Advances in Bioscience and Biotechnology
Vol.06 No.03(2015), Article ID:54711,10 pages
10.4236/abb.2015.63015
Mec1-Dependent Phosphorylation of the Scc3 Subunit of Cohesin during Mitosis in Budding Yeast
Nathalie Grandin, Michel Charbonneau*
Université François-Rabelais de Tours, CNRS, GICC UMR 7292, Tours, France
Email: *michel.charbonneau@univ-tours.fr
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 21 February 2015; accepted 8 March 2015; published 17 March 2015
ABSTRACT
Cohesin is an evolutionary conserved complex that controls chromosome segregation during mitosis. Here we show that, in response to DNA damage, Saccharomyces cerevisiae Scc3, one of the major regulatory subunits of the Smc1-Smc3-Scc1 cohesin ring, is phosphorylated on S/T-Q residues. This event depended on the Mec1 checkpoint kinase as well as on cell cycle arrest triggered by the DNA damage checkpoint network. This phosphorylation event also took place during mitosis of an unperturbed cell cycle. The present finding that S. cerevisiae Scc3 is phosphorylated during mitosis represents a potentially important new regulatory step in cohesin’s mitotic functions.
Keywords:
Cohesin, S/T-Q Phosphorylation, Mec1 and Tel1, DNA Damage, Cell Cycle, Budding Yeast

1. Introduction
Cohesin, a tripartite ring composed of the Smc1, Smc3 and kleisin (Scc1/Mcd1 in the budding yeast Saccharomyces cerevisiae, RAD21/SCC1 in humans) subunits, is essential in all eukaryotic organisms studied to date both for establishing sister chromatid cohesion during DNA replication, maintaining them together until metaphase, and allowing their timely separation during anaphase of mitosis [1] -[4] . In addition to its role in controlling cohesion between chromatids [5] -[7] , cohesin plays important roles in chromosome condensation, DNA repair, transcription regulation, chromatin structure, homolog pairing, mono-orientation of sister kinetochore and maintenance of centrosome number during mitosis [8] -[10] . Early experiments in both fission and budding yeasts have identified mutants of cohesin subunits, particularly Scc1/Rad21 and Wapl1/Rad61, which are hypersensitive to DNA damage, probably due to defective DNA repair resulting from inability of cohesin to maintain sister chromatid cohesion [5] . More recently, it has been shown that in response to DNA damage, the Scc1 budding yeast subunit of cohesin is phosphorylated by Chk1 in order to provide de novo establishment of sister chromatid cohesion [11] . In vertebrates, cohesin also appears to be regulated by phosphorylation in response to DNA damage, but in this case, phosphorylation occurs on Smc1 and Smc3 and is mediated by the ATM and ATR kinases [12] [13] .
In the present study, we have set out to detect possible phosphorylation of S. cerevisiae cohesin on S/T-Q residues, representing a consensus for phosphorylation by the MEC1 and TEL1 kinases (ATR and ATM in higher eukaryotes, respectively [14] .
2. Materials and Methods
2.1. Strains and Plasmids
Yeast strains used in this study were derivatives of BF264-15Daub (ade1 his2 leu2-3, 112 trp1-1a ura3Dns), as described previously [15] . Yeast cultures were grown at the indicated temperatures in YEP (1% yeast extract, 2% bacto-peptone, 0.005% adenine, 0.005% uracile) supplemented with 2% glucose (YEPD), sucrose or galactose, or in selective minimal medium. All strains were made isogenic by back crossing at least five times against our genetic background. Strain origins, prior to back crossing and, in some cases, modification of the selectable genetic marker, were as follows. The scc1::KanMX4/SCC1, tel1::KanMX4 and chk1::KanMX4 strains were purchased at Euroscarf (Frankfurt, Germany). The mec1::TRP1 and sml1::KanMX4 strains were from the Emili laboratory [16] . The rad24::URA3 strain was from the Friedberg laboratory [17] . The rad53-kd (K227A) strain was from the Heyer laboratory [18] . The cdc13-1 strain was from the Hartwell laboratory [19] . The mec1-kd (D2224A, N2229K) plasmid was from the Matsuura laboratory [20] . The mad2::URA3 disruption plasmid was from the Murray laboratory [21] .
All constructs for tagging genes (unique, endogenous copy) with 2 HA in 3’, 2 Myc in 5’ or 3’, 9 or 18 Myc in 3’ were made by using Polymerase Chain Reaction (PCR) to adapt the relevant restriction sites to the sequence of the genes. Details of the constructs will be made available upon request. The Myc18-MEC1 construct was from the Matsuura laboratory [20] .
2.2. Immunoprecipitation
Techniques for block and release experiments, cell extract preparation, immunoprecipitation and immunoblotting (analyzed using a FUJI-LAS4000) have been described previously [22] . Mouse monoclonal anti-HA raw ascites fluid, clone 16B12 (Covance), and mouse monoclonal anti-HA 12CA5 antibody (Roche Diagnostics) were used for immunoprecipitation and imunoblotting, respectively. Myc-tagged proteins were visualized after immunoprecipitation and Western blotting with monoclonal anti-Myc antibody, clone 9E10 (Roche Diagnostics). Anti-Phospho-(Ser/Thr) ATM/ATR substrate antibody was from Cell Signaling Technology (cat.#2851).
2.3. Mass Spectrometry Analysis
Mass spectrometry analysis was performed at the “Plate-forme d’Analyse Intégrative des Biomarqueurs” (INRA, Nouzilly, France). The proteins in polyacrylamide gel corresponding to the P-S/T-Q reactive band on the immunoblot membrane were analyzed by nanoflow liquid chromatography-tandem mass spectrometry (nanoLC- MS/MS). The gel slice was washed in water/acetonitrile (1:1) for 5 min and in acetonitrile for 10 min. Cystein reduction and alkylation were performed by successive incubations in 10 mM dithiothreitol/50 mM NH4HCO3 for 30 min at 56˚C and 55 mM iodoacetamide/50 mM NH4HCO3 for 20 min at room temperature in the dark. Gel slices were washed by incubation in 50 mM NH4HCO3/acetonitrile (1:1) for 10 min and by incubation in acetonitrile for 15 min. Proteins were digested overnight in 25 mM NH4HCO3 with 12.5 ng/µl trypsin (Sequencing Grade, Roche, Paris). The resulting peptides were extracted from gel by successive incubations in 0.1% formic acid (FA)/acetonitrile (1:1) for 10 min and in acetonitrile for 5 min. The extracts were pooled, dried and submitted to phosphopeptide enrichment using HyperSep MicroTip titanium dioxide (TiO2), (Thermo- Fisher). Enriched peptides were analyzed by nanoflow liquid chromatography-tandem mass spectrometry (nanoLC-MS/MS). The experiments were performed on a LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) coupled to an Ultimate® 3000 RSLC Ultra High Pressure Liquid Chromatographer (Dionex, Amsterdam, The Netherlands). Enriched peptides were loaded on an LCPackings trap column (Acclaim PepMap 100 C18, 100 µm inner diameter × 2 cm long, 3 µm particles, 100 Å pores). Mobile phases consisted of (A) 0.1% formic acid (FA), 97.9% water, 2% acetonitrile (v/v/v) and (B) 0.1% FA, 15.9% water, 84% acetonitrile (v/v/v). Peptides were desalted for 10 min at 5 µL/min with 4% solvent B and separated using a LCPackings nano-column (Acclaim PepMap C18, 75 µm inner diameter × 50 cm long, 3 µm particles, 100 Å pores). The gradient consisted of 4% - 55% B for 90 min. The nanoflow rate was set at 300 nl/min.
Data were acquired using Xcalibur software (version 2.1; Thermo Fisher Scientific, San Jose, CA). The instrument was operated in positive ionization mode. The Orbitrap analyzer recorded signals between 300 and 1800 m/z with a resolution set to 60,000 in MS. The minimum MS signal for triggering MS/MS was set to 500 counts. During MS/MS, fragmentation with Collision Induced Dissociation (CID) and detection occurred in the linear ion trap analyzer using the “Neutral Loss” and “MultiStage Activation (MSA)” MS methods. Neutral loss masses specified for fragmentations were 49 and 32.7 (for di and tricharged monophosphopeptide) and 98 and 65.3 (for di and tricharged diphosphopeptide). Precursor selection window was 2 m/z (1 microscan) for MS/MS. Data acquiring was automatically done with a profile MS full scan, a centroid MS/MS scan on the 5 most intense peptide ions with charge states ≥ 2 and MS3 fragmentation on the three most intense detected peaks using neutral loss detection. The maximum ion accumulation times allowed were 500 ms for full scans and 25 ms for CID-MS/MS. Target ion quantity for FT full MS was 1e6 and for MSn it was 1e4. The resulting fragment ions were scanned at the “normal scan rate” with q = 0.25 activation and activation time of 10 ms. Dynamic exclusion was activated for 30 s with a repeat count of 1. The lock mass was enabled for accurate mass measurements. Polydimethylcyclosiloxane (m/z, 445.1200025, (Si(CH3)2O))6) ions were used for internal recalibration of the mass spectra.
Raw data files were converted to MGF with Proteome Discoverer software (version 1.2; Thermo Fischer Scientific, San Jose, USA). A precursor mass range of 350 - 5000 Da and signal to noise ratio of 1.5 were the criteria used for generation of peak lists. The peptide and fragment masses obtained were matched automatically against the fungi section of a locally maintained copy of UniprotKB (download 12/2013). MS/MS ion searches were performed using MASCOT Daemon and search engine (version 2.3; Matrix Science, London, UK). The parameters used for database searches included trypsin as a protease with two missed cleavages allowed, and carbamidomethylcysteine, oxidation of methionine, N-terminal protein acetylation and S/T phosphorylation as variable modifications. The tolerance of the ions was set at 5 ppm for parent and 0.8 Da for fragment ion matches. Mascot results were incorporated in Scaffold 3 software (version 3.6, Proteome Software, Portland, USA). Peptide identifications were accepted if they could be established at greater than 95.0% probability as specified by the Peptide Prophet algorithm. Protein identifications were accepted if they could be established at greater than 95.0% probability as specified by the Protein Prophet algorithm.
3. Results and Discussion
In asynchronously growing cultures of an SCC1-HA2 strain treated with the DNA damaging agent methyl methane sulfonate (MMS) or left untreated, no S/T-Q phosphorylation of immunoprecipitated Scc1-HA2 was detected (Figure 1(a) middle panel). Strikingly, in the same experiment, a band migrating between 130 and 170 kD strongly reacted with the anti-P-S/T-Q antibody, only in the presence of MMS (Figure 1(a), top panel). On first analysis, this signal could correspond to either Smc1 or Smc3, the other two major subunits of cohesin, or, alternatively, to two of the three cohesin-associated proteins having a similar size, namely Scc3 or Pds5, the third one, Wpl1, being much smaller (Figure 1(b)). Similar observations were made in a cdc13-1 SCC1-HA2 strain, in which the temperature-sensitive cdc13-1 mutation conferred a telomeric damage [19] (Figure 1(c)). Moreover, S/T-Q phosphorylation of the same protein, presumably, was also detected in a cdc13-1 SMC3-Myc3 strain (Figure 1(d)). This indicated that the S/T-Q phosphorylated protein was associated in vivo both with Scc1 or Smc3 or was Smc3 itself.
We next set out to perform mass spectrometry analyses (Figure 2). To do this, extracts were prepared from asynchronously growing cells of the Scc1-HA2 strain, submitted to immunoprecipitation with anti-HA antibody and purified proteins separated on an SDS-acrylamide gel and revealed, following transfer onto a nitrocellulose membrane, with the anti-P-S/T-Q antibody, and the anti-HA antibody in parallel (data not shown). The acrylamide gel band corresponding to the P-S/T-Q signal was cut out after superimposing with the reactive nitrocellulose transfer membrane and subjected to trypsin digestion followed by mass spectrometry analysis. In the ana-
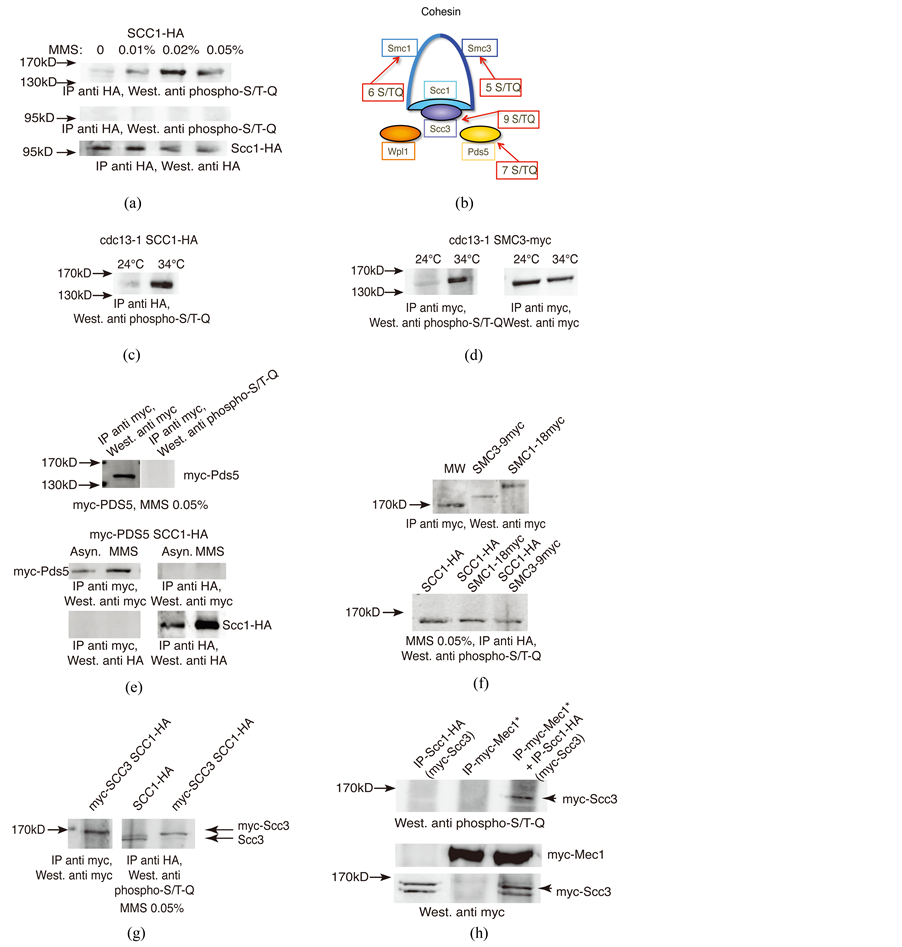
Figure 1. Scc3 is phosphorylated on S/T-Q residues following DNA damage. (a) Asynchronous cell cultures of an SCC1-HA2 strain were treated (or not, first lanes) with 0.01% - 0.05% MMS for 2 h at 29˚C. Immunoprecipitation-Western blot (IP-W) experiments were performed as indicated; (b) Schematic representation of the cohesin ring and its main associated subunits; (c)-(e) IP-W experiments on cdc13-1 SCC1-HA2 cells (c) (34˚C is a restrictive temperature for growth), cdc13-1 SMC3-Myc3 cells (d) and Myc2-PDS5 and Myc2-PDS5 SCC1-HA2 cells (e) as indicated; (f) IP-W experiments on SCC1-HA2 and SMC1-Myc18 and SMC3-Myc9 strains. MW: molecular weight markers; (g) IP-W experiments on SCC1-HA2 strains with untagged versus 2 Myc-tagged SCC3 as indicated; (h) IP from the SCC1-HA2 Myc2-SCC3 (left undamaged) and Myc18-MEC1 (treated with MMS, 2 h to activate its kinase activity symbolized by the asterisk) strains were either mixed together (right lane) or left unmixed (left two lanes as indicated). Arrowhead in top panel indicates the position of the P-S/T-Q reactive band. The SCC1-HA2 strain also harbored endogenous Myc2-SCC3 to verify production of Scc3, the P-S/ T-Q reactive protein (bottom panel). Endogenous Myc18-Mec1 was also correctly produced (middle panel).
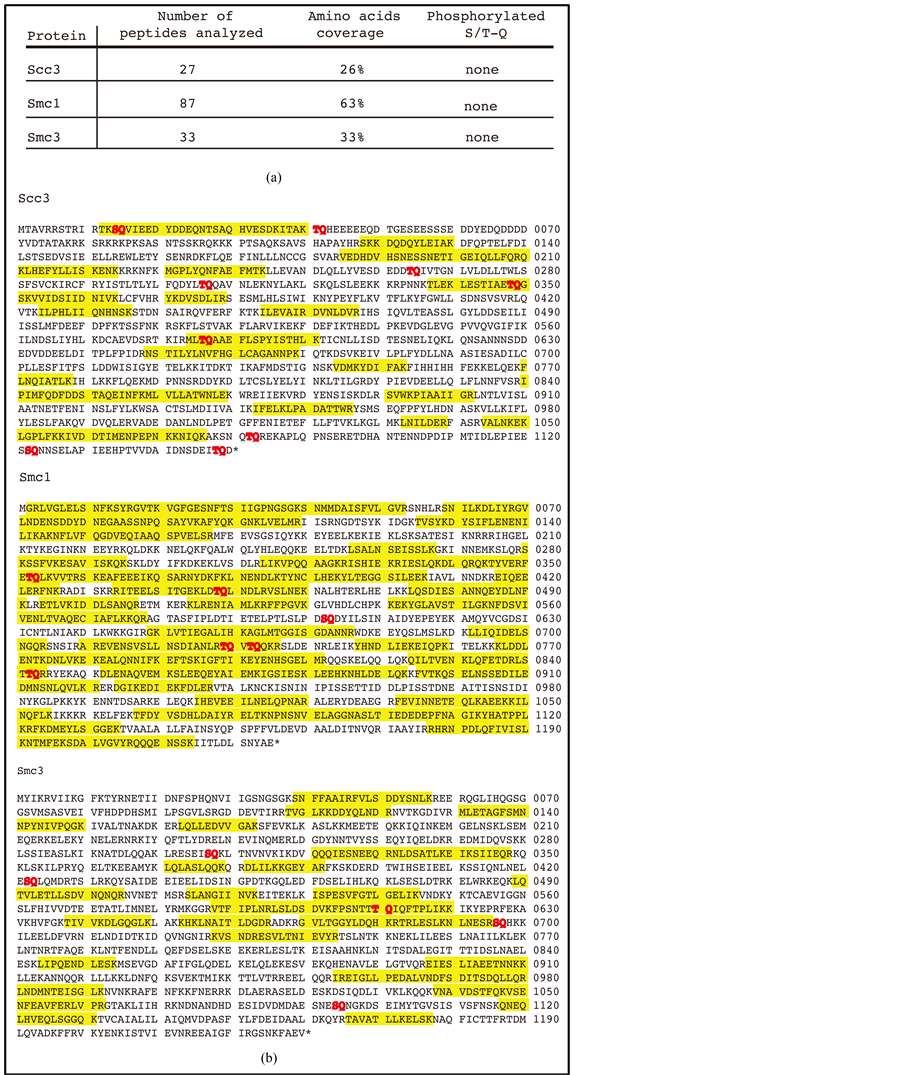
Figure 2. Mass spectrometry analysis of the anti-P-S/T-Q antibody reactive cohesin band. The acrylamide gel band corresponding to the P-S/T-Q signal was cut out after superimposing with the reactive nitrocellulose transfer membrane of another gel run in parallel and subjected to trypsin digestion followed by mass spectrometry analysis. Smc1, Smc3 and Scc3 peptides were detected in this band. (a) Summary of the results; (b) Amino acid sequences of Scc3, Smc1 and Smc3 showing in yellow the peptides recovered after trypsin digestion (from the same experiment as in (a) and analyzed, and, in red, the amino acids of the S/T-Q type. The coverage of Scc3 sequence following trypsin digestion was rather low, 26%, thus rendering identification of the residues phosphorylated upon DNA damage very difficult.
lyzed sample, three major proteins (among a total of 21) were identified, namely Smc1, Smc3 and Scc3. The number of peptides detected for each of these three proteins, as well the percentage of coverage of the complete sequences, are indicated in Figure 2(a) and their sequences shown in Figure 2(b). No event of phosphorylation could be detected in any of these peptides (Figure 2). Although Pds5 peptides were not detected, we were nevertheless concerned that it might be implicated in these events. Material immunoprecipitated from a strain bearing endogenous Myc2-Pds5 did not bring down the MMS-induced protein with the S/T-Q signal (Figure 1(e)). Moreover, no physical interactions between Myc2-Pds5 and Scc1-HA2 either under normal growth conditions or in the presence of 0.05% MMS could be detected (Figure 1(e)).
Next, SMC1 and SMC3 constructs made in fusion with 9 Myc or 18 Myc epitope tag were integrated at their respective genetic locus, in a strain that also expressed endogenous SCC1-HA2. The objective was to increase their size so as to see if the P-S/T-Q band was also increased in size (Figure 1(f), top panel). The MMS-induced S/T-Q signal was clearly migrating below the anti-Myc-Smc1-Myc18 and -Smc3-Myc9 signals (Figure 1(f), bottom panel) and was of the same size as that in the strains with untagged Smc1 or Smc3 (Figure 1(a)). This suggested that the S/T-Q signal was most probably due to Scc3. Confirming this assumption, the S/T-Q band in the Myc2-SCC3 strain was found to migrate above that in the strain with untagged SCC3 (Figure 1(g)), thus definitively establishing that this S/T-Q phosphorylation was indeed on Scc3. We noted that in both strains, the P-S/T-Q reactive material consisted of two bands (Figure 1(g)) and closer examination of left panel of Figure 1(d), as well as of Figure 3(b), also suggested the presence of two distinct P-S/T-Q bands. It is therefore possible that two distinct states of S/T-Q phosphorylation on Scc3 exist. However, we do not have at the moment any clue to further document this.
The coverage of Scc3 sequence following trypsin digestion was rather low (Figure 2(a)), thus rendering identification of the residues phosphorylated upon DNA damage very difficult. In fact, this mass spectrometric analysis just allowed us to eliminate S14, T348 and T586 (among the 9 possible residues of the S/T-Q consensus sites) as potential candidates for the S/T-Q phosphorylation observed above (Figure 2(b)).
It was formally possible that the phosphorylated protein was not Scc3 but rather Pds5 because the established strong binding between the two proteins (see for instance ref. [23] ). However, the absence of co-immunoprecipi- tation of Myc2-Pds5 by Scc1-HA2 and vice versa (Figure 1(e)) strongly argued that Pds5 was not the Scc1- HA2-attached S/T-Q phosphorylated protein.
Next, anti-Myc material IPed from a Myc18-MEC1 strain treated with MMS was mixed in vitro with anti- Scc1-HA2 IP from cells with no incurred damage (Figure 1(h)). A phosphorylated S/T-Q band clearly appeared under these conditions, but was absent from either one of the two unmixed immunoprecipitates (Figure 1(h)). Although Mec1 and Tel1 are potentially both responsible for phosphorylation on S/T-Q residues, Mec1 rather than Tel1 was chosen for these experiments because genetic inactivation of Mec1 kinase activity alone resulted in total suppression of the P-S/T-Q signal, as shown below.
To further characterize the events described above, we analyzed the S/T-Q response of the SCC1-HA2 strain to other types of damage (Figure 3(a)). Hydroxyurea (HU), which activates the replication checkpoint, different from the DNA damage checkpoint, but also dependent on Mec1 kinase, arrests cells in S phase. Nocodazole, a microtubule-poisoning drug, arrests cells in G2/M, like MMS, but in this case cells do not experience DNA damage and the arrest is independent of Mec1. As shown in Figure 3(b) (top panel), phosphorylation was detected in immunoprecipitates of Scc1-HA2 upon treatment with nocodazole, similar to MMS, but not in HU. This suggested that S/T-Q phosphorylation might be related to the G2/M arrest provoked by these drugs rather than to the damage itself. The signal totally disappeared upon genetic inactivation of both MEC1 and TEL1 (Figure 3(b), bottom panel), compatible both with the assumption that the phosphorylated residue(s) are indeed the targets of ATR-ATM-like kinases [14] and with the observation that G2/M position in the cell cycle was required for this signal. Both conditions had to be fulfilled as the S/T-Q signal was no longer detected in nocodazole-treated mec1 tel1 mutant cells even though these cells were still arrested at G2/M (Figure 3(b), bottom). Moreover, in a strain deleted for MAD2, in which the spindle checkpoint was no longer functional and which, therefore, no longer arrested at G2/M upon nocodazole-treatment [21] , the S/T-Q signal was no longer present (Figure 3(c)), thus confirming that damage without concomitant cell cycle arrest in G2/M is not sufficient to trigger phosphorylation of Scc3.
Using again the SMC1-Myc18 and SMC3-Myc9 strains, we could see that the position of the S/T-Q phosphory- lated band was at a level different from those for Smc1-Myc18 and Smc3-Myc9, confirming that the detected protein was Scc3 (Figure 3(d), top). Moreover, Myc2-Pds5 immunoprecipitate from cells treated with nocoda-

Figure 3. S/T-Q phosphorylation of Scc3 depends on G2/M arrest and can take place independently of DNA damage. (a) Determination of cell cycle stages upon different treatments by assessing cellular and nuclear morphologies under a fluorescence microscope; (b) Immunoprecipitates from a SCC1-HA2 strain (top) or from a SCC1-HA2 mec1 tel1 null strain (bottom), both treated with HU, MMS or nocodazole or left untreated (YPD) were probed with anti P-S/T-Q antibody; (c) Immunoprecipitates from a SCC1- HA2 strain or from a SCC1-HA2 mad2 null strain were probed with anti P-S/T-Q antibody; (d) In top panel, strains with the indicated relevant genotypes were treated with nocodazole, processed for SCC1-HA2 IP with anti-HA antibody and probed with anti-P-S/T-Q antibody. Positions of the Myc-tagged proteins relative to the 170 kD marker, not shown here, can be seen in Figure 1(f). In bottom panel, Myc2-PDS5 IP was probed with either anti-Myc or anti-P-S/T-Q antibody following treatment with nocodazole.
zole did not react with the anti-P-S/T-Q antibody (Figure 3(d), bottom).
Next, we assessed the dependence of the DNA damage-induced phosphorylation of Scc3’s S/T-Q residue(s) on DNA damage checkpoint genes (Figure 4(a)). First, we could observe that this signal was totally suppressed

Figure 4. S/T-Q phosphorylation of Scc3 takes place during mitosis of an unperturbed cell cycle. (a) Schematic representation of the DNA damage checkpoint kinase cascade, highlighting the major components that have been mutated in the present study, as described below (b)-(e); (b) Temperature-sensitive cdc13-1 mutant cells with the indicated relevant genotype were processed for IP-W experiments as described; (c) Alpha factor-synchronized SCC1-HA2 cells were immunoprecipitated with anti-HA antibody and probed with anti-P-S/T-Q antibody, while the main cell cycle stages were followed in parallel under the fluorescence microscope; (d) (e) Cell cycle analysis of Scc3’s S/T-Q phosphorylation following alpha factor block and release in SCC1-HA2 strains harboring the indicated construct (d) or mutations (e); (f) Cell cycle distribution of the strains with the indicated relevant genotype illustrated in panel (e) above. Ordinates indicate the percentages of the different phases of the cell cycle assessed by DAPI staining of nuclei as a function of time following release from a previous alpha factor block in G1.
after expressing a mec1 kinase-dead mutation from MEC1 genomic locus (Figure 4(b)). Therefore, Mec1 alone appears to be sufficient to phosphorylate Scc3 in response to DNA damage. The signal was also basically suppressed in a rad24 null background (Figure 4(b)), a mutation that prevents loading of the Rad17-Mec3-Ddc1 checkpoint complex and altogether prevents activation of the DNA damage checkpoint kinase cascade. It is worth recalling here that Mec1-Rad53-Chk1 and Rad24-Rad17-Mec3-Ddc1 represent two parallel pathways in the DNA damage checkpoint network and that, although both are essential for checkpoint activation, the Rad24 module is not needed for activation of the Mec1 module. Therefore, in the absence of Rad24, Mec1 was still active, yet the Scc3 S/T-Q signal was absent, thus suggesting that the absence of G2/M arrest in rad24Δ cells, rather than the absence of Mec1 activation, was likely responsible for the absence of Scc3 phosphorylation.
Simultaneous inactivation of the RAD53 and CHK1 kinases, the two downstream effectors of Mec1 during DNA damage, also resulted in the total disappearance of the Scc3’s S/T-Q phosphorylation (Figure 4(b)). However, in the single rad53 mutant, this signal was still present although less intense than in the wild type (Figure 4(b)). Activation of Rad53 and Chk1 proceed in parallel and distinct pathways, and activation of the Chk1 pathway has been shown to be particularly evident following cdc13-1-induced telomeric damage [24] , the type of damage used here (Figure 4(b)). Therefore, cdc13-1 rad53Δ mutant cells are expected to arrest at least partially because Chk1 is still present, while the cdc13-1 rad53Δ chk1Δ cells are unable to mark the G2/M arrest, thus explaining the present findings. Since Mec1 is at the top of the DNA damage checkpoint kinase cascade, it was noticeable that the S/T-Q signal, most likely to be caused by the action of Mec1, was absent in the absence of Rad24 or the combined absence of Rad53 and Chk1, two situations in which Mec1 was still present and presumably active (Figures 4(a)-(b)). Altogether, these experiments suggested that the S/T-Q signal might be more due to DNA damage-induced cell cycle arrest than to the action of Mec1 itself.
To further document these findings, we next set out to assess Scc3’s S/T-Q phosphorylation during a highly synchronous cell cycle (alpha-factor block and release) in the absence of any incurred damage (Figure 4(c)). Strikingly, an S/T-Q signal was clearly apparent at 75 - 90 min following release from the G1 block, a time that corresponded to metaphase or the metaphase-anaphase transition based on the simultaneous observation of the sampled cells by microscopy (Figure 4(c)). Noticeably, the S/T-Q signal was also present, albeit weaker, before this 75 - 90 min window, as well as after, thus suggesting that S/T-Q phosphorylation of Scc3/cohesin might be important from the onset of mitosis until around telophase (Figures 4(c)-(e)). During a synchronous mitosis, the S/T-Q signal was much less intense than that observed in MMS-treated cells or cdc13-1 cells at 34˚C, as expected from the fact that upon one of these two DNA damages the quasi total number of cells in the population eventually arrested at G2/M during the course of the block, 2 h. Again, making use of the SCC1-HA2 strains harboring either endogenous SMC1-Myc18 or SMC3-Myc9 established that, by elimination, Scc3 was most probably the protein phosphorylated on S/T-Q during mitosis of undamaged synchronized cells (Figure 4(d)). Finally, we could demonstrate that the cell cycle-associated S/T-Q phosphorylation of Scc3/cohesin was dependent on the presence of Mec1, but that simultaneous genetic inactivation of RAD53 and CHK1 did not prevent its occurrence (Figures 4(e)-(f)).
To our knowledge, in S. cerevisiae, only Scc3’s S343 has been reported to be phosphorylated upon DNA damage (ref. [25] and see also http://www.phosphogrid.org). The potential significance of this phosphorylation event has not been established. Other phosphorylation events on residues of S. cerevisiae Scc3, namely S28, S34 and S623, have been reported, but these were labeled as ambiguous; phosphorylation on Scc3’s S628 has also been found (http://www.sbeams.org). In human cells, Plk1 phosphorylates SCC3/SA2, a step essential in the cohesin dissociation pathway during prophase and prometaphase [26] . In budding yeast, the major response of cohesin to DNA damage is on its Scc1/Mcd1 subunit [11] , while in humans, in case of DNA damage, phosphorylation occurs on Smc1 and Smc3, and is thought to be mediated directly by the kinases ATM and ATR [12] [13] . Scc3 has major roles in cohesin-chromosomes association/dissociation, as well as in maintaining sister chromatid cohesion, in cooperation with the other regulatory cohesin subunits Pds5 and Wapl1 [5] [23] [27] .
In summary, we have uncovered here an event of phosphorylation of cohesin on one or several S/T-Q residues (potentially by the MEC1/ATR kinase) that most probably takes place on Scc3, a protein closely associated with the cohesin ring. Most importantly, this event takes place during mitosis of an unperturbed cell cycle, culminating in intensity around anaphase and telophase, but also present a little earlier and later. This event is of prime importance for two reasons. First, given the key position of Scc3 at the heart of the cohesin complex, this finding suggests that Scc3 phosphorylation might represent a key element in coupling the assembly/disassembly reactions of the Smc1-Smc3-Scc1 cohesin ring with the other cohesin regulatory subunits as well as with mitotic events. Second, the present findings suggest that Mec1 might intervene during mitosis independently of any incurred DNA damage and therefore independently of its checkpoint function to control cohesin through direct regulation of Scc3 by phosphorylation on S/T-Q residues. Presumably, the P-S/T-Q signal detected upon treatment with MMS or nocodazole corresponds to a G2/M enrichment of the cell population as it disappears when the damaged-induced cell cycle arrest is compromised by a mutation in a checkpoint gene. Further analyzing this Scc3 phosphorylation is crucial and represents a future challenge to fully understand cohesin biology.
Acknowledgements
We thank Akira Matsuura, Andrew Emili, Leland Hartwell, Wolf-Dietrich Heyer, Andrew Murray and Errol Friedberg for the gift of strains and plasmids. We thank Valérie Labas and Grégoire Harrichaux for performing the mass spectrometry analysis at the “Plate-forme d’Analyse des Biomarqueurs” (INRA, Nouzilly, France). The high resolution mass spectrometer was financed (SMHART project) by the European Regional Development Fund (ERDF), the “Conseil Régional du Centre”, the French National Institute for Agricultural Research (INRA) and the French National Institute of Health and Medical Research (Inserm). This work was financially supported by subventions from the “Centre National de la Recherche Scientifique” and the “Université François- Rabelais Tours” to the UMR CNRS 7292/University of Tours-GICC Research Unit.
References
- Guacci, V., Koshland, D. and Strunnikov, A. (1997) A Direct Link between Sister Chromatid Cohesion and Chromosome Condensation Revealed through the Analysis of MCD1 in S. cerevisiae. Cell, 91, 47-57. http://dx.doi.org/10.1016/S0092-8674(01)80008-8
- Michaelis, C., Ciosk, R. and Nasmyth, K. (1997) Cohesins: Chromosomal Proteins That Prevent Premature Separation of Sister Chromatids. Cell, 91, 35-45. http://dx.doi.org/10.1016/S0092-8674(01)80007-6
- Ciosk, R., Shirayama, M., Shevchenko, A., Tanaka, T., Toth, A., Shevchenko, A. and Nasmyth, K. (2000) Cohesin’s Binding to Chromosomes Depends on a Separate Complex Consisting of Scc2 and Scc4 Proteins. Molecular Cell, 5, 1-20. http://dx.doi.org/10.1016/S1097-2765(00)80420-7
- Uhlmann, F., Wernic, D., Poupart, M.A., Koonin, E.V. and Nasmyth, K. (2000) Cleavage of Cohesin by the CD Clan Protease Separin Triggers Anaphase in Yeast. Cell, 103, 375-386. http://dx.doi.org/10.1016/S0092-8674(00)00130-6
- Peters, J.M., Tedeschi, A. and Schmitz, J. (2008) The Cohesin Complex and Its Roles in Chromosome Biology. Genes and Development, 22, 3089-3114. http://dx.doi.org/10.1101/gad.1724308
- Nasmyth, K. (2011) Cohesin: A Catenase with Separate Entry and Exit Gates? Nature Cell Biology, 13, 1170-1177. http://dx.doi.org/10.1038/ncb2349
- Ocampo-Hafalla, M.T. and Uhlmann, F. (2011) Cohesin Loading and Sliding. The Journal of Cell Science, 124, 685-691. http://dx.doi.org/10.1242/jcs.073866
- Simmons-Kovacs, L.A. and Haase, S.B. (2010) Cohesin: It’s Not Just for Chromosomes Anymore. Cell Cycle, 9, 1750-1753. http://dx.doi.org/10.4161/cc.9.9.11792
- Losada, A. (2014) Cohesin in Cancer: Chromosome Segregation and Beyond. Nature Reviews Cancer, 14, 389-393. http://dx.doi.org/10.1038/nrc3743
- Mehta, G.D., Kumar, R., Srivastava, S. and Ghosh, S.K. (2013) Cohesin: Functions beyond Sister Chromatid Cohesion. FEBS Letters, 587, 2299-2312. http://dx.doi.org/10.1016/j.febslet.2013.06.035
- Heidinger-Pauli, J.M., Unal, E., Guacci, V. and Koshland, D. (2008) The Kleisin Subunit of Cohesin Dictates Damage- Induced Cohesion. Molecular Cell, 31, 47-56. http://dx.doi.org/10.1016/j.molcel.2008.06.005
- Kim, S.T., Xu, B. and Kastan, M.B. (2002) Involvement of the Cohesin Protein, Smc1, in Atm-Dependent and Independent Responses to DNA Damage. Genes and Development, 16, 560-570. http://dx.doi.org/10.1101/gad.970602
- Yazdi, P.T., Wang, Y., Zhao, S., Patel, N., Lee, E.Y. and Qin, J. (2002) SMC1 Is a Downstream Effector in the ATM/NBS1 Branch of the Human S-Phase Checkpoint. Genes and Development, 16, 571-582. http://dx.doi.org/10.1101/gad.970702
- Kim, S.T., Lim, D.S., Canman, C.E. and Kastan, M.B. (1999) Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members. The Journal of Biological Chemistry, 274, 37538-37543. http://dx.doi.org/10.1074/jbc.274.53.37538
- Grandin, N. and Reed, S.I. (1993) Differential Function and Expression of Saccharomyces cerevisiae B-Type Cyclins in Mitosis and Meiosis. Molecular and Cellular Biology, 13, 2113-2125.
- Emili, A. (1998) MEC1-Dependent Phosphorylation of Rad9p in Response to DNA Damage. Molecular Cell, 2, 183- 189. http://dx.doi.org/10.1016/S1097-2765(00)80128-8
- Siede, W., Nusspaumer, G., Portillo, V., Rodriguez, R. and Friedberg, E.C. (1996) Cloning and Characterization of RAD17, a Gene Controlling Cell Cycle Responses to DNA Damage in Saccharomyces cerevisiae. Nucleic Acids Research, 24, 1669-1675. http://dx.doi.org/10.1093/nar/24.9.1669
- Bashkirov, V.I., Bashkirova, E.V., Haghnazari, E. and Heyer, W.D. (2003) Direct Kinase-to-Kinase Signaling Mediated by the FHA Phosphoprotein Recognition Domain of the Dun1 DNA Damage Checkpoint Kinase. Molecular and Cellular Biology, 23, 1441-1452. http://dx.doi.org/10.1128/MCB.23.4.1441-1452.2003
- Garvik, B., Carson, M. and Hartwell, L. (1995) Single-Stranded DNA Arising at Telomeres in cdc13 Mutants May Constitute a Specific Signal for the RAD9 Checkpoint. Molecular and Cellular Biology, 15, 6128-6138.
- Takata, H., Kanoh, Y., Gunge, N., Shirahige, K. and Matsuura, A. (2004) Reciprocal Association of the Budding Yeast ATM-Related Proteins Tel1 and Mec1 with Telomeres in Vivo. Molecular Cell, 14, 515-522. http://dx.doi.org/10.1016/S1097-2765(04)00262-X
- Li, R. and Murray, A.W. (1991) Feedback Control of Mitosis in Budding Yeast. Cell, 66, 519-531. http://dx.doi.org/10.1016/0092-8674(81)90015-5
- Grandin, N., Corset, L. and Charbonneau, M. (2012) Genetic and Physical Interactions between Tel2 and the Med15 Subunit of Mediator in Saccharomyces cerevisiae. PLoS ONE, 7, e30451. http://dx.doi.org/10.1371/journal.pone.0030451
- Kulemzina, I., Schumacher, M.R., Verma, V., Reiter, J., Metzler, J., Failla, A.V., Lanz, C., Sreedharan, V.T., Rätsch, G. and Ivanov, D. (2012) Cohesin Rings Devoid of Scc3 and Pds5 Maintain Their Stable Association with the DNA. PLoS Genetics, 8, e1002856. http://dx.doi.org/10.1371/journal.pgen.1002856
- Sanchez, Y., Bachant, J., Wang, H., Hu, F., Liu, D., Tetzlaff, M. and Elledge, S.J. (1999) Control of the DNA Damage Checkpoint by Chk1 and Rad53 Protein Kinases through Distinct Mechanisms. Science, 286, 1166-1171. http://dx.doi.org/10.1126/science.286.5442.1166
- Chen, S., Albuquerque, C.P., Liang, J., Suhandynata, R.T. and Zhou, H. (2010) A Proteome-Wide Analysis of Kinase- Substrate Network in the DNA Damage Response. The Journal of Biological Chemistry, 285, 12803-12812. http://dx.doi.org/10.1074/jbc.M110.106989
- Hauf, S., Roitinger, E., Koch, B., Dittrich, C.M., Mechtler, K. and Peters, J.M. (2005) Dissociation of Cohesin from Chromosome Arms and Loss of Arm Cohesion during Early Mitosis Depends on Phosphorylation of SA2. PLoS Biology, 3, e69. http://dx.doi.org/10.1371/journal.pbio.0030069
- Roig, M.B., Löwe, J., Chan, K.L., Beckouët, F., Metson, J. and Nasmyth, K. (2014) Structure and Function of Cohesin’s Scc3/SA Regulatory Subunit. FEBS Letters, 588, 3692-3702. http://dx.doi.org/10.1016/j.febslet.2014.08.015
NOTES

*Corresponding author.