Journal of Biophysical Chemistry
Vol.07 No.02(2016), Article ID:66494,11 pages
10.4236/jbpc.2016.72005
Comparative Molecular Mechanics and Quantum Mechanics Study of Monohydration of Nucleic Acid Bases
Job Lino-Pérez1,2, Eduardo González-Jiménez1, Alexandra Deriabina1, Martha Velasco1, Valery I. Poltev1,3
1Faculty of Physical and Mathematical Sciences, Autonomous University of Puebla, Puebla, Mexico
2Department of Physical Engineering, University of Guanajuato, León, Mexico
3Institute of Theoretical and Experimental Biophysics, RAS, Pushchino, Russia

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 11 February 2016; accepted 14 May 2016; published 17 May 2016
ABSTRACT
DNA is the most important biological molecule and its hydration contributes essentially to the structure and functions of the double helix. We analyze the monohydration of the individual bases of nucleic acids and their methyl derivatives using methods of Molecular Mechanics (MM) with the Poltev-Malenkov (PM), AMBER and OPLS force fields, as well as ab initio Quantum Mechanics (QM) calculations at MP2/6-31G(d,p) level of theory. A comparison is made between the calculated interaction energies and the experimental enthalpies of microhydration of bases, obtained from mass spectrometry at low temperatures. Each local water-base interaction energy minimum obtained with MM corresponds to the minimum obtained with QM. General qualitative agreement was observed in the geometrical characteristics of the local minima obtained via the two groups of methods. MM minima correspond to slightly more coplanar structures than those obtained via QM methods, and the absolute MM energy values overestimate corresponding values obtained with QM. For Adenine and Thymine the QM local minima energy values are closer to those obtained by the PM potential (average of 0.72 kcal/mol) than by the AMBER force field (1.86 kcal/mol). The differences in energy between MM and QM results are more pronounced for Guanine and Cytosine, especially for minima with the water molecule forming H-bonds with two proton-acceptor centers of the base. Such minima are the deepest ones obtained via MM methods while QM calculations result in the global minima corresponding to water molecule H-bonded to one acceptor and one donor site of the base. Calculations for trimethylated bases with a water molecule corroborate the MM results. The energy profiles were obtained with some degrees of freedom of the water molecule being frozen. This data will contribute to the improvement of the molecular mechanics force fields.
Keywords:
DNA Bases, Hydration, Ab Initio Calculations, Molecular Mechanics, Hydrogen Bonding

1. Introduction
Water is one of the most abundant chemical compounds on the planet, and it constitutes a high percentage of the cell composition. To understand the role of interactions of biomolecules with water in relation to their functions, it is essential to have a detailed description of the energetic and structural aspects of interactions of the molecules involved. The first data on Deoxyribonucleic Acid (DNA) fibers obtained by X-ray diffraction showed that DNA is highly hydrated and the interactions with water are responsible for its conformational changes [1] . The microhydration of Nucleic Acid (NA) bases, i.e. interactions of the bases with separate water molecules, plays an important role in structural stabilization of the double helix. Quantum Mechanics (QM) calculations of Hartree Fock (HF), Density Functional Theory (DFT), and Second-order Møller Plesset Perturbation Theory (MP2) performed for hydrated complexes of DNA bases revealed that the geometric properties of such complexes are extremely sensitive to the interactions with one or few water molecules [2] - [4] ; the presence of just one water molecule is enough to completely change the structure of a complex of nucleic acid bases in the global minimum.
From the analysis of experimental results on hydration of oligomeric DNA duplexes, Schneider and his group [5] [6] evaluated the distribution of water molecules around the components of the NA by considering it as a “construction of hydrated blocks”. A modular scheme for the hydration was suggested. It determines the average sites of water molecules around the components of the NA, and can generate predictive patterns for the distribution of water molecules around the NA fragments. The studies of microhydration of the individual components of nucleic acids, obtained by both experimental and theoretical methods complete this scheme. Quantitative evaluation of the sites of hydration also contributes to the improvement of Molecular Mechanics (MM) force fields [7] [8] .
Experimental spectroscopy studies have provided valuable data on the hydration of the components of the NA. The first studies of water clusters with nucleic bases using mass spectrometry in a primary ionization field were made by the group of Sukhodub, who determined the enthalpies of hydration of DNA bases and some of their derivatives [9] . Important mass spectroscopy experiments were performed by Kim [10] , where the threshold ionization energies for hydrated Adenine (A) and Thymine (T) bases were reported. Studies of UV photoionization in vacuum by a supersonic molecular beam using optical spectroscopy and comparison with theoretical results enabled the determination of the ionization energies of microhydrated DNA bases [11] and of tautomers of hydrated 9-methylguanine [12] . The studies of mononucleotide complexes with individual water molecules have been reported [13] . However, all these studies do not provide direct information about the structure and stabilization energy, and theoretical interpretation of the results is necessary. The experiments of Sukhodub [9] represent the only exception, because they determine the gas-phase interaction energies of water-base complexes from the temperature dependences of the equilibrium constants of the association. However this method, in contrast to the theoretical results, does not specify the geometry of the complexes. So, despite the success of modern experimental methods, they still do not provide direct data for the detailed topology of the network of water- base hydrogen bonds (H-bonds). Thus, the computational methods of both molecular mechanics and quantum mechanics are the indispensable tools for the detailed study of the fine structure of hydration of nucleic acids.
The microhydration of the bases has been the subject of numerous theoretical studies by Monte Carlo [14] - [16] and Molecular Dynamics [17] techniques using different force fields. The hydration sites can be compared with quantum mechanics ab initio [12] [18] - [24] and DFT calculations for interactions of some water molecules with DNA bases and base pairs [4] [25] - [27] . The molecular mechanics calculations have demonstrated that the deepest minima of the interaction energy of a water molecule with nucleic bases correspond to the formation of a water bridge between two hydrophilic atoms of the base. Such a bridge can be formed in three different ways, namely between two H-bond acceptor centers of the base, between two donor centers, and one acceptor and one donor centers. The first of these scenarios was analyzed with ab intio quantum mechanical calculations more extensively (applying different basis sets), as this configuration in molecular mechanics corresponds to global minima for Guanine and Cytosine. We performed preliminary ab initio calculations using the bases with rigid geometry followed by the complete energy minimization in the space of all the degrees of freedom. We also performed the study of energy profiles, i.e. the dependences of the interaction energy on some water displacements around the base hydration sites fixing certain geometrical parameters. This information will contribute to future improvements in force fields. The comparison of theoretical MM and QM results with the experimental data, demonstrates that we need to reconsider the geometry of some minima positions for the force field parameters adjustments
2. Method of Calculation
The systems considered contain one of methylated nucleotide bases (1-methylpyrimidine or 9-methylpurine) and one water molecule. The starting geometries of the bases are the average structures obtained from X-ray experimental data in crystals, these geometries have been used in previous works [15] [28] . For simplicity we will name the above mentioned methylated bases simply as Adenine, Thymine, Cytosine, and Guanine. The calculations of the interaction energy were performed within the two schemes of Molecular Mechanics (MM) and Quantum Mechanics (QM). For MM calculations Poltev-Malenkov (PM) force field [7] [8] was used along with the potentials implemented in the AMBER program [29] [30] , in both cases the interaction energy is calculated as the sum of pair interactions of all the atoms constituting the molecules. For the PM potential, each atom-atom interaction consists of a Coulomb term and of Lennard-Jones (or 6 - 12) one (Equation (1)). To describe the interaction between the atoms capable of forming hydrogen bonds, the 6 - 12 term is replaced by a 10 - 12 term (Equation (2))
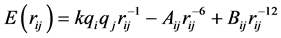 (1)
(1)
 (2)
(2)
In these equations, k is a numerical constant, qi, qj are the effective charges of atoms i and j respectively (calculated by semiempirical quantum chemistry methods and reproduced the experimental dipole moments of the molecules), rij is the distance between the atoms. The coefficients Aij, Bij and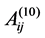 ,
, 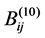 are adjustable parameters whose numerical values are the same as in previous articles [8] [28] . The AMBER potentials [29] take into account the intra-molecular terms (whose contributions are small) and do not contain the 10 - 12 terms.
are adjustable parameters whose numerical values are the same as in previous articles [8] [28] . The AMBER potentials [29] take into account the intra-molecular terms (whose contributions are small) and do not contain the 10 - 12 terms.
Quantum mechanics calculations were performed using the GAUSSIAN 03W program [31] , at MP2/6-31G(d,p). The interaction energies Eint was evaluated considering the basis set superposition error correction using the counterpoised procedure of Boys-Bernardi implemented the GAUSSIAN package [32] . After energy minimization, additional single point calculations were performed with counterpoise option to evaluate the energy of the first molecule with the basic functions of the second one, and vice versa the energy of the second molecule with the functions of the first one. These terms are subtracted from the total energy and so the corrected energy EBSSE of the system is obtained.
 (3)
(3)
All the local minima were verified by the calculations of the matrices of second derivatives of energy (Hessian) which appeared to be positive. For some local minima of Guanine and Cytosine more extensive basis set (aug-cc-pvdz) was used in order to confirm the geometry. For each base, energy scans were performed with both methods (MM and QM) by changing the position of the water molecule around the hydrophilic centers. Some geometric parameters were varied gradually, with other ones being fixed. For example azimuthal scans were made, i.e. the angle θ (Figure 1) was varied to change the position of the water molecule in the base plane around the base atom capable of forming a hydrogen bond. During these minimizations the distance r between two atoms of the two molecules and parameters φx, φy and φz which determine the rotations of the water hydrogen’s around the water oxygen were varied. The energy profiles obtained provide fine details of geometry changes which will contribute to the improvement of force fields.
3. Results and Discussion
3.1. Local Minima of Interaction Energy between DNA Bases and Single Water Molecule
The extensive calculations of the water-base systems via MM and QM methods described in the previous section enable us to reveal all the local minima for these systems. The calculated interaction energies along with those of

Figure 1. The azimuthal scans for three regions around Adenine base.
other authors are presented in Table 1. The hydrogen bond distances for these complexes are shown in Table 2.
The structures obtained with both methods are shown in Figure 2. The same number of local minima and rather close water oxygen positions were revealed in both MM and QM calculations, i.e. every MM minimum corresponds to QM minimum with the mutual water-base positions resembling those of QM minima. The PM potential functions favor the coplanar configurations of the complexes, i.e. the base ring and the 3 atoms of the water molecule located in nearly the same plane. The only exceptions are the configuration 3 for Guanine and the configuration 1 for Cytosine. Qualitatively similar structures are obtained with the AMBER potentials and the potential of Jorgensen (OPLS), the latter values were calculated in [28] and coincide with the values obtained in [14] using the method of diffusion Monte Carlo. The results of our ab initio calculations revealed both non-coplanar and coplanar conformations, for example the structures corresponding to minimum 2 for Adenine and 3 for Cytosine are completely planar whereas for structures 2 and 3 for Thymine and 3 for Guanine, one of the hydrogens of the water molecule remains in the plane of the base while the other hydrogen deviates from the plane for approximately 30˚.
Table 2 shows the inter-atomic distances of hydrogen bonds, the hydrogen bonds are shorter for MM than for QM. The inter-atomic distances N/O…HW and NH…OW for the potential PM fall in the range from 1.78 to 1.98 Å while for QM they vary from 1.94 to 2.24 Å. For AMBER potentials this region extends from 1.70 to 2.12 Å, i.e. it is larger than for PM but shorter than for the quantum-mechanical calculations deviating on average by 0.16 Å from the QM values.
Comparison of the interaction energies of the minima obtained with the MM method and those obtained with QM shows generally higher values for the former, this is true for both PM and AMBER potentials (not for all the OPLS results). This difference can be due to the MM potential adjustment to the hydration of the bases in aqueous solution [8] [28] where water molecules of the first shell are affected by the “bulk” water. The tendency for shortening the interatomic distances on including the other water molecules can be seen from comparison with other publications. This feature is reported in a DFT study [2] for complexes of Cytosine with 14 molecules of water, where for water position corresponding to minimum 4 the O-HW…O2 distance is of 1.82 Å, while our QM calculations give the value of 1.91 Å. The same tendency took place in the Hartree-Fock study [19] for Guanine with 7 to 13 water molecules.
The values of the interaction energies in minima calculated with the method MM/PM are closer to QM ones for the Adenine and Thymine (the average differences being of 0.72 kcal/mol) than for Guanine and Cytosine (2.8 kcal/mol). The reason for these differences is due to the fact that QM calculations result in rather small interaction energies for H-bonding of water molecule to two proton acceptors of the bases. This situation will be discussed in the next section.

Table 1. Energies of water-base interactions (kcal/mol) in the local minima, calculated using different MM and QM methods.
Structure numbering of the local minima corresponds to that of the Figure 1.  are the interaction energies calculated via MP2/6-31G(d,p) ab initio method.
are the interaction energies calculated via MP2/6-31G(d,p) ab initio method. 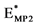 are the ab initio interaction energies calculated by other authors (MP2/6-31G(d,p) from [3] (a), RI-MP2 method from ref. [21] - [24] (b), MP2 dZ from [14] (c)). EPM, EAMBER are the MM interaction energies calculated with PM and AMBER potentials respectively. EOPLS are MM energies obtained via OPLS potentials from [28] and [14] (second column). EDFT are the interaction energy obtained with DFT method by Kim [25] [26] and from [33] (d).
are the ab initio interaction energies calculated by other authors (MP2/6-31G(d,p) from [3] (a), RI-MP2 method from ref. [21] - [24] (b), MP2 dZ from [14] (c)). EPM, EAMBER are the MM interaction energies calculated with PM and AMBER potentials respectively. EOPLS are MM energies obtained via OPLS potentials from [28] and [14] (second column). EDFT are the interaction energy obtained with DFT method by Kim [25] [26] and from [33] (d).
3.2. Interactions of Water Molecule with Two H-Bond Acceptors of the Bases
The most significant differences between MM and QM results refer to the minimum 1 of Guanine and 3 of Cytosine (Figure 2) corresponding to the interaction of water molecule with two H-bond acceptors of the base. From calculations carried out with the potential PM, we found that: The first minimum for Guanine obtained via PM potentials is the most profound one, and it is only 0.2 kcal/mol deeper than the minimum 2, which is the global one for AMBER force field (Table 1). The energy value for the minimum 1 obtained via QM calculations is less negative (−7.72 kcal/mol). The global QM minimum corresponds to the position 2 of (−10.81 kcal/mol). The interatomic distances for both QM and MM fall in the limits allowed by the geometric criterion for hydrogen bond formation [7] (Table 2), similar situation occurs for the minimum 3 for Cytosine, the water forms H-bonds via hydrogen atoms with two proton-acceptor centers of Cytosine; the QM distances N3…Hw and O2…HW resemble corresponding distances for Guanine-water complex (2.13 and 2.31 Å, respectively). With MM methods similar energy values were obtained for both force fields (Table 1). Our global QM minimum 2 with the value of −10.24 kcal/mol resembles that obtained with more extensive basis set [4] and via DFT calculations
Table 2. Hydrogen bond distances (Å) in the local minima of water-base interaction energy, calculated with MM potentials PM and AMBER and with ab initio MP2/631G(d,p) method.
The first value for each center corresponds to the N-H…OW or N-OBASE…HW distance, and the second one to N/OBASE…OW distance.

Figure 2. The positions of local energy minima for nucleic acid bases complexes with water molecule obtained using MM (PM potentials), up, and QM, down, methods.
[25] [26] (the values of −9.97 and −9.1 kcal/mol respectively). Microhydration of Cytosine and its radical anion were investigated with the DFT-B3LYP method [26] , and the deepest minimum for Cytosine was found at position 2, but for the Cytosine anion it was located at position 3, the bond lengths being shorter compared to our QM values, (Hw…O2 of 1.95 Å and Hw…N3 of 2.16 Å). The barrier between the minima 3 and 2 for cytosine-water complex is quite small; the minima 2 and 3 obtained via PM potentials have nearly the same energy (the difference of 0.1 kcal/mol). The same effect can be seen for different tautormers of Guanine via MM calculations [28] . The enol tautomer with the OH group oriented towards the N1 atom forms a complex with water molecule H-bonded to two acceptor atoms (N7 and O6) with the energy of −10.04 kcal/mol, and interatomic distances with O6 of 1.85 Å and with N7 of 1.97 Å. This minimum was also revealed using ab initio RI-MP2 method with TZVPP basis set [18] . The deepest minimum for dCMP (B3LYP/6-31G* level of DFT) corresponds to the minimum 3 of Cytosine [33] . Thus, the MP2/6-31G(d,p) level of QM calculations results in the minima of poor stability when water molecule forms H bonds with two base acceptors, and the potential barrier is rather small, however, the MM potentials show them as ones of the lowest energy.
3.3. Monohydration of Some Methylated Derivatives of Nucleic Acid Bases
There are experimental mass spectrometry data [9] for some trimethylated bases which can be useful in comparison of the results of MM and QM methods on search for water-base interaction energy minima. The substitution of the hydrogen atom capable to form H-bond by a methyl group excludes some local energy minima of water-base interactions, thus helping to refer experimental data to the definite water position. The methylation changes slightly the calculated values of water-base interaction energy minima for water positions not involved in H bonding with the hydrogen to be substituted. It was demonstrated for MM calculations earlier, and it is confirmed for QM calculations here.
The methylated bases considered in this section and compared with experimental results are: 1,4,4-trimetilci- tosine (m1,4,4Cyt), 2,2,9-trimetilguanine (m2,2,9Gua), and 6,6,9-trimetiladenine (M6,6,9Ade). The first one excludes the minima 1 and 2 for 1-methylcytosine, the second one excludes the minima 3 and 4 for 9-methylguanine, and the last one excludes the minima 1 and 3 for 9-methyladenine (Figure 3).
The calculation results obtained via MM and QM methods for trimethylated bases and the experimental enthalpies of water-base complex formation are listed in the Table 3. The values corresponding to global minima for 1-methylcytosine and 9-methylpurines (from the Table 1) are added for comparison.
The results demonstrate rather close experimental values of the enthalpies of complex formation with water molecule for m9Gua and m229Gua. The same is true for m1Cyt and m144Cyt (Table 3). Rather small differences between the values of monomethylated and trimethylated guanine and cytosine suggest the nearly same positions of the water molecule for complexes observed in experimental study and in global minima. The comparison of experimental data for m9Ade and m669Ade demonstrates less negative values for the trimethylated base, i.e. the substitution of amino group hydrogens by methyl groups changes the position of water molecule in the complex. Both MM and QM calculations suggest that the m669Ade-water complex correspond to minimum 3 for m9Ade, as the formation of other two minima for m9Ade-water complexes are blocked by methyl groups.
The calculations for m2,2,9Gua do not help to decide which minimum is more favorable, the minimum 1 (as predicted by PM potentials) or 2 (as predicted by QM and AMBER calculations). Both minima are possible for m2,2,9Gua-water complex (Table 3), and the calculated values for these minima are close for those of m9Gua (Table 1).
The calculations for m144Cyt confirm the prediction of MM calculations (both PM and AMBER versions) on more favorable for m1Cyt water position 3 (formation of two H bonds of water molecule with acceptors of the base) as compared to position 2 predicted from QM calculations. The position 2 is not possible for m144Cyt-wa- ter complex, but experimental data demonstrate very close values of the enthalpy of hydration for m1Cyt and m144Cyt.
The calculations for trimethylated bases suggest the necessity of both improvement of MM force fields and more sophisticated QM calculations to reach more adequate description of water-base interactions.
3.4. Interaction Energy Dependences on Displacement of Water Molecule from Energy Minima
Similar azimuthal scans were obtained for other bases; for Thymine-water system maximum energy difference,

Figure 3. QM interaction energy minima for three methylated bases.
Table 3. Experimental enthalpies of formation and calculated water-base interaction energies (kcal/mol) of possible complexes for some methylated derivatives of the DNA bases.
ΔHEXP, the experimentally obtained enthalpies [9] . EQM, the interaction energy calculated by ab initio MP2/6-31G(d,p) method. EPM, EAMBER, and EOPLS are designated as those values in the Table 1. The notations for the methylated bases are listed in the text. Numbers of minima according to Figure 1 are listed in parentheses.
1.83 kcal/mol, corresponds to the trajectory from minimum 1 to minimum 2. For the Guanine-water and Cytosine-water systems, there are more pronounced differences in energy, though the distances between the participating in H bonds atoms of the base and the water molecules are rather close for the two methods. It is noteworthy that for QM structures the distances of out-of-plane water hydrogen from base acceptor atom are nearly the same as for corresponding coplanar MM water-base complexes.
The second type of scans performed refer to moving a water molecule towards and away from the base starting from the minima positions (during the optimization φx, φy, and φz parameters were varied, the angle θ was fixed). When we make a radial scan such that a water molecule approaching the methyl group of the base to the distances between the oxygen and carbon shorter than 3.15 Å, the structures obtained with MM may be non- coplanar due to the repulsion of atoms. In this case the energy dependence as a function of r for the two methods show the same pattern.
The third type of scans was performed by the displacements of water molecules out of the plane of the bases… In this case the energies have the same tendency to decrease when the water moves away from the base plane (to 90˚), at the end of the scan path there can arise a marked difference (up to 3 kcal/mol for scans near amino or methyl groups).
Some MM minima refer to both water hydrogen’s in the base plane while corresponding QM minima refer to displacement of the water hydrogen not forming H-bond by 30˚ - 45˚ out of the plane (e.g. in Thymine and Guanine 2nd minima). We performed MM and QM energy scans as functions of the angle of rotation about O-H water bond (H being H-bonded to the base and the bond being in the base plane). The energy differences between QM and MM water positions fall in 0.2 kcal/mol region, thus being not great, but may be significant for some cases. More profound QM calculations and MM parameter adjustment are required for more exact water-base system description in this respect.
4. Conclusions
This paper concerns the evaluation of the interactions of nucleic acid bases with single water molecule. The calculations for such simple systems can be performed via the methods of various complexities, from simple atom-atom MM computations of the rigid molecules to correlate ab initio QM computations using extended basis sets. The comparison of the results obtained via various methods demonstrates both some common features and some differences in quantitative geometry and energy characteristics. The simulation of biomolecular systems in surrounding water is possible via MM methods only. Thus, continuous improvement of MM force fields is required for adequate reproduction and prediction of important features of the systems containing nucleic acid fragments and hundreds of water molecules (and other biologically important molecules). Such improvement is not possible using experimental data only due to the insufficient amount of such data. The high level QM computations of the simple systems can help to fill this gap.
The comparison of the results of systematic QM MP2/6-31G(d,p) level computations with different MM methods is the first step on the pathway of MM force field refinement. Our MM computations using PM and AMBER force fields have demonstrated that each local MM energy minimum can be referred to QM one. The average energy difference between corresponding minima for Adenine and Thymine complexes with one water molecule is 0.72 and 1.86 kcal/mol for PM and AMBER force fields respectively. The differences for Guanine and Cytosine are more pronounced, especially for minima which correspond to the formation of two H bonds by water molecule with two acceptors of the bases. Such minima are global ones when calculated by MM methods while QM calculations results in global minima corresponding to the formation of one H-bonds with the base acceptor and another with base donor atom. The calculations for trimethylated bases and their comparison with experimental values of the enthalpy of monohydration supply us with evidences in favor to MM results. It became evident that additional and more extended computations via both more sophisticated QM methods and MM methods with changed force-field parameters are necessary for more exact description of base hydration. The comparison of QM and MM results for both energy minimum positions and energy dependences on selected variables should help to adjust the MM force field to the construction of detailed atom-level models of DNA fragments.
Acknowledgements
This work was partially supported by the VIEP-BUAP.
Cite this paper
Job Lino-Pérez,Eduardo González-Jiménez,Alexandra Deriabina,Martha Velasco,Valery I. Poltev,1 1,1 1, (2016) Comparative Molecular Mechanics and Quantum Mechanics Study of Monohydration of Nucleic Acid Bases. Journal of Biophysical Chemistry,07,49-59. doi: 10.4236/jbpc.2016.72005
References
- 1. Franklin, R.E. and Gosling, R.G. (1953) The Structure of Sodium Thymonucleate Fibres. I. The Influence of Water Content. Acta Crystallographica, 6, 673-677.
http://dx.doi.org/10.1107/S0365110X53001939 - 2. Shishkin, O.V., Gorb, L. and Leszczynski, J. (2000) Does the Hydrated Cytosine Molecule Retain the Canonical Structure? A DFT Study. The Journal of Physical Chemistry B, 104, 5357-5361.
http://dx.doi.org/10.1021/jp993144c - 3. Shishkin, O.V., Sukhanov, O.S., Gorb, L. and Leszczynski, J. (2002) The Interaction of the Most Stable Guanine Tautomers with Water. The Structure and Properties of Monohydrates. Physical Chemistry Chemical Physics, 4, 5359- 5364.
http://dx.doi.org/10.1039/b205351a - 4. van Mourik, T., Danilov, V.I., Gonzalez, E., Deriabina, A. and Poltev, V.I. (2007) Indication of Water Droplet Formation in Hydrated Clusters of Cytosine and Adenine: An Electronic Structure Study Using B3LYP, LMP2 and PM6. Chemical Physics Letters, 445, 303-308.
http://dx.doi.org/10.1016/j.cplett.2007.07.081 - 5. Schneider, B., Cohen, D.M., Schleifer, L., Srinivasan, A.R., Olson, W.K. and Berman, H.M. (1993) A Systematic Method for Studying the Spatial Distribution of Water Molecules around Nucleic Acid Bases. Biophysical Journal, 65, 2291.
http://dx.doi.org/10.1016/S0006-3495(93)81306-7 - 6. Schneider, B. and Berman, H.M. (1995) Hydration of the DNA Bases Is Local. Biophysical Journal, 69, 2661.
http://dx.doi.org/10.1016/S0006-3495(95)80136-0 - 7. Poltev, V.I., Grokhlina, T.I. and Malenkov, G.G. (1984) Hydration of Nucleic Acid Bases Studied Using Novel Atom- Atom Potential Functions. Journal of Biomolecular Structure and Dynamics, 2, 413-429.
http://dx.doi.org/10.1080/07391102.1984.10507576 - 8. González, E., Cedeno, F.I., Teplukhin, A.V., Malenkov, G.G. and Poltev, V.I. (2000) Refinamiento de la metodología de la simulación de la hidratación de los áícidos nucleicos. Revista Mexicana de Física, 46, 142-147.
- 9. Sukhodub, L.F. (1987) Interactions and Hydration of Nucleic Acid Bases in a Vacuum. Experimental Study. Chemical Reviews, 87, 589-606.
http://dx.doi.org/10.1021/cr00079a006 - 10. Kim, S.K., Lee, W. and Herschbach, D.R. (1996) Cluster Beam Chemistry: Hydration of Nucleic Acid Bases; Ionization Potentials of Hydrated Adenine and Thymine. The Journal of Physical Chemistry, 100, 7933-7937.
http://dx.doi.org/10.1021/jp960635d - 11. Belau, L., Wilson, K.R., Leone, S.R. and Ahmed, M. (2007) Vacuum-Ultraviolet Photoionization Studies of the Microhydration of DNA Bases (Guanine, Cytosine, Adenine, and Thymine). The Journal of Physical Chemistry A, 111, 7562-7568.
http://dx.doi.org/10.1021/jp0705929 - 12. Chin, W., Mons, M., Piuzzi, F., Tardivel, B., Dimicoli, I., Gorb, L. and Leszczynski, J. (2004) Gas Phase Rotamers of the Nucleobase 9-Methylguanine Enol and Its Monohydrate: Optical Spectroscopy and Quantum Mechanical Calculations. The Journal of Physical Chemistry A, 108, 8237-8243.
http://dx.doi.org/10.1021/jp048492f - 13. Liu, D., Wyttenbach, T. and Bowers, M.T. (2006) Hydration of Mononucleotides. Journal of the American Chemical Society, 128, 15155-15163.
http://dx.doi.org/10.1021/ja062418o - 14. van Mourik, T., Benoit, D.M., Price, S.L. and Clary, D.C. (2000) Ab Initio and Diffusion Monte Carlo Study of Uracil- Water, Thymine-Water, Cytosine-Water, and Cytosine-(Water) 2. Physical Chemistry Chemical Physics, 2, 1281-1290.
http://dx.doi.org/10.1039/a909183a - 15. González, E., Deriabina, A., Teplukhin, A., Hernández, A. and Poltev, V.I. (2003) Monte Carlo Study of Three- Dimensional Organization of Water Molecules around DNA Fragments. Theoretical Chemistry Accounts, 110, 460- 465.
http://dx.doi.org/10.1007/s00214-003-0502-y - 16. Coutinho, K., Ludwig, V. and Canuto, S. (2004) Combined Monte Carlo and Quantum Mechanics Study of the Hydration of the Guanine-Cytosine Base Pair. Physical Review E, 69, Article ID: 061902.
http://dx.doi.org/10.1103/physreve.69.061902 - 17. Kabelác, M. and Hobza, P. (2007) Hydration and Stability of Nucleic Acid Bases and Base Pairs. Physical Chemistry Chemical Physics, 9, 903-917.
http://dx.doi.org/10.1039/B614420A - 18. Hanus, M., Ryjácek, F., Kabelác, M., Kubar, T., Bogdan, T.V., Trygubenko, S.A. and Hobza, P. (2003) Correlated ab Initio Study of Nucleic Acid Bases and Their Tautomers in the Gas Phase, in a Microhydrated Environment and in Aqueous Solution. Guanine: Surprising Stabilization of Rare Tautomers in Aqueous Solution. Journal of the American Chemical Society, 125, 7678-7688.
http://dx.doi.org/10.1021/ja034245y - 19. Shukla, M.K. and Leszczynski, J. (2008) Hydration-Dependent Structural Deformation of Guanine in the Electronic Singlet Excited State. The Journal of Physical Chemistry B, 112, 5139-5152.
http://dx.doi.org/10.1021/jp7100557 - 20. Crews, B., Abo-Riziq, A., Grace, L., Callahan, M., Kabelác, M., Hobza, P. and de Vries, M.S. (2005) IR-UV Double Resonance Spectroscopy of Guanine-H2O Clusters. Physical Chemistry Chemical Physics, 7, 3015-3020.
http://dx.doi.org/10.1039/b506107e - 21. Kabelac, M., Zendlova, L., Reha, D. and Hobza, P. (2005) Potential Energy Surfaces of an Adenine-Thymine Base Pair and Its Methylated Analogue in the Presence of One and Two Water Molecules: Molecular Mechanics and Correlated ab Initio Study. The Journal of Physical Chemistry B, 109, 12206-12213.
http://dx.doi.org/10.1021/jp045970d - 22. Hanus, M., Kabelác, M., Rejnek, J., Ryjácek, F. and Hobza, P. (2004) Correlated ab Initio Study of Nucleic Acid Bases and Their Tautomers in the Gas Phase, in a Microhydrated Environment, and in Aqueous Solution. Part 3. Adenine. The Journal of Physical Chemistry B, 108, 2087-2097.
http://dx.doi.org/10.1021/jp036090m - 23. Trygubenko, S.A., Bogdan, T.V., Rueda, M., Orozco, M., Luque, F.J., Sponer, J., Slavícek, P. and Hobza, P. (2002) Correlated ab Initio Study of Nucleic Acid Bases and Their Tautomers in the Gas Phase, in a Microhydrated Environment and in Aqueous Solution. Part 1. Cytosine. Physical Chemistry Chemical Physics, 4, 4192-4203.
http://dx.doi.org/10.1039/B202156K - 24. Rejnek, J., Hanus, M., Kabelác, M., Ryjácek, F. and Hobza, P. (2005) Correlated ab Initio Study of Nucleic Acid Bases and Their Tautomers in the Gas Phase, in a Microhydrated Environment and in Aqueous Solution. Part 4. Uracil and Thymine. Physical Chemistry Chemical Physics, 7, 2006-2017.
http://dx.doi.org/10.1039/B501499A - 25. Kim, S., Wheeler, S.E. and Schaefer, H.F. (2006) Microsolvation Effects on the Electron Capturing Ability of Thymine: Thymine-Water Clusters. The Journal of Chemical Physics, 124, Article ID: 204310.
http://dx.doi.org/10.1063/1.2197828 - 26. Kim, S. and Schaefer, H.F. (2007) Microhydration of Cytosine and Its Radical Anion: Cytosine·(H2O)n (n = 1 - 5). The Journal of Chemical Physics, 126, Article ID: 064301.
http://dx.doi.org/10.1063/1.2432123 - 27. Close, D.M., Crespo-Hernández, C.E., Gorb, L. and Leszczynski, J. (2006) Influence of Microhydration on the Ionization Energy Thresholds of Thymine: Comparisons of Theoretical Calculations with Experimental Values. The Journal of Physical Chemistry A, 110, 7485-7490.
http://dx.doi.org/10.1021/jp061064k - 28. Poltev, V.I., Malenkov, G.G., Gonzalez, E.J., Teplukhin, A.V., Rein, R., Shibata, M. and Miller, J.H. (1996) Modeling DNA Hydration: Comparison of Calculated and Experimental Hydration Properties of Nuclic Acid Bases. Journal of Biomolecular Structure and Dynamics, 13, 717-725.
http://dx.doi.org/10.1080/07391102.1996.10508884 - 29. Cornell, W.D., Cieplak, P., Bayly, C.I., Gould, I.R., Merz, K.M., Ferguson, D.M., Spellmeyer, D.C., Fox, T., Caldwell, J.W. and Kollman, P.A. (1995) A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. Journal of the American Chemical Society, 117, 5179-5197.
http://dx.doi.org/10.1021/ja00124a002 - 30. Case, D.A., Cheatham, T.E., Darden, T., Gohlke, H., Luo, R., Merz, K.M., Onufriev, A., Simmerling, C., Wang, B. and Woods, R.J. (2005) The Amber Biomolecular Simulation Programs. Journal of Computational Chemistry, 26, 1668- 1688.
http://dx.doi.org/10.1002/jcc.20290 - 31. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery Jr., J.A., Vreven, T., Kudin, K.N. and Burant, J.C. (2004) Gaussian 03, Revision D. 01. Gaussian Inc., Wallingford, 26.
- 32. Boys, S.F., and Bernardi, F.D. (1970) The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Molecular Physics: An International Journal at the Interface between Chemistry and Physics, 19, 553-566.
http://dx.doi.org/10.1080/00268977000101561 - 33. Hunter, K.C., Rutledge, L.R. and Wetmore, S.D. (2005) The Hydrogen Bonding Properties of Cytosine: A Computational Study of Cytosine Complexed with Hydrogen Fluoride, Water, and Ammonia. The Journal of Physical Chemistry A, 109, 9554-9562.
http://dx.doi.org/10.1021/jp0527709