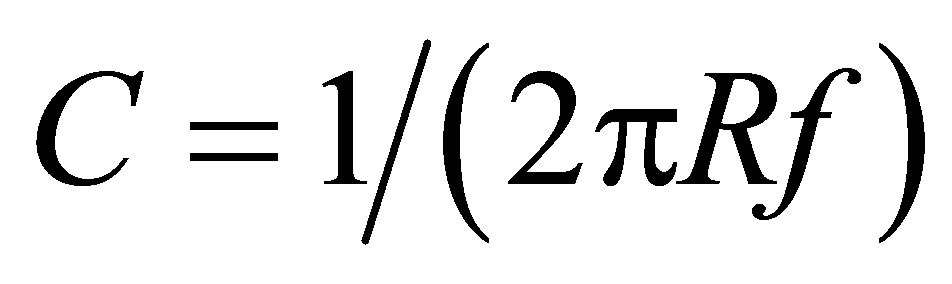 , where R is the measuring resistance, f is the frequency of quasi-resonance.
, where R is the measuring resistance, f is the frequency of quasi-resonance.
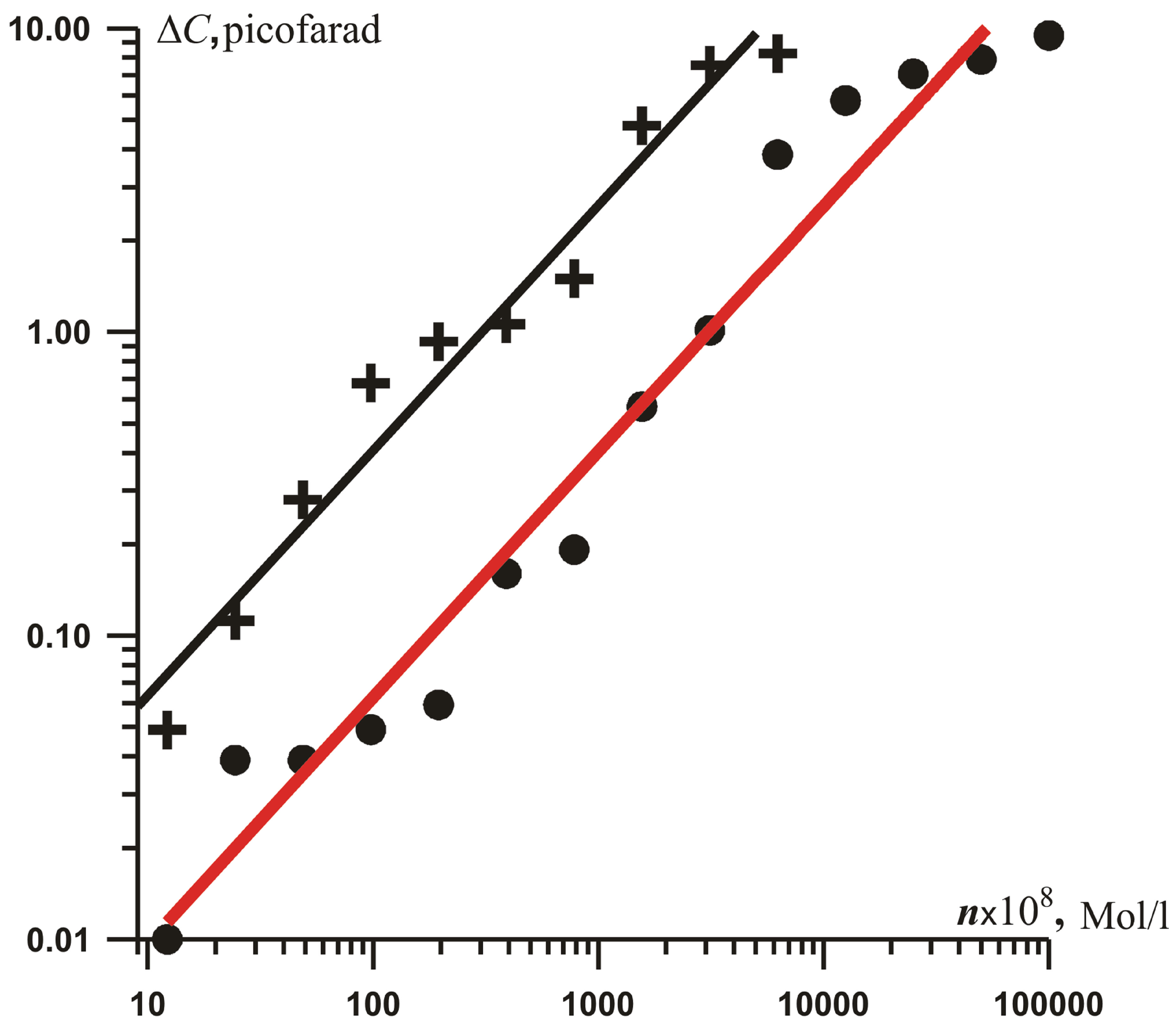
As one can see in Figure 2, a concentration depen-
 (a)
(a) (b)
(b)
Figure 2. Concentration dependencies of (a) the increment ΔC of the electrically charged double layer capacitance (coefficient of fitting determination R2 is 0.965) and (b) conductivity 1/RX of ionic current (coefficient of fitting determination R2 is 0.93 and 0.88 for the “ ” and “+”) for salts solutions: FeSO4 (
” and “+”) for salts solutions: FeSO4 ( ) and Fe(NO3)3 (+), in logarithmic scale.
) and Fe(NO3)3 (+), in logarithmic scale.
dence of increment DC of electrically charged double layer capacitance is approximately linear at low carrier concentrations, and DC is proportional to the metal valence in accordance with the Garnett formula for the dielectric permittivity  of a composite material [30- 32]:
of a composite material [30- 32]:
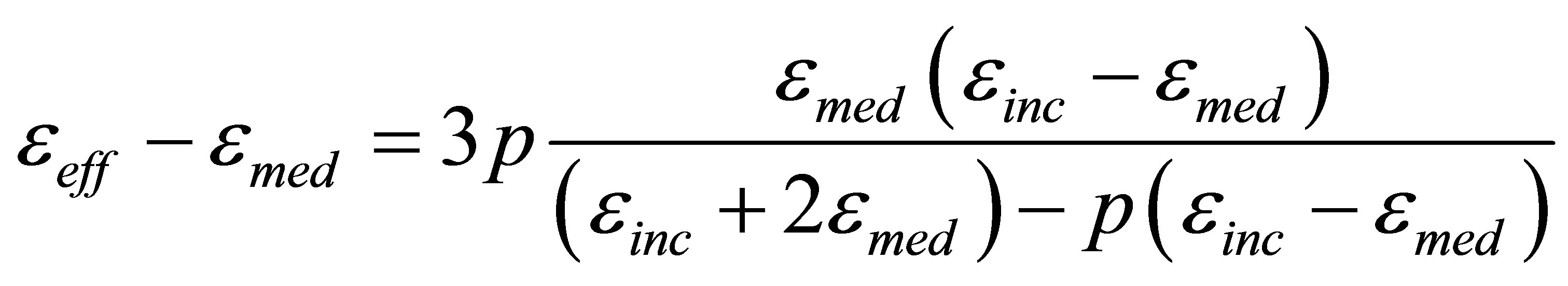 , (1)
, (1)
where 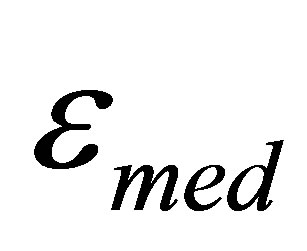 is the dielectric permittivity of the media,
is the dielectric permittivity of the media,  is the dielectric permittivity of inclusions, р is the volume fraction of inclusions:
is the dielectric permittivity of inclusions, р is the volume fraction of inclusions: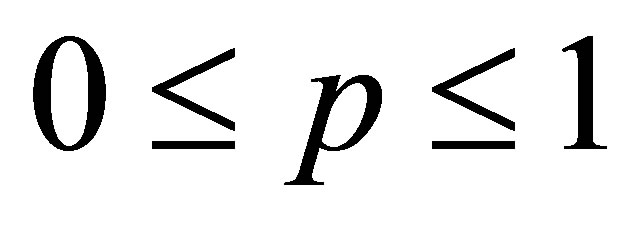 .
.
3. Model of Compressed LB Monolayer
We analyze a structure that arises in the compressed Langmuir monolayer before and after first-order phase transition into solid state on different water subphase surfaces.
Let us consider a case when the monolayer which is located on the surface of the water solution with threevalence iron, is compressed up to surface pressure not exceeding a value at which the first-order phase transition occurs. Then one can suppose that the compression is accompanied by forming of organo-metallic compound  via the following reaction:
via the following reaction:
 (2)
(2)
where hydrogen bonds are indicated as “ ”.
”.
Now, one can make an assumption about a Langmuir monolayer structure after the first-order phase transition. To do it, we utilize the fact that chloride anions template a pentanuclear helicate formation in a reaction of octahedral metal center Fe(II) with, for example, tris(bipyridyl) ligand [33]. We also take into account the existence of such types of Fe nitrides, as Fe16N2, Fe4N, Fe3N, Fe2N. Then, one can suggest the following mechanism of complexification of Fe with derivatives of thiophene pyrrole series at excess concentrations of transition metal [Fe] ? 1. When compressing monolayer at surface pressure  on the surface of ferric salt solution with hydrochlorous acid molecules, the octahedral iron complex is synthesized during the phase transition due to interaction with
on the surface of ferric salt solution with hydrochlorous acid molecules, the octahedral iron complex is synthesized during the phase transition due to interaction with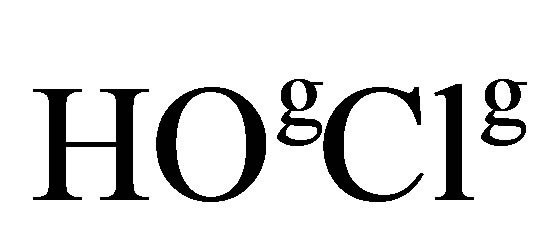 :
:
 (3)
(3)
Here, the complex with octahedral 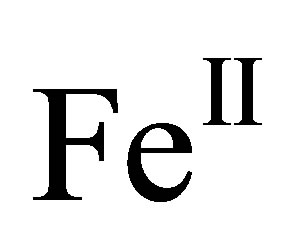 is formed by hydrogen bonds between the pentanuclear helicate
is formed by hydrogen bonds between the pentanuclear helicate  with the chloride anion and the complex
with the chloride anion and the complex , dithiopyrrole is an ion of H-dithiopyrrole molecule obtained by deprotonation, at which the loss of H+ takes place in the dissociation process. Then a reaction of forming monomer
, dithiopyrrole is an ion of H-dithiopyrrole molecule obtained by deprotonation, at which the loss of H+ takes place in the dissociation process. Then a reaction of forming monomer  follows:
follows:
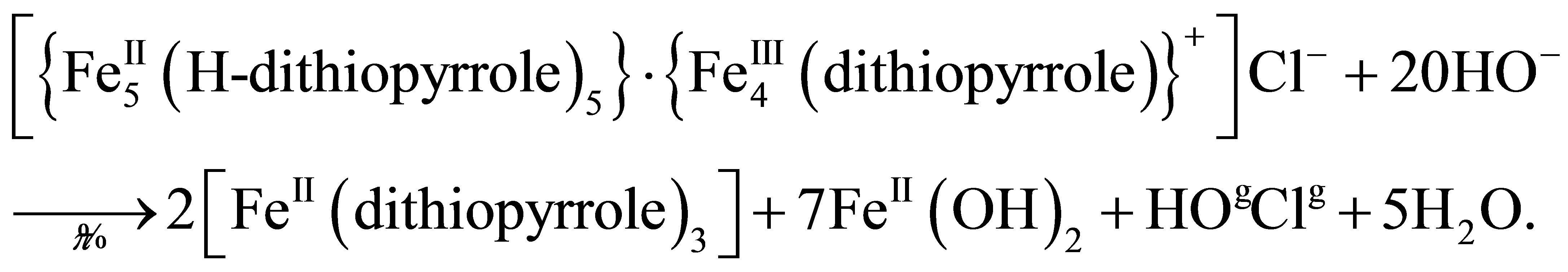 (4)
(4)
Additional hydrochlorous acid molecules produced in the reaction (4) are further involved in the synthesis of monomers. Under the action of the surface pressure, two monomer are bonded into dimer .
.
Further repetition of reactions (3, 4) and monolayer compression lead to the formation of the polymer network  with hexagonal cells in which there are six coordinating centers of Fe(II).
with hexagonal cells in which there are six coordinating centers of Fe(II).
According to the Weiss model [34], a macrocycle with a high-spin Fe(II)-state has a convex shape, and the lowspin Fe(III)-state forms a flat macrocycle, for example, Fe in the tetrapyrrole complex. In this case, the energy loss due to the appearance of dome structure at the transition to high-spin Fe2+ state is compensated by the gain due to ferromagnetic ordering.
In subsequent, we present the results of thermodynamic and electro-chemical investigations confirming that the complexes in the reaction (4) with the  center in a high-spin state form the structure of LB monolayer.
center in a high-spin state form the structure of LB monolayer.
4. Thermodynamic Analysis
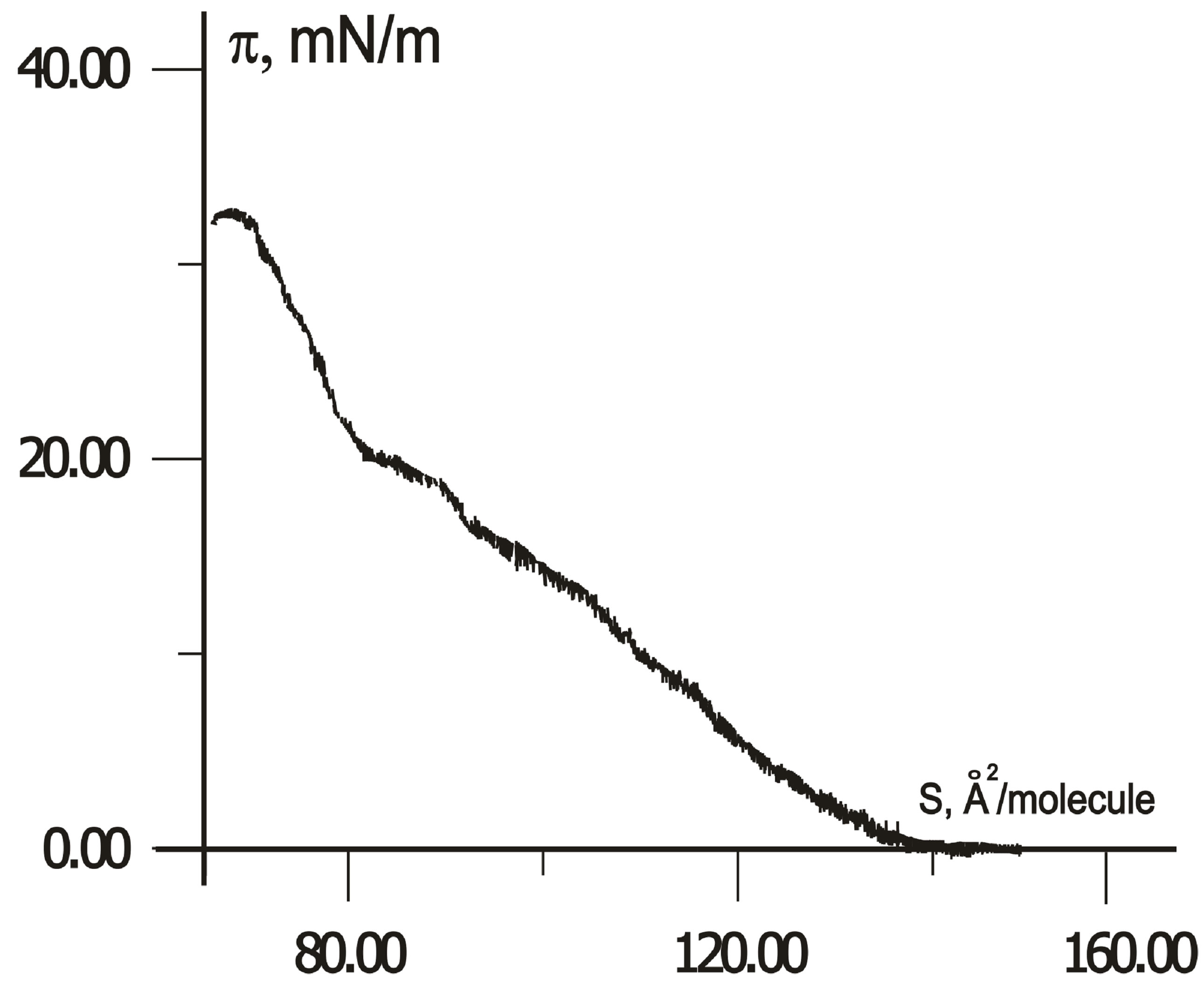
When compressing, the first-order phase transitions from two-dimensional gas (liquid) to a crystalline state (solid) can occur in Langmuir monolayers. However, the LB monolayer of thiophene pyrrole derivatives has no crystalline state in distilled water and on a subphase with rare earth salts only. In contrast, the compression isotherms of H-dithiopyrrole on subphase with trivalent ions of cerium, samarium, and erbium, and/or ferric nitrate have a plateau, as shown in Figure 3. An area SLn per H-dithiopyrrole molecule in crystalline monolayer depends
 (a)
(a)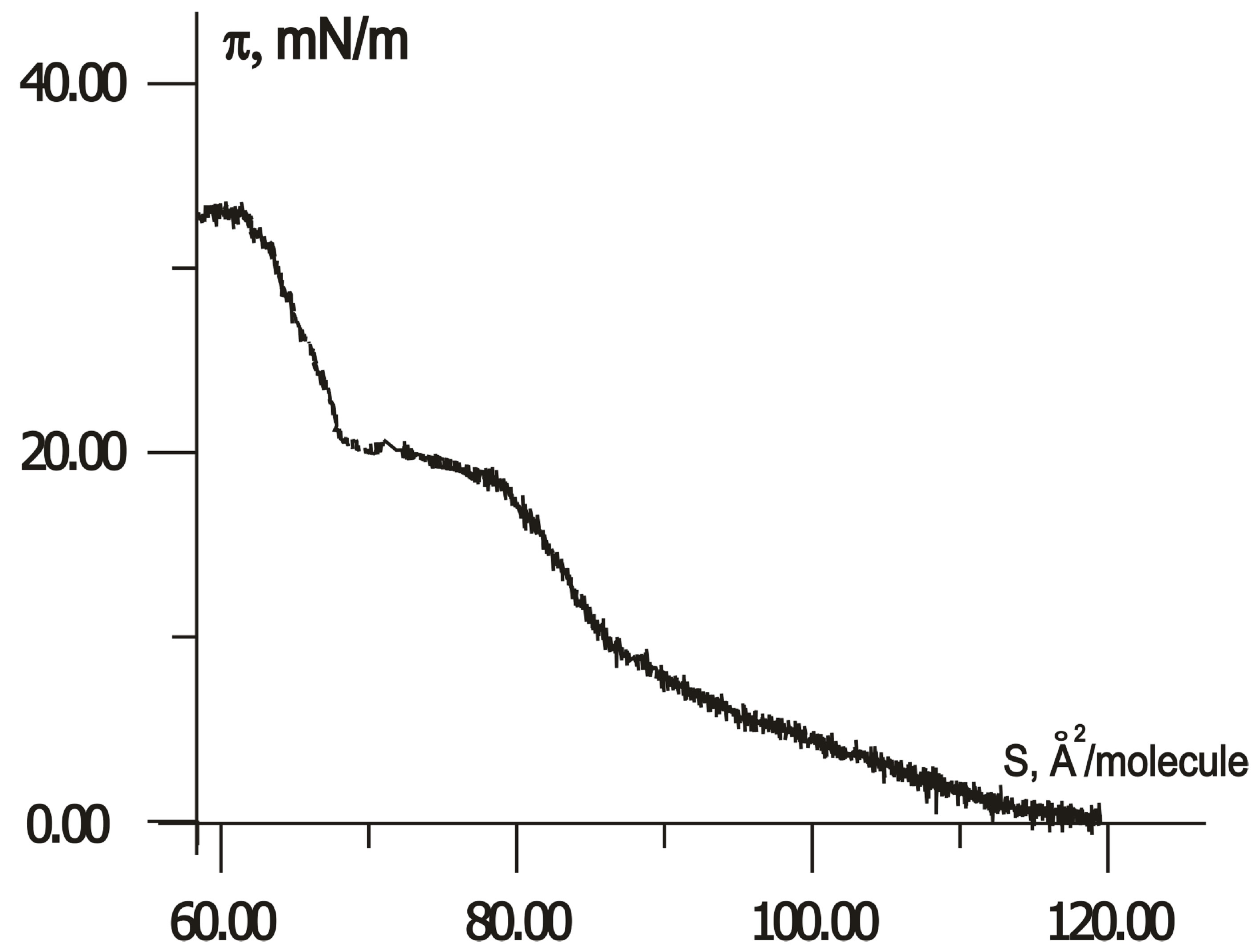 (b)
(b) (c)
(c)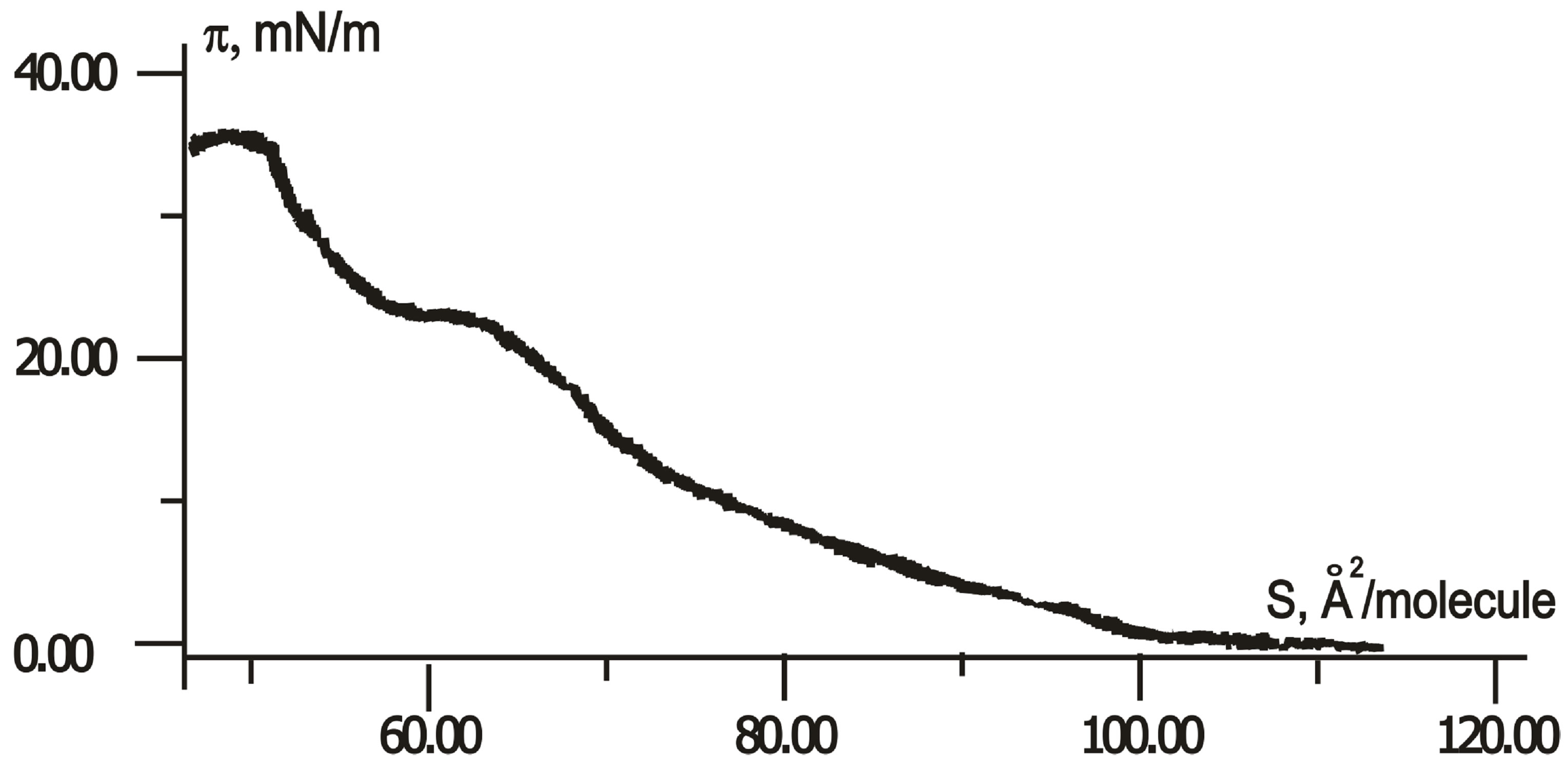 (d)
(d)
Figure 3.  -S isotherms for compressed H-dithiopyrrole monolayers on a surface of Fe(NO3)3 solution with Ce (a), Sm (b) and Er (c) at concentration ratio [Ln]/[Fe] = 1 and without rare earth elements (d).
-S isotherms for compressed H-dithiopyrrole monolayers on a surface of Fe(NO3)3 solution with Ce (a), Sm (b) and Er (c) at concentration ratio [Ln]/[Fe] = 1 and without rare earth elements (d).
on the type of rare earth element in water subphase with Fe(NO3)3. According to isotherms in Figure 3, SCe = 87.5 Å2/molecule, SSm = 80 Å2/molecule, SEr = 72.5 Å2/molecule with ratio [Ln]/[Fe] of rare-earth metals and Fe concentrations equals to unity, without rare-earth element-SFe = 62 Å2/molecule.
In Figure 4 the dependence of area SLn per Hithiopyrrole molecule on the relative concentration [Ln]/ [Fe] is shown. S-shaped character of the area SLn dependence upon the concentration of rare earth ion is manifested in monolayers formed with cerium and erbium ions. For samarium there is no such inflection point.
Thermodynamic studies carried out for monolayers of thiophenepyrrole compounds with ferric ions and rare earth elements allow to establish the dependence of the area S per H-dithiopyrrole molecule upon ionic radius of rare-earth element shown in Figure 5. In Figure 5 the

Figure 4. Dependencies of the area SLn, per H-dithiopyrrole molecule in crystalline monolayer on concentration [Ln] of rare earth ion being in subphase: for cerium (D), samarium (ƒ), erbium (O).
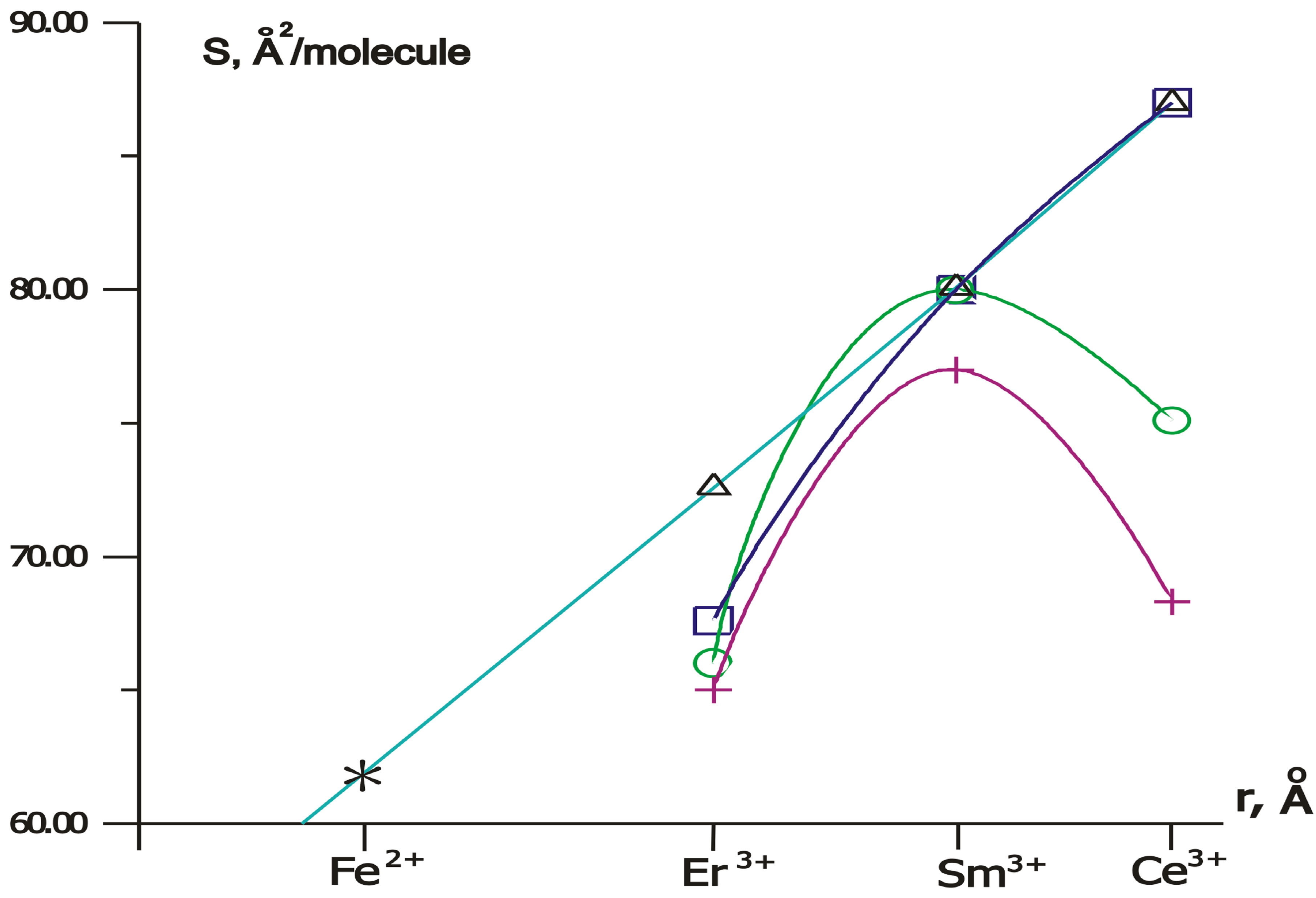
Figure 5. Dependencies of the area S per H-dithiopyrrole molecule in crystalline LB monolayer upon the radius r, Å of metal ion at concentration ratio [Ln]/[Fe] =1 (D); 0.2 (ƒ); = 0.1 (O); 0.01 (+). “*” denots SFe (rFe).
radius of high-spin iron ion Fe2+, 3d6 is 0.78 Å [35], and the radii of trivalent ions are 1.081 (Ce3+), 1.002 (Sm3+), 0.912 (Er3+) Å [36]. Data fitting was performed with coefficient of determination R-squared = 0.993892. According to the data shown in Figure 5, we conclude that the area SLn is proportional to the radius of the rare earth ion rLn at high concentrations of rare-earth atoms ([Ln]/[Fe] = 1). So, in this case the size of the area S correlates with metal ionic radius r because the sequence
 for [Ln]/[Fe] = 1 (5)
for [Ln]/[Fe] = 1 (5)
is in direct correspondence with a sequence
 .
.
For lower concentrations ([Ln]/[Fe] = 0.01, 0.1, 0.2), the proportional dependence of SLn on the rare earth ion radius rLn is not observed, and we have the following sequences: 1)  for [Ln]/[Fe] = 0.012)
for [Ln]/[Fe] = 0.012)  for [Ln]/[Fe] = 0.13)
for [Ln]/[Fe] = 0.13) 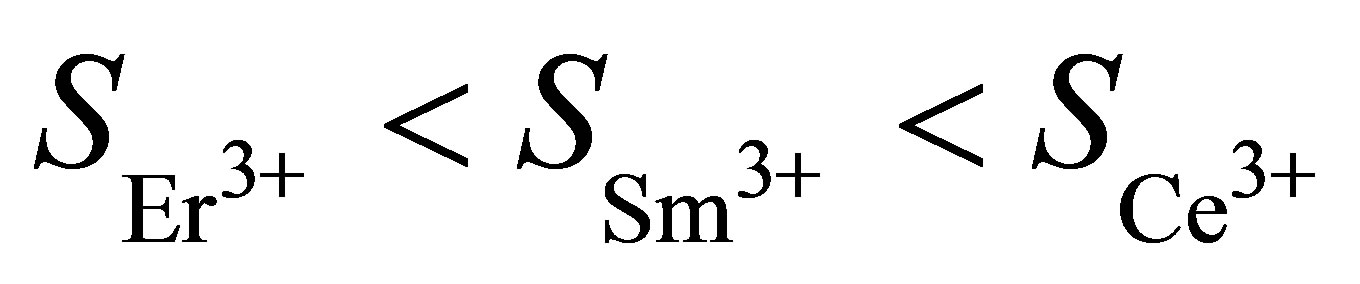 for [Ln]/[Fe] = 0.2.
for [Ln]/[Fe] = 0.2.
Further, let us find metal valences in thin LB film for different concentration of rare-earth metals.
5. Valence States of 2D-Metal Centers in Dithiopyrrole Thin LB Films
The capacitance C of the near electrode Helmholtz double layer comprising an ultra-thin LB film depends on the charge state of its constituents. Therefore, the valence state of metal atoms in the film was determined from the increment of the impedance of a sensor placed into deionized water.
Let  be increments of capacitances
be increments of capacitances
 ,
,  of the charged double layer of the sensor with LB film formed before and after the first-order phase transition in respect to the capacitance of a pure sensor (without LB-film) in deionized water:
of the charged double layer of the sensor with LB film formed before and after the first-order phase transition in respect to the capacitance of a pure sensor (without LB-film) in deionized water:
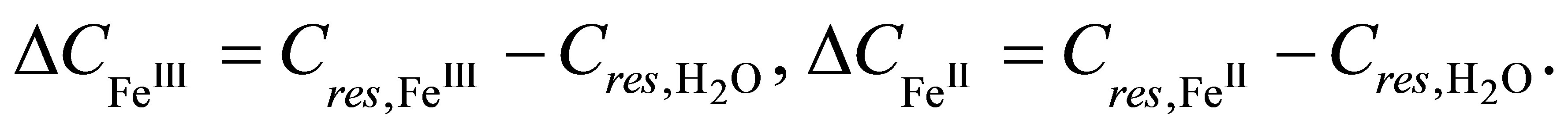
We estimate the ration 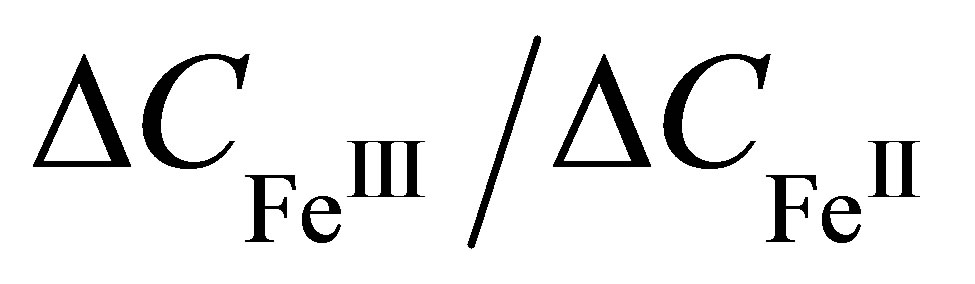 using Garnett formula (1). Let us suppose that
using Garnett formula (1). Let us suppose that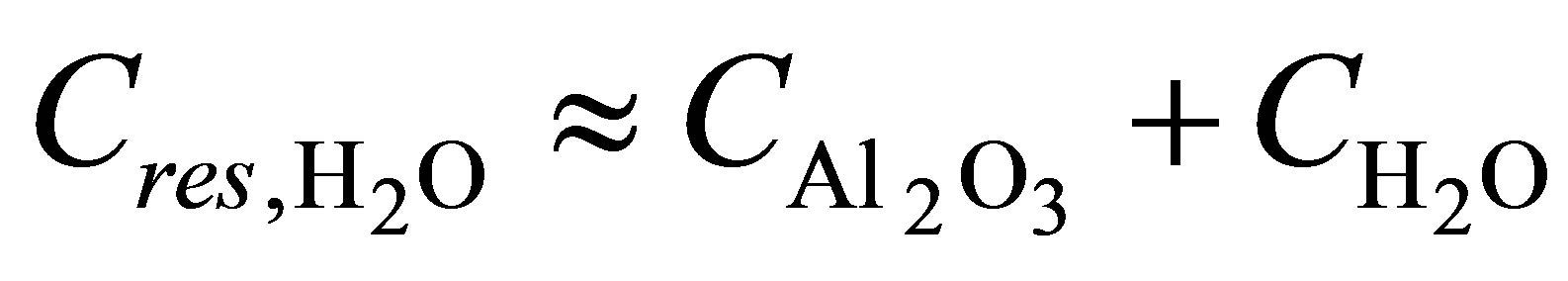 , where
, where  are capacitance of the nanoporous AOA and pure water in pores respectively. Then, in the case of low concentrations
are capacitance of the nanoporous AOA and pure water in pores respectively. Then, in the case of low concentrations  , taking into account a relationship of the dielectric permeability e with polarizability a and dipole moment
, taking into account a relationship of the dielectric permeability e with polarizability a and dipole moment 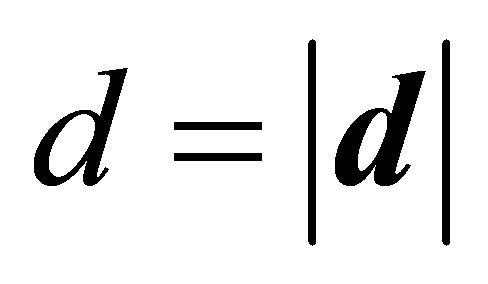 in an electric field with strength
in an electric field with strength :
:
 the Formula (1) allows to evaluate the relationship
the Formula (1) allows to evaluate the relationship 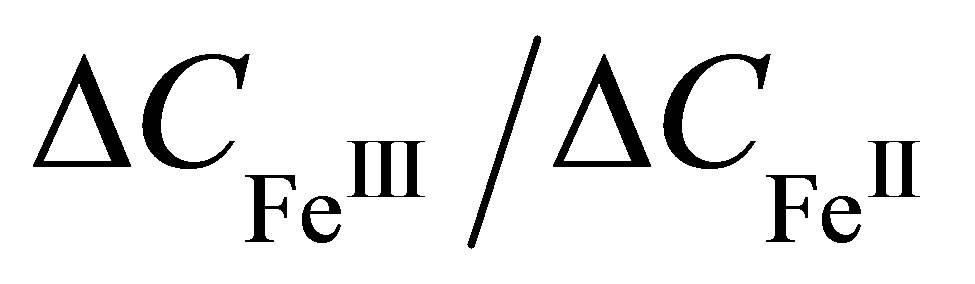 as
as
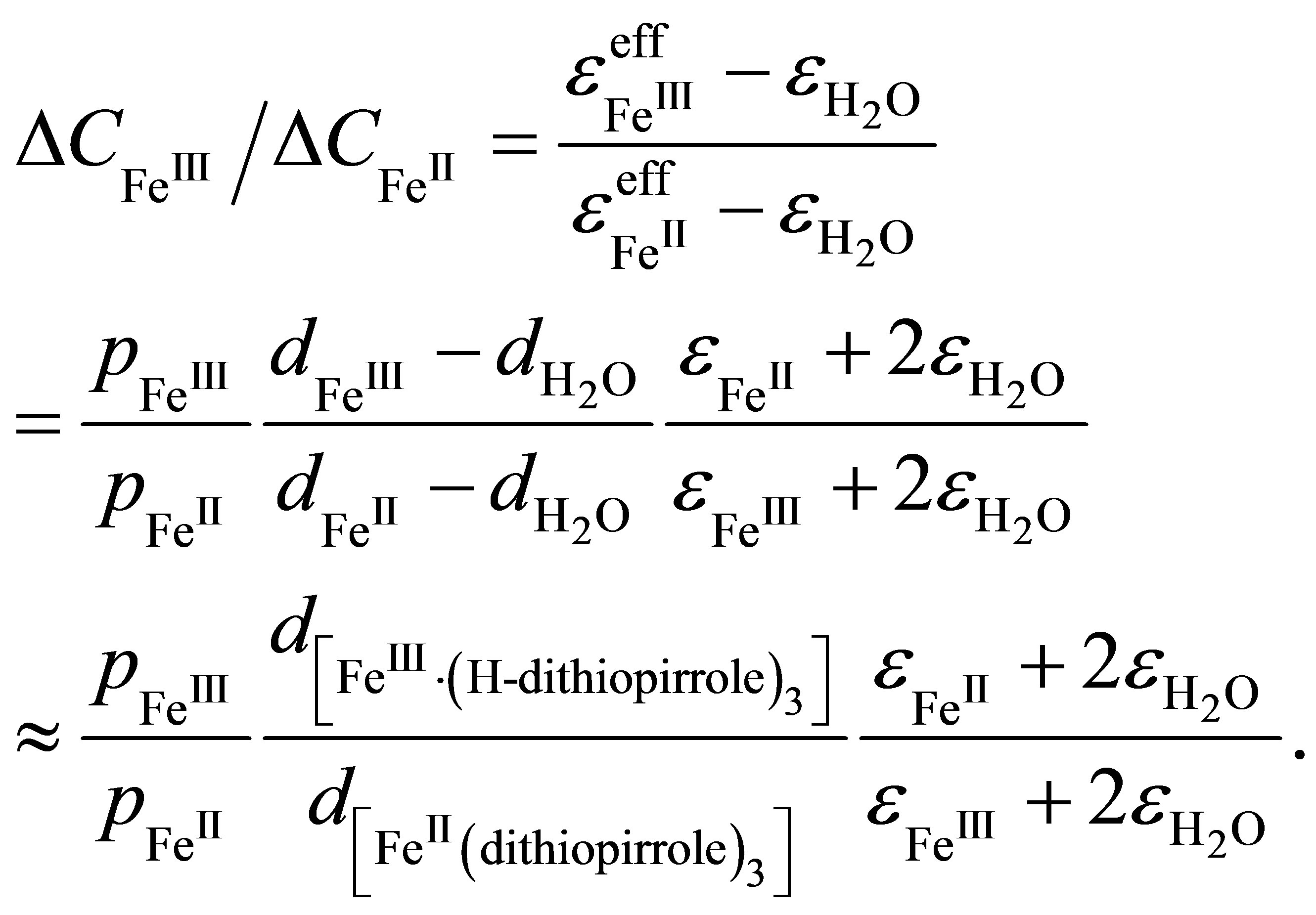 (6)
(6)
Here it is assumed that the differences between the dipole moments of the hydrated metal centers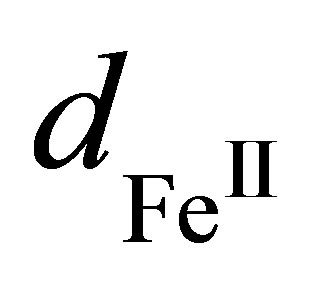 ,
,  and water
and water  are dipole moments
are dipole moments
 and
and  of dithionilepyrrole complexes of bivalent and trivalent Fe, respectively. Since the sizes of the complexes and their dielectric permeabilities can be considered as approximately equal, and the volume fraction p is not changed after the phase transition, according to Formula (6) the mean value of ratio of the capacitances increment
of dithionilepyrrole complexes of bivalent and trivalent Fe, respectively. Since the sizes of the complexes and their dielectric permeabilities can be considered as approximately equal, and the volume fraction p is not changed after the phase transition, according to Formula (6) the mean value of ratio of the capacitances increment  is approximately equal to the ratio
is approximately equal to the ratio
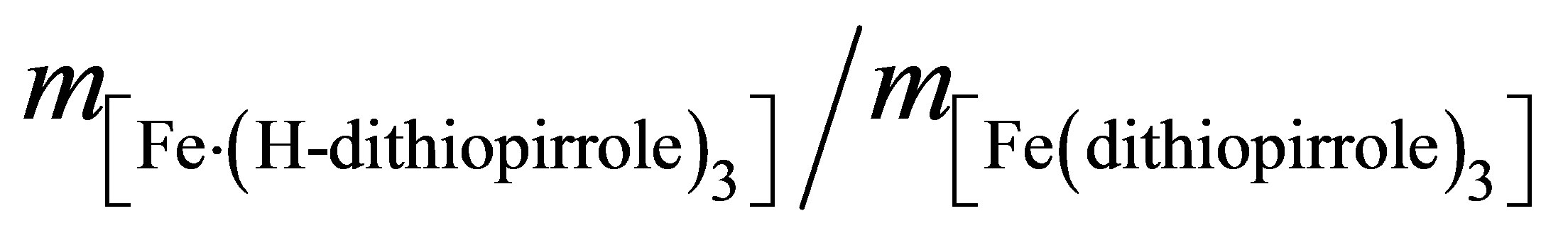 of the metal center valences before and after the phase transition:
of the metal center valences before and after the phase transition:
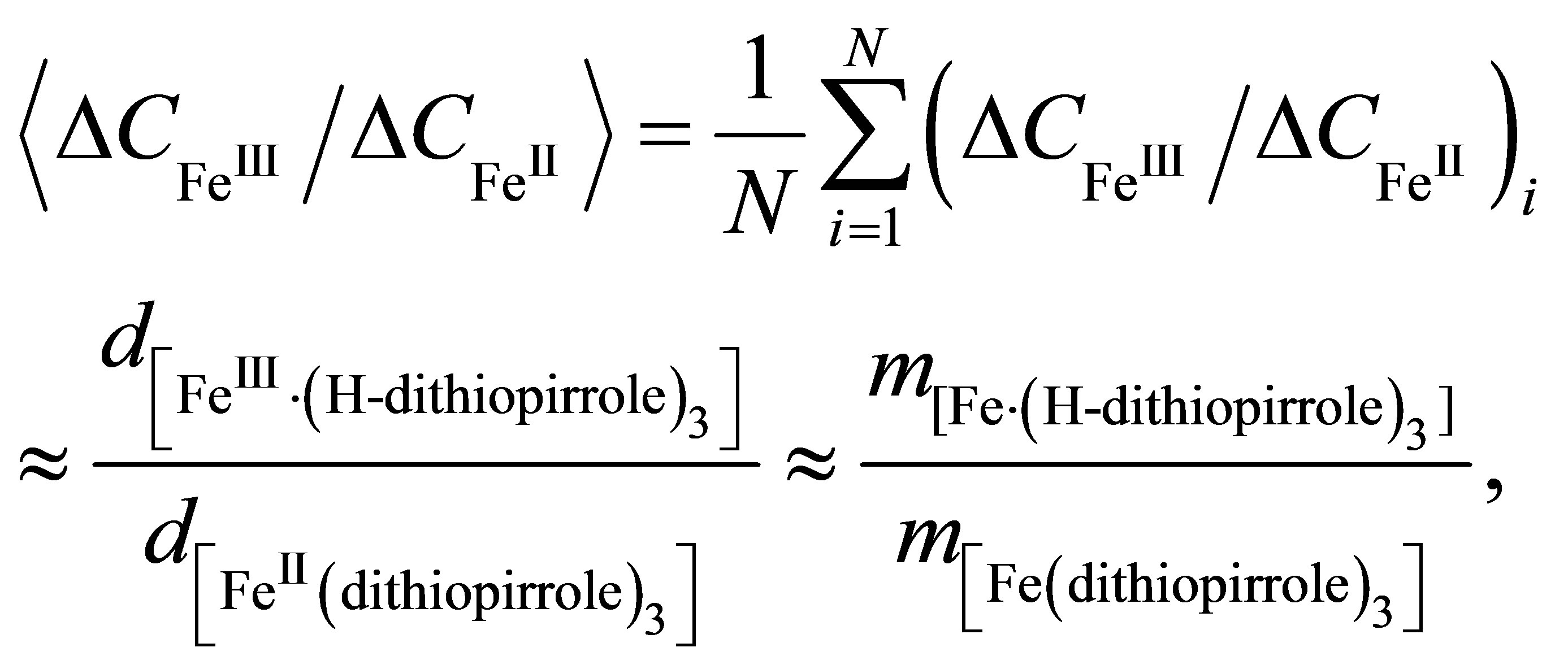 (7)
(7)
where N is the number of experimental data (N = 50). Averaging of the experimental data gives a following value of the ratio (7):
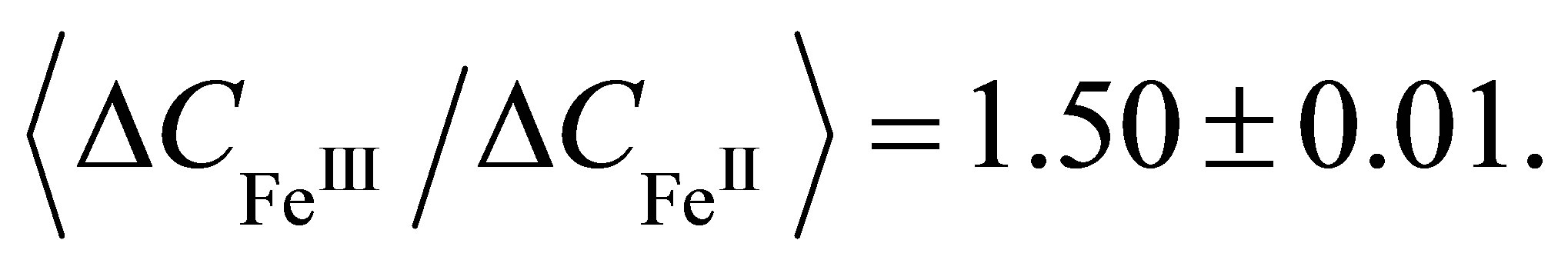 (8)
(8)
It follows from the ratio (8) that the amorphous LB film, which is formed before the phase transition, contains trivalent ions Fe(III). And valence mFe of the metal center in the crystalline iron-containing dithionilepyrrole LB film which is formed on the subphase without rare earth elements after the phase transition, is equal to 2 (Fe(II)). Therefore, a change in Fe valence state from trivalent to bivalent one takes place at the first-order phase transition in the Langmuir monolayer [37].
Valence mv of the Fe-containing dithionilepyrrole LB films at inclusion of rare-earth ions in different concentrations [Ln] is calculated by the Formula (7). The valence mv of the Fe-rare-earth-containing dithionilepyrrole LB films after the phase transition is represented in Figure 6. One can see that the dependences of valence mv for crystalline phase on the concentrations ratio [Ln]/[Fe] have extrema at values [Ln]/[Fe] < 0.2 for Ln =
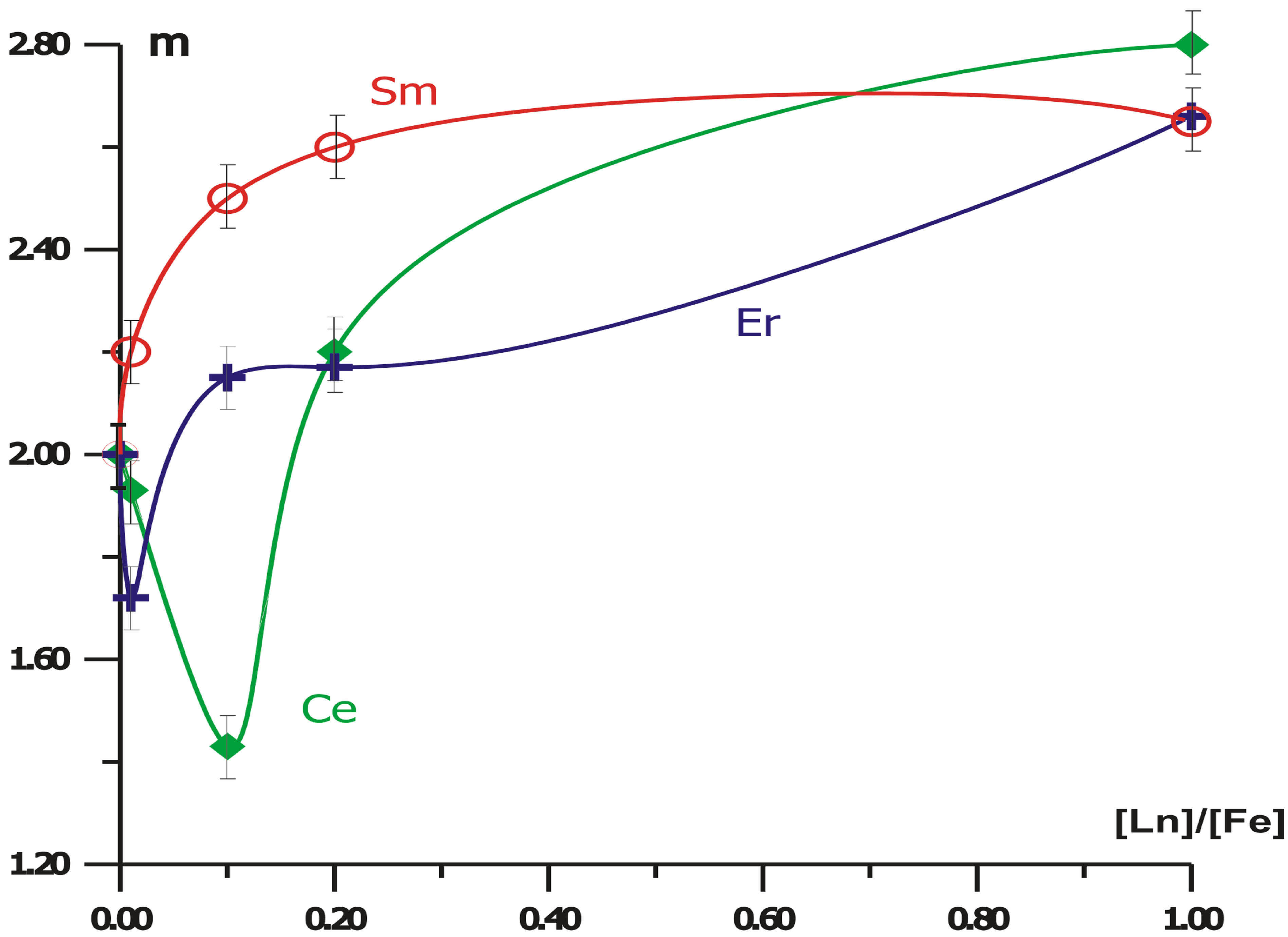
Figure 6. Dependencies of the metal center valence m on rare-earth salt concentration for crystalline iron-containing dithionilepyrrole LB monolayers forming in sub-phase with Ce (¨), Sm (O), Er (+).
Ce, Er. The extremum is not observed for Sm. Probably, the dependences are related to the electronic structure of rare-earth metals and their integer valence states, namely +3, +4 for Се; +3, +2 for Sm; +3, +1 for Er.
6. Mechanism of Complex Formation
6.1. Excitonic Nature of the Transition Metal Mixed Valence State in Organometallic LB Complexes
The distinctive feature of the crystalline iron-containing dithionilepyrrole LB monolayers are exciton mechanism of metal atoms excitation in nano-cavities of organic diamagnetic matrix and magnetic ordering [8]. These properties are stipulated by paramagnetism of iron-containing complexes of aromatic macrocyclic ligands [38], from which during the self-assembly process (provided deformation of their electron density) the multi-nuclear organometallic 2D-complexes of thiophene pyrrole oligomer are formed. Nonlinear optical properties of the Langmuir-Blodgett films are caused by the transfer of energy released at localization of π-electron states of the organic ligand environment to a three-particle excitation in the conduction band which consists of two electrons and a hole (negative exciton). The long lifetime of the spin-polarized electron states of the metal atoms leads to the Hanle effect [8]. During excitation of negative exciton, the transition of localized d-electron of transition metal (Fe) into delocalized p(d)-orbitals of the carbon subsystem takes place.
According to the above considered experimental and theoretical data, the increase in the valence mFe of Fe from (+2) to (+3) is related with the transition of localized d-electrons of valence band to the conduction band. Respectively, this phase of the mixed valence state is a metallic one. The presence of hybridization processes testifies strong exchange interactions for the excited levels, which lead to the appearance of the resonant band gap [39,40] and, as a consequence, to the transfer of electrons from excited d-levels of carbon subsystem on ground s-, p-levels.
Strong correlation interactions in the ground state tighten the energy gap [8,39], that leads to the vanishing of the forbidden resonance band and the return of electrons from hybridized electronic orbitals on localized valent levels of the metal subsystem. Since the electron returns to the valent band and is localized on Fe atom, its valence decreases from (+3) to (+2). Due to the fact that in the ground state there are no conditions for hybridization of electronic orbitals of the carbon and metal subsystems, and p-electrons of undoped chain of double bonds in the carbon matrix are localized, the metallic phase of the mixed valence state is changed to semiconductor phase with bivalent ions Fe2+.
Let us assume that the semiconductor state lifetime trg or what is the same, the time of resonance exchange gap existence is larger than the time of the hybridization process tH: trg > tH. With such a relation of relaxation times tH and trg, the hybridization process running in the excited state is virtual. Therefore, the electron transitions from the localized d-levels of metal to delocalized d-levels of the carbon subsystem are accompanied by only virtual, unobservable process of the change in the valence state of Fe, without appearance of other ions, except of Fe2+, in electrochemical measurements.
Thus, the mixed valence state is formally a coexistence of domains of semiconductor (Fe2+) and metal phases (Fe3+). The charge carrier in such a system is a negatively charged exciton, which consists of a coherent electronhole pair and an unpaired electron.
6.2. The Intermediate Valence of the Transition Metal in the Ln/Fe-Center with Nano-Cyclic Thiophenepyrrole Ligands
In this subsection we discuss the mechanism of metastabilization of hybrid electron orbitals through the indirect exchange-exchange interaction between f-and delectrons.
The exchange interactions in the sub-lattice of Ln atoms and between the Ln sublattices and transition-metal atoms (indirect exchange) are weak [12,20-25,40-42]. Sign of the value of the exchange interactions determines magnetic properties of the compound. Negative exchange leads to diamagnetism, and the positive one—to paramagnetism. For lanthanides the negative exchange takes place in the lower valence state (II), and the positive one —for the high valence (III). For example, compounds of ytterbium (III, 4f13) demonstrate distinct paramagnetism and ytterbium compounds (II) are diamagnetic [43-46]. Interaction of local spin moments of f-electrons and negatively charged coherent exciton results in alignment of spins of rare-earth atoms and, hence, to the emergence of a magnetic field for rare-earth sublattice. The direction of this alignment depends on the sign of the exchange interactions.
Let us discuss experimental study of the valence state which is represented in Figure 5. One can observe a linear dependence of the area per molecule upon ionic radii for relative concentration ratio [Fe]/[Ln] = 1, when taking into account radius of the high-spin iron (Fe2+) and the radii of trivalent rare-earth atoms. Then, taking into account the correlation between the sign of the exchange interactions and valence of lanthanide, the metal-polymer complexes in the dithionilepyrrole LB films are paramagnetic, that is also consistent with the conclusion made on the results of researches of magnetic atomic force microscopy on paramagnetism of the ironcontaining dithionilepyrrole LB films [38].
Due to paramagnetism of the films, a direction of magnetic field  produced by magnetic moments of the rare-earth atoms coincides with the direction of nonequilibrium spin Sex of the exciton state. Since the field
produced by magnetic moments of the rare-earth atoms coincides with the direction of nonequilibrium spin Sex of the exciton state. Since the field  is weak, the Zeeman splitting exhibits itself in removal of the levels degeneracy of trivalent Fe(III) in the metal phase of the mixed valence state.
is weak, the Zeeman splitting exhibits itself in removal of the levels degeneracy of trivalent Fe(III) in the metal phase of the mixed valence state.
Let us assume that for lower Zeeman sublevels of Fe3+-center, as well as for the ground state of Fe2+-center, the direction of spins is “down”, and for the top is “up”. Since the relaxation time tS of nonequilibrium spin Sex is large by virtue of nano-size of metal center, the lifetime of semiconductor phase of the mixed state, having the order of the resonance exchange gap lifetime trg in the band structure, is less than tS: tS > trg. Therefore, the Pauli principle forbids the transition from the conduction band to the valence band. As a consequence, the hybridization of d-orbitals for C and Fe atoms stops to be a virtual. Therefore, a part of Fe atoms transfers into metastable trivalent state. The presence of both Fe2+ and Fe3+ gives intermediate “fractional” valence observed in the electrochemical experiments (Figure 6).
In conclusion of this subsection we mention the naivety of an idea about presence in Langmuir-Blodgett film both coordination iron centers Fe(II), Fe(III) due to balance shift towards Fe3+-centers at coordination interaction of rare-earth metals, mainly with S, but not with N. The consequence of such a naive explanation of “fractional” valence would be multi-component chemical composition and heterogeneity, which are not observed according to the experimental data above presented.
6.3. The Valence of the Rare Earth Center of Complex with Dithionilepyrrole Ligand in Fe-Containing Polymer LB Monolayer of Thiophenepyrrole Series Derivatives
In this subsection, using the experimental data and the model representations about the mixed-valence state of iron, we estimate the valence of the rare-earth center intercalated into the thin dithionilepyrrole LB film.
LB technique allows to grow epitaxially sulfide nanocrystals, such as PbS, ZnS [47-50]. To do this, it is necessary to find such experimental conditions that active thiophene groups of monolayer near the interface would create a lattice with parameters close to the corresponding sulfide lattice ones. Therefore, accounting the presence of trivalent ions Er3+, or Sm3+, or Ce3+ in subphase, we can make an assumption about an epitaxial growth of 2D-crystals of rare earth sulfide derivatives (Ln2S3) on the surface of the dithionilepyrrole Langmuir monolayer. One should bear in mind that thermodynamic stability of sulfides Ln2S3 is different. So, Ce sulfide is decomposed in acids [51], Er sulfide is thermodynamically unstable [52], whereas SmS is resistant to aggressive media at room temperatures [53].
Besides sulphides, Ln can also form intermetallic compounds with Fe according the chemical formula of Ln2Fe17. Since the formula of intermetallic compound contains a large number of Fe atoms, the chemical equilibrium between sulfides and Ln intermetallic compounds depends not only on the stability of sulfide, but also on concentration of the transition metal. Therefore, it is necessary to consider separately cases of low and high iron concentrations.
The case of relatively low concentrations of Fe ([Fe]/[Ln] ≤ 1).
At low concentrations of Fe in respect to rare earths, the probability to find in nano-cavities a number of Fe atoms sufficient to form intermetallic compound is small. Consequently, in this case one can neglect the possibility of chemical reactions with formation of intermetallic compounds and propose the complexification reaction in the form:
 (9)
(9)
where Ln = Sm, Er, Ce; ligand thionyl is one of two dithionilepyrrole parts, pyrrole is another part of dithionileperrole molecule.
As established above, during the compression, a coordinational-polymeric hexagonal network of nano-cyclic ligands with transition metal centers is formed at the phase transition. The linear dependence of the area per molecule on the radii of Ln3+ and Fe2+ in Figure 5 and the absence of the first-order phase transition on the subphase without Fe ions indicate that steric hindrance to find a large rare-earth atoms in LB monolayer disappear with the emergence of nanocavities. Therefore, the complexes of trivalent rare-earth silicides type can be produced in ligand nano-cavities.
In accordance with peculiarities of Ln intermediate valence state manifestation, the probability for conduction-band delocalized s(p)-electrons to transit to the valence band f-orbitals of Sm and Er with decreasing their valence to (+2) or (+1) is negligible small in the compressed monolayer. And, contrary, f-electrons of Ce can be delocalized with increasing the valence of Ce to (+4) at the compression.
Blue color LB film preserves at inclusion of Ln atoms into it. Moreover, in UV and visible wavelength range spectra for Fe-containing LB-films with Er, the transitions between levels of rare earth atoms are in red spectral region [54]. These spectral peculiarities can be explained by trivalence of Ln ions intercalated in the monolayer, because trivalent rare earth compounds are usually weakly colored [55-57] in contrast to bivalent ones [55,58].
Also, there is a weak dependence of the experimental values of intermediate valence (mex) (see Figure 6) upon the Ln type for concentration ratio [Ln]/[Fe] ≥ 1:  and
and 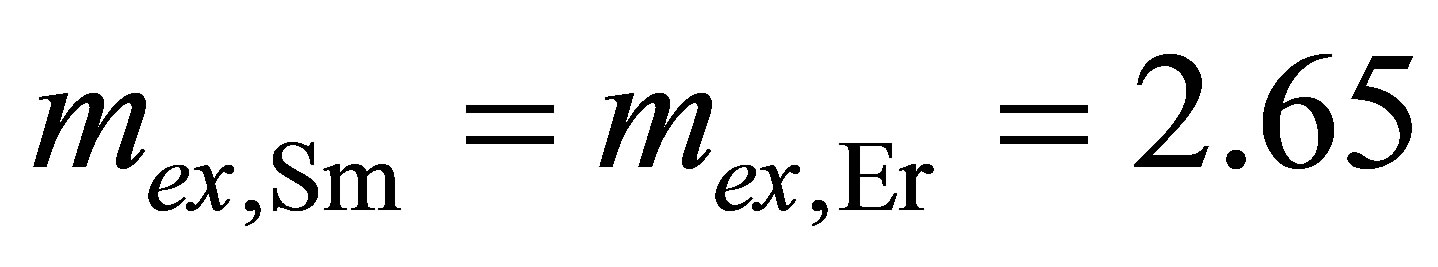 for Ceor Er-, Sm-containing LB films with octahedral Fe(II) centers, respectively.
for Ceor Er-, Sm-containing LB films with octahedral Fe(II) centers, respectively.
From the aforesaid, and in combination with 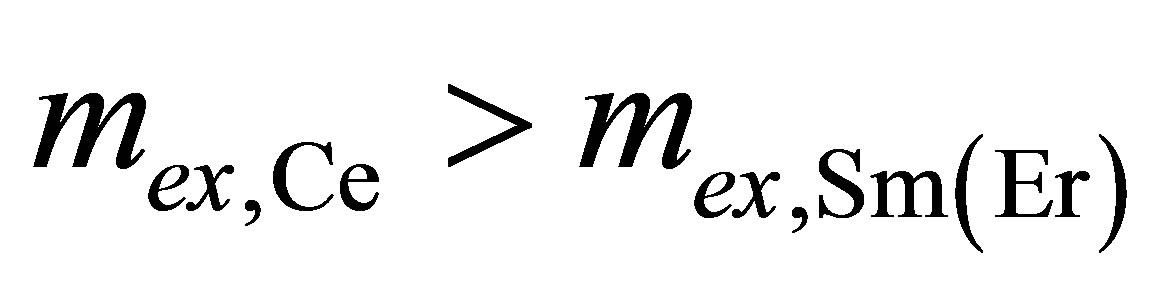 it follows that Sm and Er atoms preserve their original valence (+3) whereas Ce atoms may be in the intermediate valence state in the LB monolayer.
it follows that Sm and Er atoms preserve their original valence (+3) whereas Ce atoms may be in the intermediate valence state in the LB monolayer.
Now, given the above established valence of Sm and Er, we can estimate a mixed valence mFe of iron Fe in the dithionilepyrrole LB film using the formula
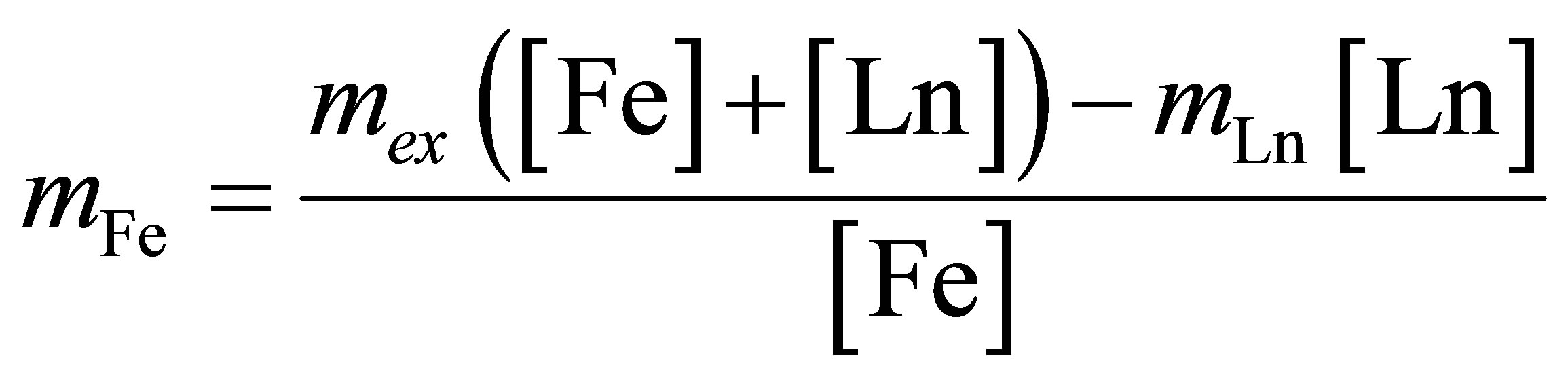 , (10)
, (10)
where mLn is the valence of lanthanide. Substituting mex into this formula, we obtain mFe ≈ +2.3 for Er-, Smcontaining LB films.
Assuming mFe to be a constant for a given ligand environment, we can estimate intermediate valence of Ce using the Formula (10). Substituting the value mFe in the Formula (10), we find mСe ≈ +3.3 which is the same as for bulk intermetallide Се2Fe17 [8].
The case of relatively high Fe concentrations ([Fe]/[Ln] > 10).
Chemical equilibrium can be shifted toward forming of 2D-intermetallic compounds such as Ln2Fe17 at such low concentrations of trivalent Ln ions in the subphase. Let us consider the conditions under which it occurs.
The experimental data: 1) S-shaped dependence of the area per dithionilepyrrole molecule on concentration ratio [Fe]/[Ln] for Er and Ce and its absence for Sm (see Figure 4).
2) The indermediate valence mex < 2 for the Ce-or Er-containing LB films if [Fe]/[Ce] ≥ 10 or [Fe]/[Er] ≥ 100, respectively, and the intermediate valence mex > 2 for the Sm-containing LB films for arbitrary values of [Fe]/[Sm] (Figure 6), and 3) thermodynamic instability of Er and Ce sulphides, indicate a change of complexification mechanism for Er and Ce, but not for Sm at the high concentrations of Fe.
In addition, in metal complexes with aromatic ligands in the presence of S the reduction of ferric chloride up to Fe1+ and Fe0 is possible [59-61]. Fe reduction is caused by its higher electronegativity, which is equal to 1.64, in comparison with the Ln, for example, the electronegativity of Ce is 1.08 [20].
Then, taking into account the existence of such types of Fe nitrides as Fe16N2, Fe4N, Fe3N, Fe2N and on the basis of the above written, we propose the following mechanism of Fe complexification with thiophene pyrrole series oligomer at [Fe]/[Ln] > 10.
Intercalation of Ce and Er, protruding as electron donor in the coordinating compound , provides reduction of Fe ions in the LB monolayer with formation of organo-intermetallic complex, e.g. in reaction of the form:
, provides reduction of Fe ions in the LB monolayer with formation of organo-intermetallic complex, e.g. in reaction of the form:
 (11)
(11)
where p, r, q are number of atoms Fe in organointermetallide (11) with valence +2, +1, 0 respectively; p + r + q = 17.
Mean or “fractional” valence  is determined by the formula
is determined by the formula
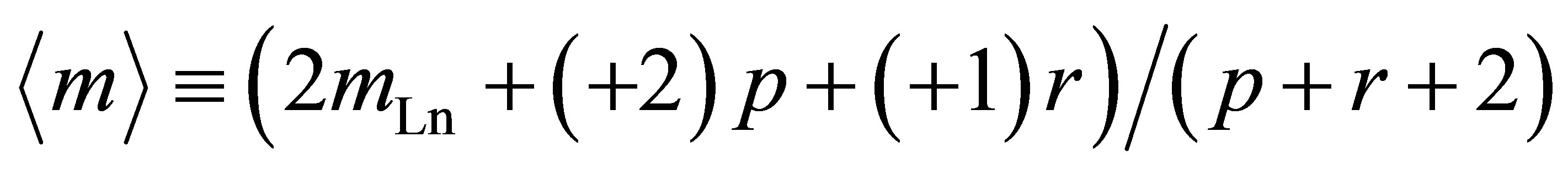 . (12)
. (12)
Now, from comparison of 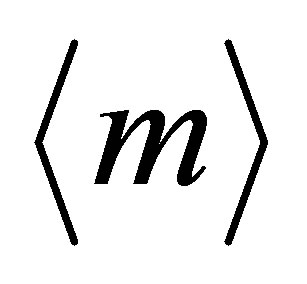 and experimentally observable values mex, we find the number p, r, q of Fe atoms of appropriate valence in the complex (11). The Formula (12) gives values
and experimentally observable values mex, we find the number p, r, q of Fe atoms of appropriate valence in the complex (11). The Formula (12) gives values  = +1.71, +1.91 close to mex ≈ +1.72, +1.92 for Er (mEr = +3), Ce (mCe = +3.7), respectively, when p = 1, r = 4, q = 12. This means that at [Fe]/[Ln] > 10 the 2D-LB-complexes are produced in the form
= +1.71, +1.91 close to mex ≈ +1.72, +1.92 for Er (mEr = +3), Ce (mCe = +3.7), respectively, when p = 1, r = 4, q = 12. This means that at [Fe]/[Ln] > 10 the 2D-LB-complexes are produced in the form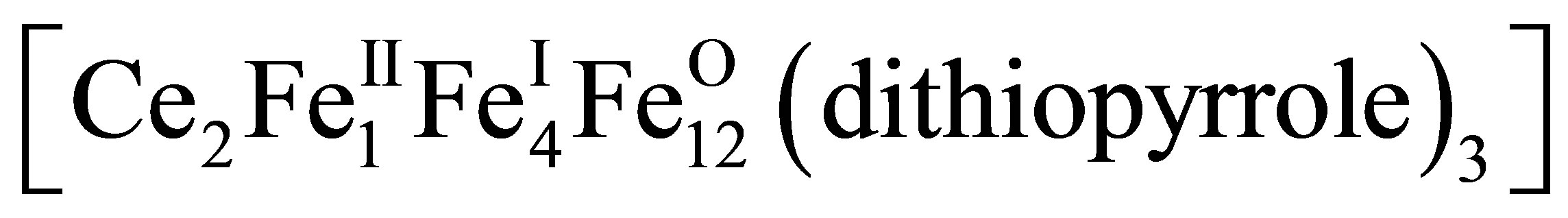 ,
, 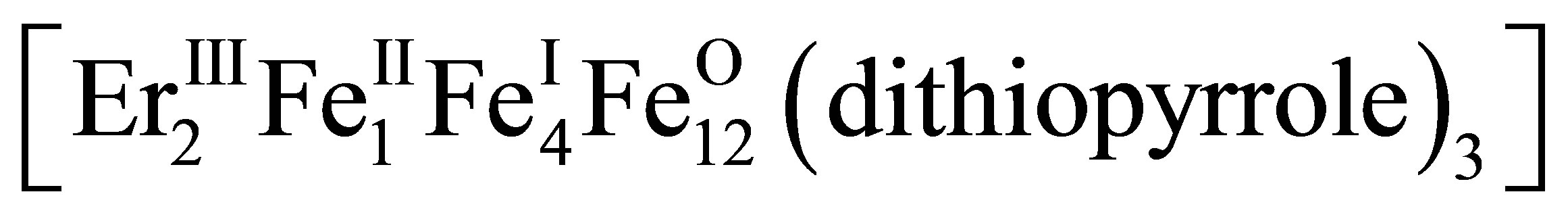 with trivalent Er and Ce being in the state of intermediate valence +3.7.
with trivalent Er and Ce being in the state of intermediate valence +3.7.
For generality, it is necessary to note the following. Other organometallic complexes were synthesized, such as lanthanide complexes of bipyridine (bipy) [Ln(bipy)4], which also form infinite coordination polymeric chains in crystalline state [62-66]. In contrast to the proposed approach, their nature is considered within the concept of polymeric complexes with charge transfer.
7. Conclusion
Thus, a coordinational-polymeric hexagonal network of nano-cyclic ligands with metal centers is formed at the phase transition when compressing the Langmuir monolayers of thiophene pyrrole series oligomer with alkyl fragment on surface of subphase with rare-earth and/or Fe ions. Intermediate valence states in the thin Fe-containing LB film for different concentration of rare-earth metals Er, Ce, Sm have been established.
8. Acknowledgements
The present work was supported by the National Academy of Sciences of Belarus through the Program “Convergence”.
REFERENCES
- V. Arslanov, Russian Chemical Advances, Vol. 69, 2000, p. 839. http://dx.doi.org/10.1016/j.snb.2005.04.021
- M. Kalinina, N. Golubev, O. Rightmen, et al., Sensor & Actuators, B, Chemical, Vol. 114, 2006, p. 19.
- A. Kakkar, Chemical Reviews, Vol. 102, 2002, p. 3579. http://dx.doi.org/10.1021/cr010360k
- A. Pfeil and J.-M. Lehn, “Helicate Self-Organization— Positive Cooperativity in the Self-Assembly of DoubleHelical Metal-Complexes,” Journal of the Chemical Society, Chemical Communications, 1992, p. 838.
- V. Kaganer, H. Möhwald and P. Dutta, Reviews of Modern Physics, Vol. 71, 1999, pp. 779-819l. http://dx.doi.org/10.1103/RevModPhys.71.779
- M. Rikukawa and M. Rubner, Langmuir, Vol. 10, 1994, p. 519. http://dx.doi.org/10.1021/la00014a030
- E. Punkka, M. Rubner, J. Hettinger, J. Brooks and S. Hannahs, Physical Review B: Condensed Matter, Vol. 43, 1991, p. 9076. http://dx.doi.org/10.1103/PhysRevB.43.9076
- H. Krylova and L. Hurski, “Spin Polarization in Strong Correlated Nanosystems,” LAP LAMBERT Academic Publishing, AV Akademikerverlag GmbH & Co., Germany, 2013.
- J. Steed and J. Atwood, “Supramolecular Chemistry,” John Wiley & Sons, New York, Singapore City, 2000.
- M. Alfimov, “Photonics of Supermolecular Nanosize Structures,” Izv. RAN, Ser. Chem., 2004, p. 1303.
- D. Khomski, Uspekhi Phys. Nauk, Vol. 129, 1979, pp. 443-485. http://dx.doi.org/10.3367/UFNr.0129.197911c.0443
- V. Shaburov, et al., JETPh Letters, Vol. 41, 1985, pp. 213-215.
- B. Johansson, Philosophical Magazine, Vol. 30, 1974, p. 469. http://dx.doi.org/10.1080/14786439808206574
- L. Finkelshtain, Phys. Metal & Material Sci., Vol. 57, 1984, p. 402.
- S. Vonsovski, M. Katsnelson and А. Тrefilov, Phys. Metal & Material Sci., Vol. 76, 1993, p. 3.
- E. Tonkov and I. Aptekar, Physics of the Solid State, Vol. 16, 1974, pp. 1507-1508.
- U. Schärer and P. Wachter, Solid State Communications, Vol. 96, 1995, pp. 497-501. http://dx.doi.org/10.1016/0038-1098(95)00418-1
- A. Sovetnov, et al., Physics of the Solid State, Vol. 44, 2002, pp. 1498-1500.
- A. Sovetnov, et al., Physics of the Solid State, Vol. 39, 1997, pp. 1017-1019.
- A. Lukoyanov, et al., Physics of the Solid State, Vol. 49, 2007, pp 95-101. http://dx.doi.org/10.1134/S1063783407010179
- N. Duc, “Handbook on the Physics and Chemistry of Rare Earths,” Vol. 24, 1997, p. 369. http://dx.doi.org/10.1016/S0168-1273(97)24008-0
- M. Brooks, L. Nordstrom and B. Johansson, Physica B, Vol. 172, 1991, p. 95. http://dx.doi.org/10.1016/0921-4526(91)90421-A
- J. Liu, F. de Boer, P. de Chatel, R. Coehoorn and K. Buschow, Journal of Magnetism and Magnetic Materials, Vol. 132, 1994, p. 159. http://dx.doi.org/10.1016/0304-8853(94)90310-7
- T. Jacobs, K. Buschow, G. Zhou, X. Li and F. de Boer, Journal of Magnetism and Magnetic Materials, Vol. 116, 1992, p. 220. http://dx.doi.org/10.1016/0304-8853(92)90166-L
- A. Liechtenstein, M. Katsnelson, V. Antropov and V. Gubanov, Journal of Magnetism and Magnetic Materials, Vol. 67, 1987, p. 65. http://dx.doi.org/10.1016/0304-8853(87)90721-9
- A. Kel’in and O. Kulinkovich, Folia Pharmaceutica Universitatis Carolinae, Vol. 18, 1995, pp. 96-97.
- H. Krylova, A. Drapeza and A. Khmelnitski, Devices and Control Systems, No. 6, 1993, pp. 43-44.
- I. Abramov, et al., St. Petersburg J. Electronics, Vol. 4, 2012, pp. 59-67.
- “RC-Generator of Harmonic Oscillations,” http://www.gaw.ru/html.cgi/txt/doc/op/funop_14_2_2.htm
- A. Vinogradov, “Electrodynamics of Composite Materials,” URSS, Moscow, 2001.
- J. Garnett, Philosophical Transactions of the Royal Society, Vol. 205, 1906, p. 237. http://dx.doi.org/10.1098/rsta.1906.0007
- V. Hrushevski, et al., Vestnik BSU. Seria 1: Phy. Math. Inform., No. 2, 2012, pp. 23-28.
- B. Hasenkopf, J.-M. Lehn, B. Kneisel, et al., Angewandte Chemie International Edition, Vol. 35, 1996, p. 1838. http://dx.doi.org/10.1002/anie.199618381
- W. Kaim and B. Schewederski, “Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life,” Wiley, Chichester, 1994.
- R. Shannon and C. Prewitt, Acta Crystallographica Section B, Vol. 25, 1969, p. 925; Vol. 26, 1970, p. 1046.
- A. Ryabukhin, High Temperature Melts, No. 1, 1997, pp. 58-63.
- V. Hrusheski, et al., Proceedings of the 6th International Conference ‘Medical Social Ecology of Personality’, Minsk, 4-5 April 2008, Vol. 1, pp. 120-123
- V. Anishchik, N. N. Dorozhkin, H. V. Grushevskaya, V. V. Hrushevsky, G. G. Krylov, L. V. Kukharenko and M. A. Senyuk, Proceedings SPIE, Vol. 5219, 2003, pp. 141- 150. http://dx.doi.org/10.1117/12.506173
- G. V. Grushevskaya, L. I. Komarov and L. I. Gurskii, Physics of the Solid State, Vol. 40, 1998, pp. 1802-1805. http://dx.doi.org/10.1134/1.1130660
- M. Brooks, L. Nordström and B. Johansson, Physica, B, Vol. 172, 1991, p. 95.
- J. P. Liu, F. R. Boer, P. Chatel, R. Coehoorn and K. H. J. Buschow, Journal of Magnetism and Magnetic Materials, Vol. 132, 1994, pp. 159-179. http://dx.doi.org/10.1016/0304-8853(94)90310-7
- T. H. Jacobs, K. H. J. Buschow, G. F. Zhou, X. Li and F. R. Boer, Journal of Magnetism and Magnetic Materials, Vol. 116, 1992, pp. 220-230. http://dx.doi.org/10.1016/0304-8853(92)90166-L
- W. J. Evans and M. Horzbor, Journal of Organometallic Chemistry, Vol. 326, 1987, pp. 299-306. http://dx.doi.org/10.1016/0022-328X(87)87001-8
- P. Scott and P. Hitchcock, Journal of the Chemical Society, Chemical Communications, 1995, pp. 579-580.
- I. Gavrilov and V. Kravchenko, The Journal of Structural Chemistry, Vol. 14, 1972, p. 345.
- O. Yoshihiro and T. Isobe, Inorganica Chimica Acta, Vol. 144, 1988, pp. 143-146. http://dx.doi.org/10.1016/S0020-1693(00)80978-0
- J. Fendler, “Membrane-Mimetic Approach to Advanced Materials,” Springer, Berlin, 1994. http://dx.doi.org/10.1007/BFb0020989
- B. Bunker, P. Rieke, B. Tarasevich, A. A. Campbell, G. E. Fryxell, G. L. Graff, L. Song, J. Liu, J. W. Virden and G. L. McVay, Science, Vol. 264, 1994, pp. 48-55. http://dx.doi.org/10.1126/science.264.5155.48
- V. Klechkovskaya and L. Feygin, Crystallography, Vol. 41, 1998, pp. 975-982.
- N. Novikova, S. Zheludeva, O. Konovalov, M. Kovalchuk, N. D. Stepina, I. V. Myagkov, Y. K. Godovsky, N. N. Makarova, E. Yu. Tereschenko and L. G. Yanusova, Journal of Applied Crystallography, Vol. 36, 2003, pp. 727-731. http://dx.doi.org/10.1107/S0021889803000189
- B. Nekrasov, “Foundation of General Chemistry,” Chemistry, Moscow, 1973.
- “Erbium Sulfide,” ESPI Metals, CAS Number: 12159-66-9. www.espimetals.com.
- A. Visokikh, et al., Vestnik of Tumen State University, No. 3, 2007, pp. 124-129.
- A. Drapesa, H. V. Grushevskaya, V. V. Hrushevsky, V. M. Parkun, M. A. Senuk and M. V. Parkun, Proceedings SPIE, Vol. 6258, 2006.
- W. J. Evans, Polyhedron, Vol. 6, 1987, pp. 803-835. http://dx.doi.org/10.1016/S0277-5387(00)80921-3
- T. Moeller, “Comprehensive Inorganic Chemistry,” Pergamon Press, Oxford, 1973, Vol. 4, pp. 44.
- W. Carnall, “Handbook on the Physics and Chemistry of Rare Earths,” Elsevier, Amsterdam, 1979, Vol. 3, pp. 24.
- G. Meyer, Chemical Reviews, Vol. 88, 1988, pp. 93-107. http://dx.doi.org/10.1021/cr00083a005
- H. Brintzinger, G. Palmer and R. Sands, Journal of American Chemical Society, Vol. 88, 1966, p. 623. http://dx.doi.org/10.1021/ja00955a053
- [61] I. Alexandrov, et al., High Molecular Compounds, Ser.B, Vol. 51, 2009, pp. 1573-1577.
- [62] R. R. Rakhimov, E. M. Jackson, J. S. Hwang, A. I. Prokof’ev, I. A. Alexandrov, A. Y. Karmilov and A. I. Aleksandrov, Journal of Applied Physics, Vol. 95, 2004, p. 7133. http://dx.doi.org/10.1063/1.1668613
- [63] I. Fedushkin, M. Bochkarev, S. Dechert and H. Schumann, Chemistry-A European Journa, Vol. 7, 2001, pp. 3558-3563. http://dx.doi.org/10.1002/1521-3765(20010817)7:16<3558::AID-CHEM3558>3.0.CO;2-H
- [64] Н. Sitzmann, О. Schmitt, F. Weber and G. Wolmershauser, Zeitschrift fur Anorganische und Allgemeine Chemie, Vol. 627, 2001, pp. 1-3.
- [65] I. Fedushkin, S. Dechert and H. Schumann, Angewandte Chemie, Vol. 113, 2001, pp. 584-587. http://dx.doi.org/10.1002/1521-3757(20010202)113:3<584::AID-ANGE584>3.0.CO;2-7
- [66] I. Fedushkin, М. Bochkarev and Н. Schumann, “Synthesis, Molecular Structures and Some Phenomena of Bipyridyl Complexes of Lanthanoides,” Proceedings of the International Conference ‘IX Tage der Seltenen Erden’, Gommern, December 1996.
- [67] M. Bochkarev, A. A. Fagin, I. L. Fedushkin, T. V. Petrovskaya, W. J. Evans, M. A. Greci and J. W. Ziller, Russian Chemical Bulletin, Vol. 48, 1999, pp. 1782-1785. http://dx.doi.org/10.1007/BF02494829