Journal of Biophysical Chemistry
Vol.5 No.1(2014), Article ID:43166,8 pages DOI:10.4236/jbpc.2014.51002
Geometrical criteria for left-handed twists within protein beta-strands
1Centre d’ Informatique pour la Biologie, Institut Pasteur, Paris, France; bernard.caudron@pasteur.fr
2Unité de Bioinformatique Structurale, Département de Biologie Structurale et Chimie, Institut Pasteur, Paris, France; *Corresponding Author: jjestin@pasteur.fr
Copyright © 2014 Bernard Caudron, Jean-Luc Jestin. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Bernard Caudron, Jean-Luc Jestin. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received 7 January 2014; revised 7 February 2014; accepted 14 February 2014
Keywords:Beta-Sheet; Protein Data Bank; Protein Backbone; Protein Structure; Model Quality Assessment; CASP
ABSTRACT
Using a statistical analysis on beta-sheet structures from the Protein Data Bank, characteristic angles within beta-strands were correlated to the nature of the side chains. The twists were computed from the atomic coordinates of five consecutive amino acids’ alpha carbons from single beta-strand sequences. Conditions on the angles for twists to be mainly left-handed are given together with the frequency of occurrence for these non-standard geometrical properties within protein beta-strands. Applications in protein structure prediction and CASP challenges in particular are envisioned by making use of the probabilities of occurrence in protein structures of angle value ranges for given amino acids.
1. INTRODUCTION
The number of protein sequences is growing at an ever increasing pace along with the developing DNA sequencing facilities [1-3]. Determination of the threedimensional structures of these proteins at atomic resolution relies mainly on time consuming approaches such as NMR or X-ray diffraction. Protein structure prediction made remarkable progress over the last decades [4,5]. In particular, template-based methods provide an efficient approach to obtain three-dimensional models for proteins. Contact maps, correlated mutations and large sets of homologous sequences were also used to establish protein structure models [6-10]. Based on known three-dimensional structures from the Protein Data Bank (PDB) [11,12], protein structural characteristics derived from statistical analyses facilitate the quality assessment of models which is essential in protein structure prediction.
Beta-sheets and alpha-helices represent the major structural elements composing proteins and their folds [13-18]. Within protein sequences, the location of betastrands composing the sheets can be predicted [19,20]. In three dimensions, beta-pleated sheet structures are often represented as planes over large scales; their curvature and geometrical properties were precisely described [21-26]. Beta-bulges define further irregular structures within beta-sheets [27]. The right-handed twist of betastrands was studied structurally and energetically [28-30]; the dihedral angles Phi and Psi in particular have values within narrow ranges characterizing beta-strands within the sheets and their twist [31]. The amino acid composition of beta-strands was found to depend on the parallel or antiparallel nature of beta-strands as well as on the position within beta-strands [32-34]. It allowed the definition of a rule based on the protein sequence defining the anti-parallelism of beta-strands within sheets [35]. Other rules linking beta-strand properties and protein topology were identified [26,36-41]. The packing within protein domains was analyzed for sheets [42]. Good prediction accuracies of protein beta-sheets’ topology were achieved [43-45].
In an attempt to facilitate protein structure prediction, we investigated geometrical properties of protein structures at a scale which is sufficiently large to be connected to topological properties of the proteins and sufficiently small to be linked to amino acid side chains characterizing protein sequences. In this report, we focus on the geometry of beta-strands within beta-sheets by measuring angles within sets of consecutive alpha carbons in beta-strands.
2. METHODS
Pdb21 available at the www address http://mobyle.pasteur.fr/cgi-bin/portal.py#forms::pdb21 is a program written in perl. It uses as entry files, PDB files (pdbxxxx.ent), lists of PDB files corresponding to proteins or a protein domains’ structures which may be bound to other molecules or files of protein structural models written in a PDB file format. The program eliminates files associated to polypeptides of less than fifty amino acids from the lists. It takes into consideration the first protein chain of each file. The output file (.xls) defines for each amino acid of the proteins within the list, its number defining the position within the sequence, its location within the secondary structure elements and the set of angles given as integers in degrees as defined in Figure 1.
So as to avoid biases due to closely related conformations of a same protein with different PDB references, the proteins were chosen randomly among more than 70,000 protein structures of the PDB and two amino acids with identical chemical formulas with the same position numbering within a protein domain starting at the same position and ending at the same position with identical values for the angle α’ were considered as a single amino acid for the statistical analysis. To eliminate biases such as redundancy in the experimental data extracted from the PDB [46], the sorting of information from the PDB files was not carried out at the level of proteins according to their sequence identity, but was done at the level of individual amino acids. Each list was composed of a set of about 800 randomly chosen protein structures from the PDB. The probabilities were derived from the analysis of lists. The errors on the probabilities given in the tables were calculated from two distinct lists.
Another list considered resulted from the removal of protein sequences with significant sequence identity [47]: known as the 25% list, it is available online as the “recent.pdb_select25.nsigma3” file. All angles were annotated with primes in this work to avoid any confusion with earlier work [48,49].
3. RESULTS AND DISCUSSION
3.1. Characteristic Angles within Beta-Strands
Alpha carbons of amino acids are numbered along the protein sequence and their coordinates in the three-dimensional space are given in PDB files. Links are drawn between adjacent alpha carbons. Two links allow an angle to be defined (Figure 1). For each amino acid alpha
 P is the plane containing the atoms 1, 2 and 3 and Q is the plane containing the atoms 3, 4 and 5. α’ is the angle between these two planes. The other angles are defined by two links between two alpha carbons (cf. text). The amino acid side-chains and the angle β’ are not represented in this scheme for clarity; the amino acid in the single-letter code as given in the tables is located at position 3 and therefore part of both planes P and Q.
P is the plane containing the atoms 1, 2 and 3 and Q is the plane containing the atoms 3, 4 and 5. α’ is the angle between these two planes. The other angles are defined by two links between two alpha carbons (cf. text). The amino acid side-chains and the angle β’ are not represented in this scheme for clarity; the amino acid in the single-letter code as given in the tables is located at position 3 and therefore part of both planes P and Q.
Figure 1. Scheme of alpha carbons within a betastrand highlighting the angles calculated.
carbon located at position noted 3 within a beta-strand (Figure 1), the five angles α’, β’, γ’, δ’ and ε’ are calculated from alpha carbons’ atomic coordinates using the scalar product of the corresponding normed vectors. While the angle α’ is defined as an oriented angle between two planes (i.e. between their normal vectors) with values between −180˚ and 180˚, the other four angles are defined by values between 0 and 180˚ using the vectors corresponding to the following pairs of alpha carbons.

The two planes P and Q are defined by the carbon atoms 1, 2, 3 and 3, 4, 5 respectively. The sign of the angle α’ was defined as the sign of the scalar product (.) noted below by making use of the vector product (^) between vectors p and q, which are respectively normal to the planes P and Q.
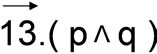
A positive sign for the angle α’ (Figures 1 and 2, Tables 1(a) and 1(b)) then corresponds to the righthanded twist well known for beta-strands within betasheets. Conversely, a left-handed twist is characterized by a negative angle α’. The angles between these virtual bonds defined at a coarser scale than the known dihedral angles Phi and Psi between chemical bonds were necessary to define the notion of twist for amino acids found in a single beta-strand within a sheet. Unique atoms within amino acids such as the alphaor beta-carbon were used in other works for the comparison of protein structures [35,50,51].
3.2. Statistical Analysis of the Angles within Protein Beta-Strands
The twist was defined by the angles Phi and Psi [28] or by a dihedral angle (theta) between alpha and betacarbons of odd residues [31]. Here, the twist was defined for five alpha carbons on a single beta-strand within a beta-sheet by measuring the angle α’ between the two planes P and Q (Figure 1). The distribution of the angles
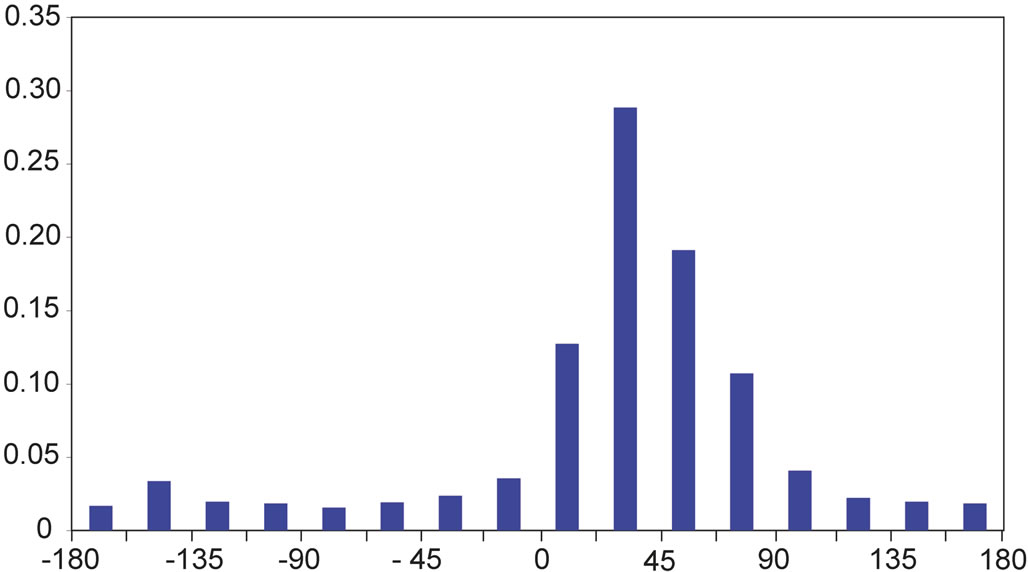
α’ is similar for all amino acids, except for proline. The right-handed twist is characterized here by the distribution of the angle α’ typically found around +30˚ (Figure 2(a)). The probability for finding a proline within a beta-strand associated to an angle α’ between the two planes P and Q between 0˚ and 22˚ is low (0.03), while it is about 3 to 5 times higher for the other amino acids (Table 2(a)).
The classical notion of right-handed twist is not valid
 (a)
(a) 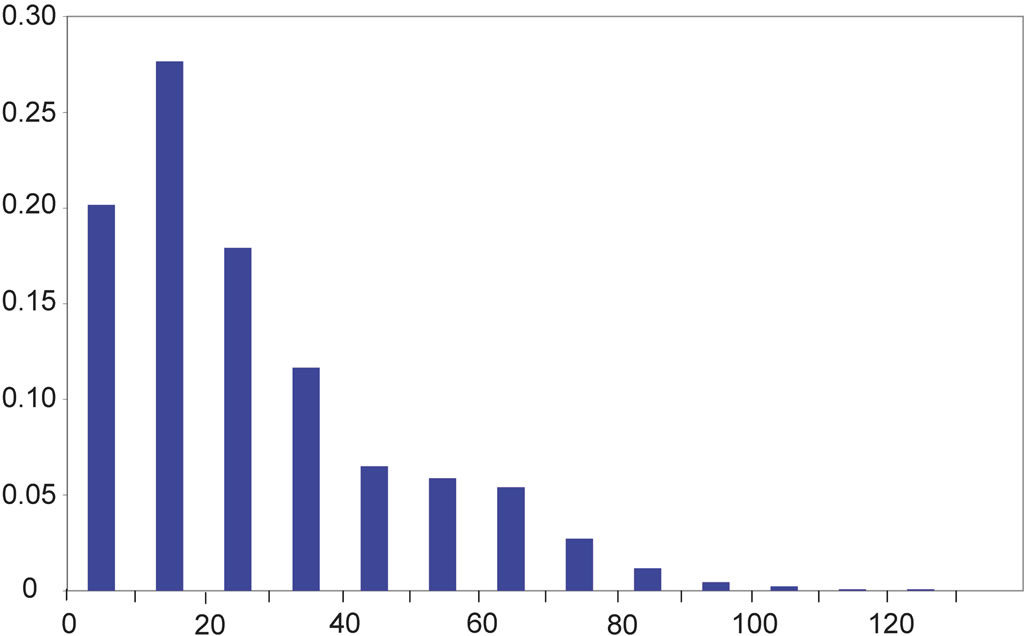 (b)
(b) 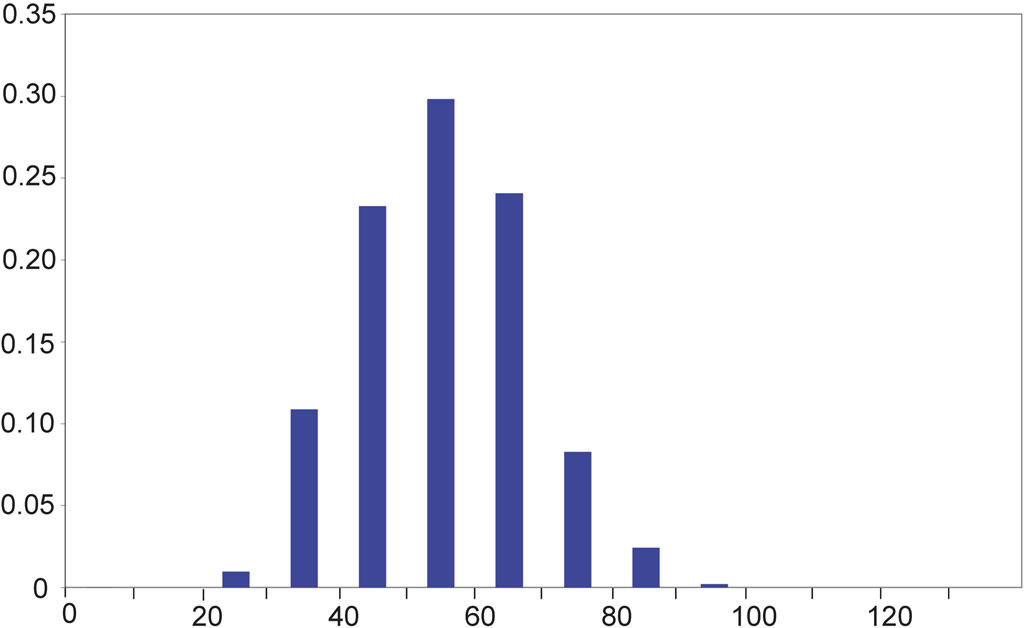 (c)
(c)
On the y-axis is the probability for an angle to be found within ranges of 22.5˚ for α’ and within ranges of 10˚ for the angles β’ and δ’ (x-axis). On the x-axis, α’ ranges from −180˚ to +180˚ and β’ and δ’ range from 0˚ to +140˚.
Figure 2. Distribution of the values of the angles α’ (a), β’ (b), and δ’ (c).

Table 1. Distributions of the angle α’ for different ranges of values for β’, γ’ and ε’.
anymore in two extreme cases characterized by high values for β’ (β’ > 67.5˚) or by low values for ε’ (ε’< 33.5˚). These situations occur in respectively in 6.0% or 6.1% (688/11365 or 5457/89130) and 2.1% or 2.2% (236/11365 or 1961/89130) of the amino acids within beta-strands (Table 1(a)). The angle α’ is then found to have most frequently negative values: the local twist is generally left-handed for about 8% of the amino acids within beta-strands characterized here by extreme values for the angles β’ and ε’ (Table 1(a)). Another relation between the distribution of α’ and the values of γ’ was noted: more than half of the amino acids (56% or 59%) are associated to an α’ value of less than 45˚; but in the case of minimal values for the angle γ’ (γ’ < 33.5˚), more than three quarters of the α’ values are found to be above 45˚ (Table 1(b)).
The angle β’ described earlier was used for protein secondary structure determination in the program DSSP in particular [48,52]. Its distribution (Figure 2(b)) is anomalous for both amino acids glycine and proline, when compared to the other 18 canonical amino acids found within proteins: the probability for β’ to have a value between 0 and 22.5˚ is almost twice lower than for most other amino acids (Table 2(a)). Noticeably, almost half of the β’ values for the amino acid proline (47%) are found between 22.5˚ and 45˚ (Table 2(a)). This observation has to be linked to the distribution of the angle δ’ (Figure 2(c)), which has values within the same range (22.5˚ to 45˚) for only 1% of the prolines within betastrands, i.e. ten to sixty times less than for other amino acids (Table 2(b)).
In beta-strands, the angle δ’ for glycine is found to be in more than half of the cases (57%) between 22.5˚ and 45˚, that is about two to six times more frequently than most other amino acids (Table 2(b)). Accordingly, we noted that glycine represents 5.0% of the amino acids in beta-strands, while glycine represents 56% of the amino acids within beta-strands satisfying the condition (δ’ < 31˚). Glycine and proline whose conformation in proteins were extensively described using the dihedral Phi and Psi angles [53,54] appear also as special cases at a coarser scale among the other canonical amino acids because of their altered δ’ and β’ distributions within beta-strands. As illustrated in Figure 3, three successive alpha carbons centered around glycine tend to be almost aligned more frequently than for other amino acids.
3.3. Application in Structural Model Quality Assessment
Coarse-grained structural models of proteins may consist of their alpha carbon coordinates for all or most amino acids. It is then of interest to have an estimate of the twist values which do not rely on the angles Phi and Psi in particular, but only on alpha carbon coordinates.
The statistical analysis of protein structures reported above may be further used to evaluate the quality of structural models. As an example, given that the angle δ’ value at prolines is statistically less than 45˚ in about 1% of the occurrences within structures reported in the Protein Data Bank, it is unlikely that the δ’ value at prolines is less than 45˚ in a predicted structural model of interest. The higher the number of prolines for which the δ’ value at prolines is less than 45˚, the less likely will be the predicted structural model.
This approach combined with the use for the twenty amino acids of further statistically relevant observations within beta-sheets and within other structures such as those reported recently [25,40] will allow the likelihood of structural models to be estimated using new criteria: it shall improve the assessment of protein structure model quality.
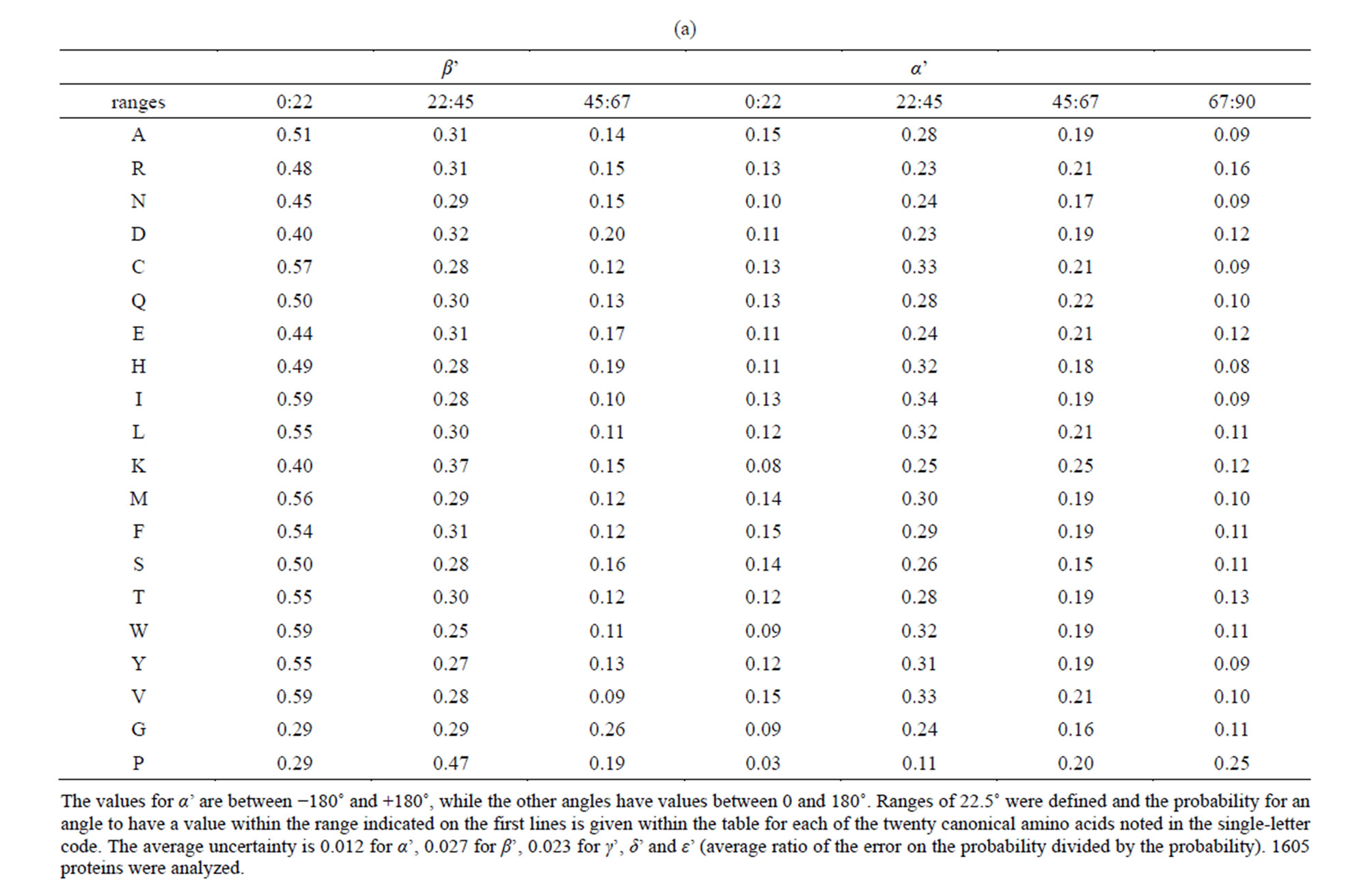
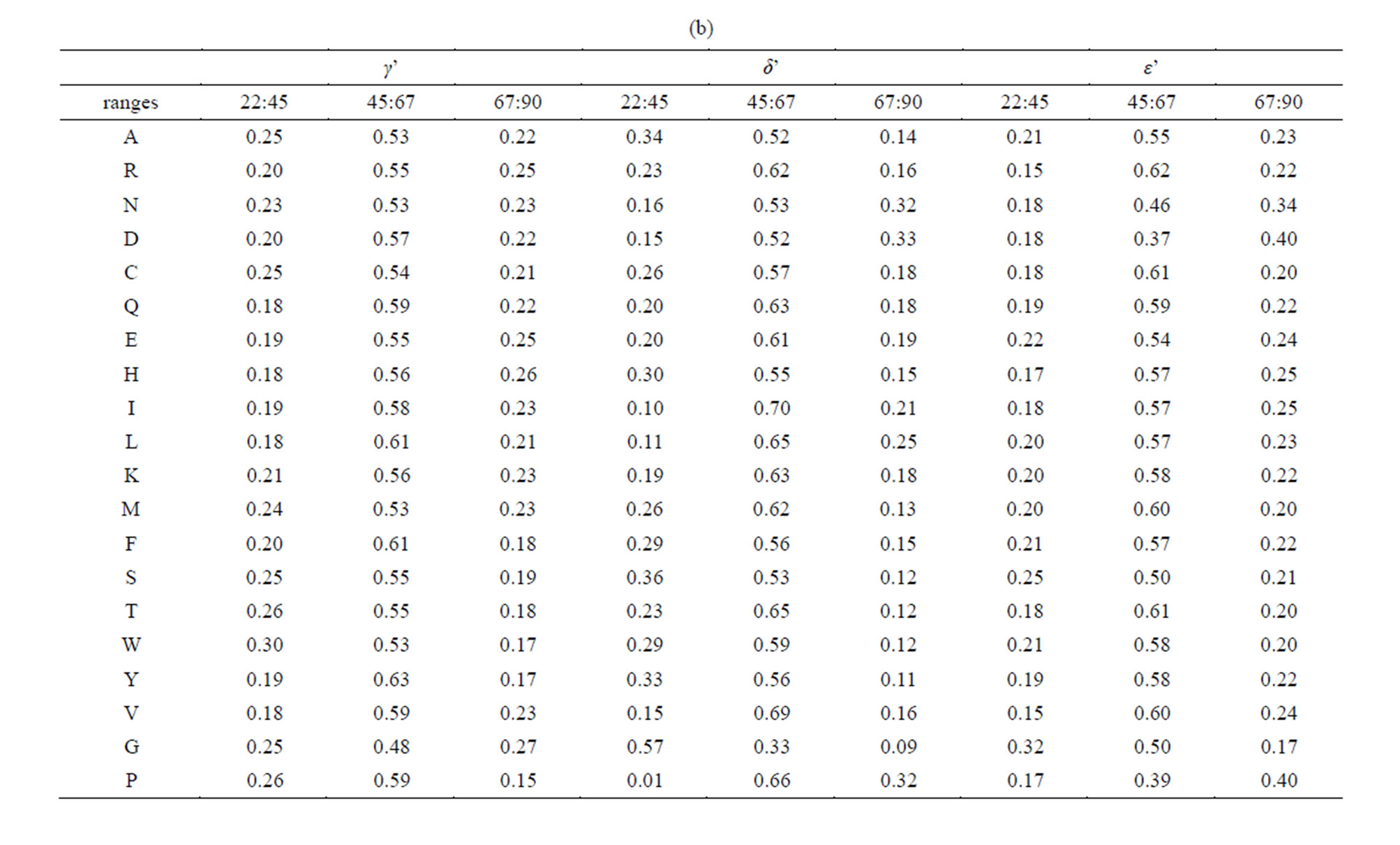
Table 2. Distribution of the angles α’, β’, γ’, δ’ and ε’.
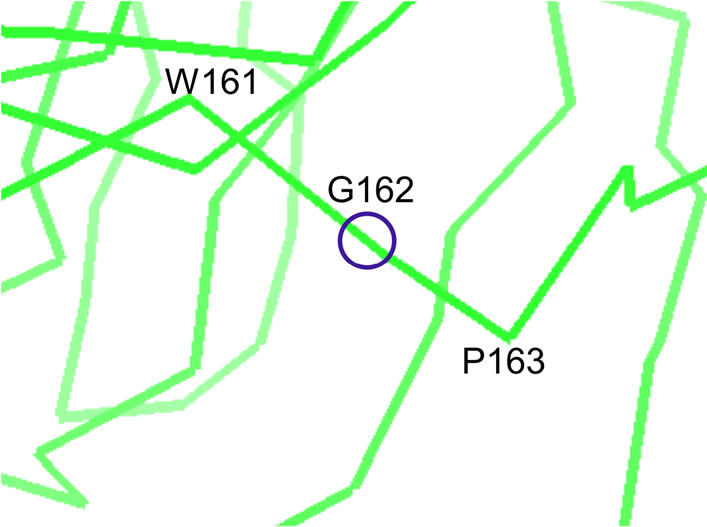 In Dactylium dendroides galactose oxidase protein structure [61] (PDB reference: 1gog) within the betastrand extending from amino acids 160 to 166, the values for α’, β’, γ’, δ’ and ε’ at glycine 162 are respectively −173˚, 20˚, 58˚, 9˚ and 70˚.
In Dactylium dendroides galactose oxidase protein structure [61] (PDB reference: 1gog) within the betastrand extending from amino acids 160 to 166, the values for α’, β’, γ’, δ’ and ε’ at glycine 162 are respectively −173˚, 20˚, 58˚, 9˚ and 70˚.
Figure 3.A typical structure of glycine within a beta-strand.
4. CONCLUSION
A statistical analysis of a large number of protein structures from the Protein Data Bank allowed the definition of geometrical criteria generally associated to lefthanded twists in protein beta-sheets, while a righthanded twist is common for protein beta-sheets. These statistical results on protein structures may be further implemented as probabilities of occurrence for given sets of angles within structure prediction algorithms or used as restraints in protein modeling approaches [55]. These statistical results may be also used within programs for the evaluation of structural model quality [56-58] and contribute to the improvement of protein structure prediction strategies which are evaluated every second year by the critical assessment of protein structure prediction methods (CASP) [59,60].
ACKNOWLEDGEMENTS
The authors thank F. Rey and M. Delarue for their comments and B. Néron for interfacing the program.
REFERENCES
- Iliopoulos, I., Tsoka, S., Andrade, M.A., et al. (2003) Evaluation of annotation strategies using an entire genome sequence. Bioinformatics, 19, 717-726. http://dx.doi.org/10.1093/bioinformatics/btg077
- Juncker, A.S., Jensen, L.J., Pierleoni, A., et al. (2009) Sequence-based feature prediction and annotation of proteins. Genome Biology, 10, 206. http://dx.doi.org/10.1186/gb-2009-10-2-206
- Blicher, T., Gupta, R., Wesolowska, A., et al. (2010) Protein annotation in the era of personal genomics. Current Opinion in Structural Biology, 20, 335-341. http://dx.doi.org/10.1016/j.sbi.2010.03.008
- Moult, J., Hubbard, T., Bryant, S.H., et al. (1997) Critical assessment of methods of protein structure prediction (CASP): Round II. Proteins, 29, 2-6. http://dx.doi.org/10.1002/(SICI)1097-0134(1997)1+<2::AID-PROT2>3.0.CO;2-T
- Kryshtafovych, A., Fidelis, K. and Moult, J. (2011) CASP9 results compared to those of previous CASP experiments. Proteins, 79, 196-207. http://dx.doi.org/10.1002/prot.23182
- Seno, F., Trovato, A., Banavar, J.R. and Maritan, A. (2008) Maximum entropy approach for deducing amino acid interactions in proteins. Physical Review Letters, 100, Article ID: 078102. http://dx.doi.org/10.1103/PhysRevLett.100.078102
- Vassura, M., Margara, L., Di Lena, P., et al. (2008) Reconstruction of 3D structures from protein contact maps. IEEE-ACM Transactions on Computational Biology and Bioinformatics, 5, 357-367. http://dx.doi.org/10.1109/TCBB.2008.27
- Sulkowska, J.I., Morcos, F., Weigt, M., et al. (2012) Genomics-aided structure prediction. Proceedings of the National Academy of Sciences of USA, 109, 10340-10345. http://dx.doi.org/10.1073/pnas.1207864109
- Taylor, W.R., Jones, D.T. and Sadowski, M.I. (2012) Protein topology from predicted residue contacts. Protein Science, 21, 299-305. http://dx.doi.org/10.1002/pro.2002
- Hopf, T.A., Colwell, L.J., Sheridan, R., et al. (2012) Three-dimensional structures of membrane proteins from genomic sequencing. Cell, 149, 1607-1621. http://dx.doi.org/10.1016/j.cell.2012.04.012
- Abola, E.E., Sussman, J.L., Prilusky, J. and Manning, N.O. (1997) Protein Data Bank archives of three-dimensional macromolecular structures. Methods in Enzymology, 277, 556-571. http://dx.doi.org/10.1016/S0076-6879(97)77031-9
- Sussman, J.L., Lin, D., Jiang, J., et al. (1998) Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallographica Section D, 54, 1078-1084. http://dx.doi.org/10.1107/S0907444998009378
- Brenner, S.E., Koehl, P. and Levitt, M. (2000) The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Research, 28, 254-256. http://dx.doi.org/10.1093/nar/28.1.254
- Bateman, A., Birney, E., Cerruti, L., et al. (2002) The Pfam protein families database. Nucleic Acids Research, 30, 276-280. http://dx.doi.org/10.1093/nar/30.1.276
- Banavar, J.R., Maritan, A., Micheletti, C. and Trovato, A. (2002) Geometry and physics of proteins. Proteins, 47, 315-322. http://dx.doi.org/10.1002/prot.10091
- Pal, L., Dasgupta, B. and Chakrabarti, P. (2005) 3(10)- Helix adjoining alpha-helix and beta-strand: Sequence and structural features and their conservation. Biopolymers, 78, 147-162. http://dx.doi.org/10.1002/bip.20266
- Greene, L.H., Lewis, T.E., Addou, S., et al. (2007) The CATH domain structure database: New protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Research, 35, D291-D297. http://dx.doi.org/10.1093/nar/gkl959
- Andreeva, A., Howorth, D., Chandonia, J.M., et al. (2008) Data growth and its impact on the SCOP database: New developments. Nucleic Acids Research, 36, D419-D425. http://dx.doi.org/10.1093/nar/gkm993
- Rost, B. and Sander, C. (1993) Prediction of protein secondary structure at better than 70% accuracy. Journal of Molecular Biology, 232, 584-599. http://dx.doi.org/10.1006/jmbi.1993.1413
- Martin, J., Letellier, G., Marin, A., et al. (2005) Protein secondary structure assignment revisited: A detailed analysis of different assignment methods. BMC Structural Biology, 5, 17. http://dx.doi.org/10.1186/1472-6807-5-17
- Pauling, L. and Corey, R.B. (1951) Configurations of polypeptide chains with favored orientations around single bonds: two new pleated sheets. Proceedings of the National Academy of Sciences of USA, 37, 729-740. http://dx.doi.org/10.1073/pnas.37.11.729
- Sternberg, M.J. and Thornton, J.M. (1977) On the conformation of proteins: An analysis of beta-pleated sheets. Journal of Molecular Biology, 110, 285-296. http://dx.doi.org/10.1016/S0022-2836(77)80073-9
- Salemme, F.R. (1983) Structural properties of protein beta-sheets. Progress in Biophysics and Molecular Biology, 42, 95-133. http://dx.doi.org/10.1016/0079-6107(83)90005-6
- Koh, E., Kim, T. and Cho, H.S. (2006) Mean curvature as a major determinant of beta-sheet propensity. Bioinformatics, 22, 297-302. http://dx.doi.org/10.1093/bioinformatics/bti775
- Jestin, J. and Caudron, B. (2013) Characterizing the topology of protein beta-sheets by an axis. 162-1466-1-SP, HAL-Pasteur 00907789.
- Guilloux, A., Caudron, B. and Jestin, J.L. (2014) A method to predict amino acids at proximity of beta-sheet axes from protein sequences. Applied Mathematics, 5, 79- 89. http://dx.doi.org/10.4236/am.2014.51009
- Chan, A.W., Hutchinson, E.G., Harris, D. and Thornton, J.M. (1993) Identification, classification, and analysis of beta-bulges in proteins. Protein Science, 2, 1574-1590. http://dx.doi.org/10.1002/pro.5560021004
- Chothia, C. (1973) Conformation of twisted beta-pleated sheets in proteins. Journal of Molecular Biology, 75, 295- 302. http://dx.doi.org/10.1016/0022-2836(73)90022-3
- Chou, K.C., Pottle, M., Nemethy, G., et al. (1982) Structure of beta-sheets-origin of the right handed twist and of the increased stability of anti-parallel over parallel sheets. Journal of Molecular Biology, 162, 89-112. http://dx.doi.org/10.1016/0022-2836(82)90163-2
- Lasters, I. (1990) Estimating the twist of beta-strands embedded within a regular parallel beta-barrel structure. Protein Engineering, 4, 133-135. http://dx.doi.org/10.1093/protein/4.2.133
- Shamovsky, I.L., Ross, G.M. and Riopelle, R.J. (2000) Theoretical studies on the origin of beta-sheet twisting. The Journal of Physical Chemistry B, 104, 11296-11307. http://dx.doi.org/10.1021/jp002590t
- Lifson, S. and Sander, C. (1979) Antiparallel and parallel beta-strands differ in amino acid residue preferences. Nature, 282, 109-111. http://dx.doi.org/10.1038/282109a0
- Farzadfard, F., Gharaei, N., Pezeshk, H. and Marashi, S.A. (2008) Beta-sheet capping: Signals that initiate and terminate beta-sheet formation. Journal of Structural Biology, 161, 101-110. http://dx.doi.org/10.1016/j.jsb.2007.09.024
- Bhattacharjee, N. and Biswas, P. (2009) Structural patterns in alpha helices and beta sheets in globular proteins. Protein and Peptide Letters, 16, 953-960. http://dx.doi.org/10.2174/092986609788923239
- Caudron, B. and Jestin, J.L. (2012) Sequence criteria for the anti-parallel character of protein beta-strands. Journal of Theoretical Biology, 315, 146-149. http://dx.doi.org/10.1016/j.jtbi.2012.09.011
- Sternberg, M.J. and Thornton, J.M. (1977) On the conformation of proteins: Towards the prediction of strand arrangements in β-pleated sheets. Journal of Molecular Biology, 113, 401-418. http://dx.doi.org/10.1016/0022-2836(77)90149-8
- Sternberg, M.J. and Thornton, J.M. (1977) On the conformation of proteins: Hydrophobic ordering of strands in β- pleated sheets. Journal of Molecular Biology, 115, 1-17. http://dx.doi.org/10.1016/0022-2836(77)90242-X
- Wang, W. and Hecht, M.H. (2002) Rationally designed mutations convert de novo amyloid-like fibrils into monomerric β-sheet proteins. Proceedings of the National Academy of Sciences of the United States of America, 99, 2760-2765. http://dx.doi.org/10.1073/pnas.052706199
- Richardson, J.S. and Richardson, D.C. (2002) Natural β- sheet proteins use negative design to avoid edge-to-edge aggregation. Proceedings of the National Academy of Sciences of the United States of America, 99, 2754-2759. http://dx.doi.org/10.1073/pnas.052706099
- Koga, N., Tatsumi-Koga, R., Liu, G., Xiao, R., Acton, T.B., Montelione, G.T. and Baker, D. (2012) Principles for designing ideal protein structures. Nature, 491, 222-227. http://dx.doi.org/10.1038/nature11600
- Guilloux, A., Caudron, B. and Jestin, J.L. (2013) A method to predict edge strands in beta-sheets from protein sequences. Computational and Structural Biotechnology Journal, 7, 1-7. http://dx.doi.org/10.5936/csbj.201305001
- Chothia, C., Levitt, M. and Richardson, D. (1977) Structure of proteins: Packing of alpha-helices and pleated sheets. Proceedings of the National Academy of Sciences of the United States of America, 74, 4130-4134. http://dx.doi.org/10.1073/pnas.74.10.4130
- Cheng, J. and Baldi, P. (2005) Three-stage prediction of protein β-sheets by neural networks, alignments and graph algorithms. Bioinformatics, 21, i75-i84. http://dx.doi.org/10.1093/bioinformatics/bti1004
- Zafer, A., Yucel, A. and Hakan, E. (2011) Bayesian models and algorithms for protein β-sheet prediction. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 8, 395-409. http://dx.doi.org/10.1109/TCBB.2008.140
- Subramani, A. and Floudas, C.A. (2012) β-sheet topology prediction with high precision and recall for β and mixed α/β proteins. PLoS ONE, 7, Article ID: e32461. http://dx.doi.org/10.1371/journal.pone.0032461
- Peng, K., Obradovic, Z. and Vucetic, S. (2004) Exploring the bias in the PDB using contrast classifiers. In: Altman R.B., Dunker, K., et al., Eds., Pacific Symposium Biocomputing, World Scientific, Singapore.
- Hobohm, U., Scharf, M., Schneider, R. and Sander, C. (1992) Selection of representative protein data sets. Protein Science, 1, 409-417. http://dx.doi.org/10.1002/pro.5560010313
- Kabsch, W. and Sander, C. (1983) Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577-2637. http://dx.doi.org/10.1002/bip.360221211
- Efimov, A.V. (1986) Standard conformations of a polypeptide chain in irregular protein regions. Molekuliarnaia Biologiia, 20, 250-260.
- Richards, F.M. and Kundrot, C.E. (1988) Identification of structural motifs from protein coordinate data: Secondary structure and first-level supersecondary structure. Proteins, 3, 71-84. http://dx.doi.org/10.1002/prot.340030202
- Taylor, W.R. and Orengo, C.A. (1989) A holistic approach to protein structure alignment. Protein Engineering Design & Selection, 2, 505-519. http://dx.doi.org/10.1093/protein/2.7.505
- Rose, G.D. and Seltzer, J.P. (1977) A new algorithm for finding the peptide chain turns in a globular protein. Journal of Molecular Biology, 113, 153-164. http://dx.doi.org/10.1016/0022-2836(77)90046-8
- MacArthur, M.W. and Thornton, J.M. (1991) Influence of proline residues on protein conformation. Journal of Molecular Biology, 218, 397-412. http://dx.doi.org/10.1016/0022-2836(91)90721-H
- Ho, B.K. and Brasseur, R. (2005) The Ramachandran plots of glycine and pre-proline. BMC Structural Biology, 5, 14. http://dx.doi.org/10.1186/1472-6807-5-14
- Skolnick, J., Kolinski, A. and Ortiz, A.R. (1997) Monsster: A method for folding globular proteins with a small number of distance restraints. Journal of Molecular Biology, 265, 217-241. http://dx.doi.org/10.1006/jmbi.1996.0720
- Costantini, S., Facchiano, A.M. and Colonna, G. (2007) Evaluation of the structural quality of modeled proteins by using globularity criteria. BMC Structural Biology, 7, 9. http://dx.doi.org/10.1186/1472-6807-7-9
- DeRonne, K.W. and Karypis, G. (2009) Improved estimation of structure predictor quality. BMC Structural Biology, 9, 41. http://dx.doi.org/10.1186/1472-6807-9-41
- Benkert, P., Biasini, M. and Schwede, T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics, 27, 343-350. http://dx.doi.org/10.1093/bioinformatics/btq662
- Moult, J., Fidelis, K., Kryshtafovych, A., Rost, B. and Tramontano, A. (2009) Critical assessment of methods of protein structure prediction—Round VIII. Proteins, 77, 1-4. http://dx.doi.org/10.1002/prot.22589
- [61] Moult, J., Fidelis, K., Kryshtafovych, A. and Tramontano, A. (2011) Critical assessment of methods of protein structure prediction (CASP)—Round IX. Proteins, 79, 1-5. http://dx.doi.org/10.1002/prot.23200
- [62] Ito, N., Phillips, S.E., Stevens, C., Ogel, Z.B., Mcpherson, M.J., Keen, J.N., Yadav, K.D.S. and Knowles, P.F. (1991) Novel thioether bond revealed by a 1.7 A crystal structure of galactose oxidase. Nature, 350, 87-90. http://dx.doi.org/10.1038/350087a0