Green and Sustainable Chemistry
Vol.09 No.01(2019), Article ID:90635,15 pages
10.4236/gsc.2019.91002
Conference Paper: Green Industry Adapted to Recycling Needs of Lubumbashi City and Surrounding Areas in Democratic Republic of the Congo
Topwe Milongwe Mwene-Mbeja1, Céline Vaneeckhaute2
1Department of Chemistry, Faculty of Science, University of Lubumbashi, Lubumbashi, Democratic Republic of the Congo
2BioEngine-Research Team on Green Process Engineering and Biorefineries, Department of Chemical Engineering, Faculty of Science and Engineering, Université Laval, Québec City, Canada
![]()
Copyright © 2019 by author(s) and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: December 5, 2018; Accepted: February 18, 2019; Published: February 21, 2019
ABSTRACT
The interest of this conference is agricultural, environmental, bioenergetics, and sanitary. In that context, domestic, agricultural and industrial environments produce organic waste, which needs to be collected, selected, stored and recycled properly in order to avoid environmental pollution and promote agriculture. The green Industry proposed involves the conversion of natural, non-toxic organic waste in order to efficiently produce organic fertilizers for agriculture. These types of fertilizers from biological origin are suitable because they are not toxic for human and the environment. Enzymatic reactions described in this presentation concern mainly the hydrolysis of proteins, sugars and lipids, the acidification of intermediate products from hydrolysis, the formation of acetate, and the production of methane. In other words, this review is timely as it discusses for the chemical behavior or the reactivity of different functional groups to better understand the enzymatic catalysis in the transformations of residual proteins, carbohydrates, and lipids to generate biomethane and fertilizers. In the same perspective, this review is to enrich the documentation related to organic reactions catalyzed by enzymes, which occur in the anaerobic degradation of residual organic substances, with emphasis on the structures of organic compounds and reaction mechanisms. This will allow understanding the displacement of the electrons of a reactive entity rich in electrons to another reactive entity that is poor in electrons to form new bonds in products.
Keywords:
Organic Wastes, Organic Fertilizers, Bioenergetics, Pollution, Environment, Biocatalysts

1. Introduction
The green industry is an industry based on green chemistry. The latter can be defined as the design, development and application of chemical processes and products to reduce or eliminate the use and formation of harmful substances for human health and the environment. Bio-catalytic processes fully participate in the concept of green chemistry, which was introduced in the 1990s [1] - [6]. Biocatalysts constitute an alternative more green than traditional organic synthesis. They are adequate tools for the industrial transformation of naturals or synthetic compounds because bio-catalytic processes take place under mild reaction conditions with less energy and fewer problems of epimerization and rearrangement. In addition, biocatalysts are biodegradable, regio-, chemo- or stereospecific, which minimizes the formation of secondary products [1] - [6].
When organic residues are not recycled or processed efficiently, they are broken down by microorganisms and this favors the multiplication of insects, which can prove to be true vectors of disease. Odors from organic decomposition sites also affect quality of life. To solve the problem of organic waste, biomethanation is an ecological and efficient solution for converting organic matter to produce methane as well as organic fertilizers under the action of microorganisms. The valorization of organic waste through biomethanation contributes to the improvement of hygiene, and reduces bad odors and the use of non-natural fertilizers and pesticides.
Biomethanation also helps to avoid incineration and the dumping of organic waste in landfills in order to reduce emissions of greenhouse gases. Indeed, landfills and the incineration of organic waste essentially produce greenhouse gases such as carbon dioxide and nitrogen dioxide [7]. Energy from biogas is very important especially in regions lacking energy infrastructure because it reduces the use of fossil fuels, limits deforestation and improves people’s livelihoods who prepare food with firewood. Such green recycling is a good strategy to clean up the environment and improve agriculture, particularly in underdeveloped countries, such as Democratic Republic of Congo.
2. Enzymatic Reactions
Biomethanation of organic waste is a collection of organic reactions such as hydrolysis, acidification, synthesis of acetate (ethyl acetate or methyl acetate) and synthesis of methane catalyzed by enzymes. Various substrates (proteins, sugars and lipids) are metabolized or degraded in an anaerobic fashion by specific enzymes and intermediate products serve as starting materials for subsequent enzymatic conversions to produce methane and fertilizers.
3. Hydrolysis
The residual organic substances consist mainly of proteins, sugars and lipids. The enzymatic hydrolysis of these macromolecules is the first step in the production of methane and fertilizers. Enzymes are proteins or organic catalysts secreted within the cells of microorganisms. However, enzymes are also capable of acting outside the original cell. Normally, different reactions are catalyzed by different enzymes, despite the fact that a small number of enzymes are capable of catalyzing several substrates. In general, the amino acids in active centre of the enzyme act as acids, bases, nucleophiles or electrophiles and form hydrogen bonds with the substrates or with other amino acid residues in order to stabilize the transition state (Figure 1) [8] [9] [10]. From this point of view, a triad composed of serine, histidine and aspartate forms a catalytic centre of certain proteases and these three amino acids work together to carry out the expected catalytic activity. In the catalytic triad, histidine plays the role of a base (Figure 1) [8] [9] [10].
Amino acids are small molecules with an amine group, carboxylic acid group and a prosthetic group (R), which differentiate the amino acids from each other. The amino acids are constituents of proteins. They are generally levorotatory (L) and the amine functional group is on the left (Figure 2). Polar amino acids are more frequently present in the active centre of the enzyme and depending on the reaction conditions, they can be ionized. That is why they release protons as acids and they capture protons as bases and being nucleophiles they transfer electrons to electrophiles during catalysis (Figure 2) [8] [9] [10]. In the active centre, the side chains of the amino acids participate in the acid and base chemical catalysis and covalent catalysis. In other words, they can give or accept protons and thereby the active centre is thus the biological equivalent of a strong acid or strong base solution [8] [9] [10].
Nucleophilic N-terminal hydrolases constitute a family of enzymes specialized in the breakdown of amide bonds, for instance, serine protease characterized by the presence of serine, histidine and aspartic acid in the active centre [11] [12]. The strategic step in the serine protease mechanism is the nucleophilic attack at the carbonyl group of the polypeptide (protein) by the oxygen ion from the

Figure 1. Catalytic activity of serine, histidine and aspartate.

Figure 2. Ionized amino acids.
hydroxyl group of the serine (Scheme 1, reaction 2). The role of aspartate is to remove the proton bound to the nitrogen atom of histidine, while histidine removes the acid proton from the hydroxyl group of serine (Scheme 1, reaction 1) [11] [12].
Sugars or carbohydrates are organic compounds that act as an energy reservoir at the level of higher plants. Because humans consume plants, sugars turn out to be essential constituents of human food. In general, carbohydrates are one of the major chemical groups containing carbon, hydrogen and oxygen, such as starch, which is an energy reserve substance in the plant cell [13] - [23]. Starch is a long chain made up of glucose molecules and non linear glucose molecules constitute amylopectin (non linear polymer). On the other hand, glucose molecules that are linear form amylose (linear polymer). The glucose molecules within the starch can range from a few hundred to a few thousand linked together by alpha-glycoside linkages [13] - [23].
During the hydrolysis of starch, it was found that the alpha stereochemistry is maintained in the final product and the plausible catalytic mechanism involves the protonation of the glycosidic oxygen by the proton donor glutamate amino acid (Scheme 2, reaction 2) [24] - [34]. This step is followed by the ejection of the leaving group, due to an electron lone pair oriented antiperiplanar to the leaving group, to produce a corresponding cyclic oxocarbonium ion (Scheme 2, reaction 2). The latter reacts with a water molecule to generate an intermediate compound, which yields a proton to aspartate and this results in the formation of glucose molecules in the reaction medium (Scheme 2, reaction 3) [24] - [34].
Cellulose is an organic polymer formed by several units of glucose molecules linked together by beta-(1-4)-glycosidic bonds (Scheme 3) [24] - [34]. It is associated with lignin and hemicellulose, which form a protective layer or a shield of the cellulose. It has been observed that lignin and hemicellulose are difficult to hydrolyse with enzymes. For this reason, the cellulosic waste must be treated beforehand, for instance, in using dilute acids to degrade lignin and hemicellulose and thus promote the efficiency and rapidity of the enzymatic hydrolysis of cellulose (Scheme 3) [24] - [34]. In this perspective, it has been reported that the enzyme derived from the fungus Trichoderma viride was capable of transforming
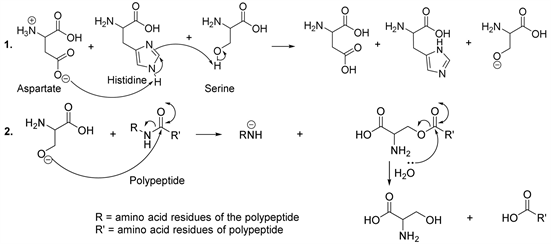
Scheme 1. Serine protease mechanism.
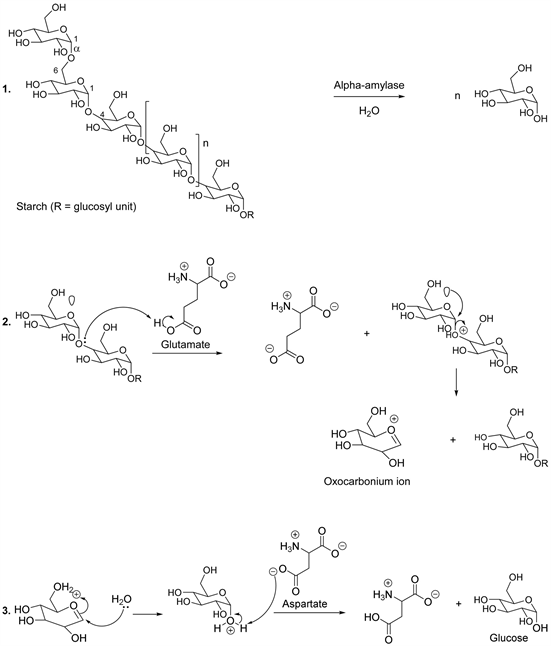
Scheme 2. Catalytic mechanism of starch.

Scheme 3. Enzymatic hydrolysis of cellulose.
the cellulosic waste and the liquid glucose was cleanly obtained [24] - [34]. The authors mentioned that the natural fungus Trichoderma viride produces cellulase, which reacts with a crystalline fraction of a cellulose molecule (Scheme 3). From this point of view, it has been reported that the hydrolysis of cellulosic residues, from sugar cane, with cellulase produced glucose with excellent yield (Scheme 3) [24] - [34].
Cellulase is a group of different enzymes, in particular, the endoglucanase enzymes which hydrolyze beta-(1-4)-glycosidic internal bonds, exoglucanase enzymes that hydrolyze beta-(1-4)-glycosidic external bonds and the cellobiase that hydrolyzes the disaccharide cellobiose to produce two molecules of glucose. Experimental studies showed that two amino acids (glutamate and aspartate) constitute the root of the catalytic activity in the hydrolysis of cellulose. In this regard, the glutamate amino acid yields a proton to glycosidic oxygen and the aspartate amino acid acts as a base by accepting the proton of a water molecule, which acts as a nucleophile (Scheme 4) [24] - [34]. According to the theory of stereo-electronic control, the carbon-hydrogen bond of beta-glycoside (equatorial isomer) is activated by two lone pairs of electrons oxygen antiperiplanar to the carbon-hydrogen bond (two electronic effects) [24] - [34]. In this context, the proposed hydrolysis mechanism evolved the formation of a twist-boat conformation, which allows the substrate to line up a lone pair of electrons oxygen in an antiperiplanar way to the leaving group, before adopting a chair-like transition state (cyclic oxocarbonium ion) (Scheme 4) [24] - [34]. The nucleophilic attack at the oxocarbonium ion leads to the production of an intermediate adduct, which donates a proton to aspartate to produce the axial isomer (alpha-glycoside) and the equatorial isomer (beta-glycoside) (Scheme 4) [24] - [34].
The best known disaccharide is sucrose obtained from sugarcane juice or sugar beet. During enzymatic hydrolysis, sucrose degrades to generate D-(+)-glucose and D-(-)-fructose (Scheme 5, reaction 1) [35] - [42]. Concerning the reaction mechanism, Reddy and Maley have demonstrated the role of the aspartate amino acid as well as that of the glutamate amino acid in the catalytic activity of the enzyme invertase (Scheme 5) [35] - [42]. In this perspective, the carboxylate group of the side chain of aspartate acts as nucleophile, while the carboxylic group of the glutamate amino acid acts as an acid (donor of the proton) (Scheme 5, reaction 2) and the conjugate base accepts the proton of the water (Scheme 5,
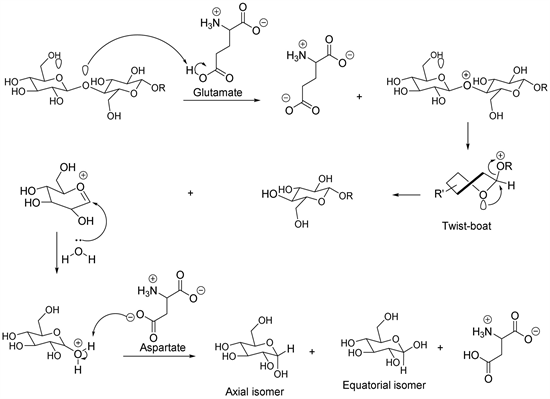
Scheme 4. Cellulose enzymatic hydrolysis mechanism.
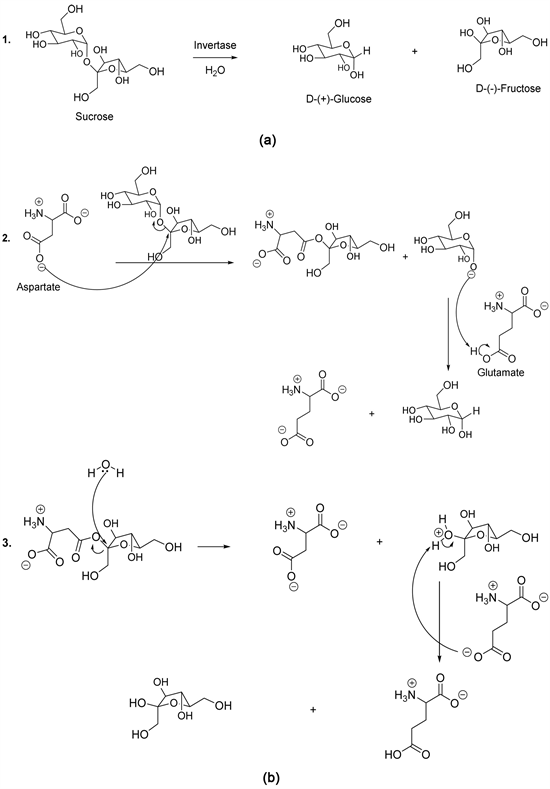
Scheme 5. (a) Enzymatic hydrolysis of sucrose; (b) Sucrose enzymatic hydrolysis mechanism.
reaction 3). The latter acts also as a nucleophile (Scheme 5, reaction 3) [35] - [42].
Lipids are triglycerides or esters of fatty acids and glycerol containing long (R) chains which are insoluble in water (Scheme 6). From the environmental point of view, lipids are important organic constituents in wastewater, which contribute enormously to environmental pollution. In this regard, if they are not removed, the lipids form a layer on the surface of the water and thus prevent the diffusion of oxygen from the air into the water. Consequently, the result is the death of aquatic living beings. From this perspective, it has been shown that the lipases are potential catalysts used to hydrolyze oils from industrial effluents,
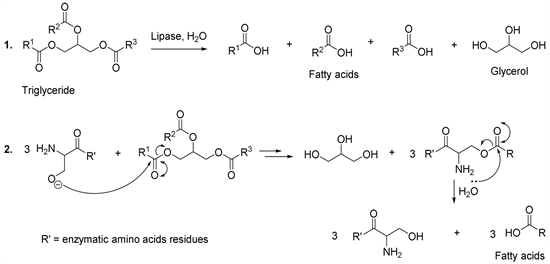
Scheme 6. Triglyceride enzymatic hydrolysis mechanism.
slaughterhouses and frying oils [43]. Experimental observations have shown that the lipases catalyze the hydrolysis of the ester bonds to produce fatty acids and glycerol (Scheme 6, reaction 1) [44]. These enzymes contain serine, histidine and aspartate amino acid in the active centre. It has been found that the aspartate amino acid can be replaced by the glutamate amino acid in the case of Geotrichum candidum lipase [44]. This enzyme uses the glutamate residue to specifically hydrolyze the fatty acids having cis double bonds in the hydrocarbon side chains [44]. With this in mind, the reaction mechanism of the lipases is thus similar to that of serine protease (Scheme 1, reaction 2).
The catalytic activity of the lipases is influenced by the water molecules present in the structure of these enzymes. From this point of view, it has been observed that when the lipases are dehydrated, they lose the ability to catalyze esterification reactions involving congested alcohols. This could be explained by the fact that the absence of water affects the mobility necessary to bind the substrates in the active centre [45] [46] [47]. This is why lipases are less active in solvents which are miscible with water because they extract water integrated into the structure of the enzyme [45] [46] [47].
4. Acidification
The products from hydrolysis are used by microorganisms as substrates to produce acids, alcohols and carbon dioxide (Scheme 7). At this level of biomethanation, amino acids could be used as a source of energy for microorganisms. From this point of view, Rupasinghe and his colleagues reported an enzymatic method for producing glucose, ethanol and acetic acid from apple residues (Scheme 7) [48] [49] [50] [51]. Indeed, the cellulose was pretreated under acidified aqueous conditions and the hydrolysis was optimized in using commercial enzymes (beta-glucosidase and pectinase). The glucose was fermented using Saccharomyces cerevisiae to produce ethanol. The bioconversion of ethanol to acetic acid was carried out using Acetobacter aceti [48] [49] [50] [51].
The enzymatic oxidation of ethanol to acetaldehyde is catalyzed by alcohol
![]()
Scheme 7. Synthesis of acetic acid.
dehydrogenase (Scheme 7, reaction 2). On the other hand, the oxidation of acetaldehyde to acetic acid is catalyzed by the aldehyde dehydrogenase (Scheme 7, reaction 3). These enzymes use nicotinamide adenine dinucleotide (NAD) as co-enzyme and glutamate plays the role of a base in the active centre of the enzyme (dehydrogenase). Regarding the dehydrogenation of ethanol, the mechanism involves the transfer of the hydride ion to NAD+ (oxidized form of NAD) and this results in the formation of NADH (reduced form of NAD) and the desired product (acetaldehyde) (Scheme 7, reaction 2), while dehydrogenation of acetaldehyde involves nucleophilic attack by water molecule upon the electrophilic carbon of acetaldehyde. This step is followed by the removal of the proton by glutamate and the transfer of the hydride ion at the level of the oxidized form of NAD (Scheme 7, reaction 3) [52] [53] [54].
5. Synthesis of Acetate
It has been observed that the major part of the methane comes from the acetate (Scheme 8) [55] [56] [57]. In this context, it has been demonstrated that the lipase catalyses the reaction between acetic acid and ethanol to produce volatile natural ethyl acetate and water as by product [55] [56] [57]. Because of the reversibility of the reaction, it is important to remove water in order to optimize the formation of the product. Concerning the mechanism, the lipase utilized serine, histidine and aspartate amino acid to catalyze the production of acetate from acetic acid and alcohol (ROH). In this perspective, aspartate amino acid gives a proton to the hydroxyl group of acetic acid (Scheme 8, reaction 2). This step is followed by serine nucleophilic attack and the result is the loss of one
![]()
Scheme 8. Synthesis of acetate.
molecule of water and forming an intermediate adduct, which in turn reacts with the alcohol (ethanol or methanol) to produce the corresponding acetate (Scheme 8, reaction 3) [55] [56] [57].
6. Synthesis of Methane
Bacteria degrade ethyl acetate to produce ethanol and acetic acid. The latter is then metabolized to produce methane and carbon dioxide with the assistance of decarboxylase as catalyst. From the mechanism point of view, it has been reported that decarboxylation involves the formation of negatively charged intermediate compounds. In this perspective, the electrophilic substitution of carbon dioxide by a proton involves the loss of carbon dioxide to form a carbanion (methyl anion), which accepts a proton from the catalytic amino acid residue, in this case lysine, to generate the desired product (methane) (Scheme 9) [58] [59]. It has been observed that lysine and cysteine are at the centre of catalytic activity of decarboxylase (Scheme 9) [60].
7. Conclusion
This conference paper has showed the importance of recycling residual organic substances using biomethanation as an ecological strategy that minimizes environmental degradation. In fact, energy from biogas is very important especially in regions lacking energy infrastructure, for instance in Democratic Republic of the Congo, because it reduces the use of fossil fuels, limits deforestation and improves people’s livelihoods. In the same regard, natural fertilizers produced from domestic and non hazardous industrial waste are benefic to promote agriculture in order to minimize the use of synthetic fertilizers, which could harm the
![]()
Scheme 9. Synthesis of methane
environment. The reaction mechanisms have been also proposed to better understand the formation of products, which occur during different steps of biomethanation.
Conflicts of Interest
I declare that I do not have conflict of interest regarding the publication of this conference paper.
Cite this paper
Mwene-Mbeja, T.M. and Vaneeckhaute, C. (2019) Conference Paper: Green Industry Adapted to Recycling Needs of Lubumbashi City and Surrounding Areas in Democratic Republic of the Congo. Green and Sustainable Chemistry, 9, 11-25. https://doi.org/10.4236/gsc.2019.91002
References
- 1. Yin, C., Zhang, C. and Gao, M. (2011) Enzyme-Catalyzed Synthesis of Vitamin E Succinate Using a Chemically Modified Novozym-435. Chinese Journal of Chemical Engineering, 19, 135-139. https://doi.org/10.1016/S1004-9541(09)60189-0
- 2. Iemhoff, A., Sherwood, J., McElroy, C.R. and Hunt, A.J. (2018) Towards Sustainable Kinetic Resolution, a Combination of Bio-Catalysis, Flow Chemistry and Bio-Based Solvents. Green Chemistry, 20, 136-140. https://doi.org/10.1039/C7GC03177G
- 3. Birringer, M., EyTina, J.H., Salvatore, B.A. and Neuzil, J. (2003) Vitamin E Analogues as Inducers of Apoptosis: Structure-Function Relation. British Journal of Cancer, 88, 1948-1955. https://doi.org/10.1038/sj.bjc.6600981
- 4. Devine, P.N., Howard, R.M., Kumar, R., Thompson, M.P., Matthew D., Truppo, M.D. and Turner, N.J. (2018) Extending the Application of Biocatalysis to Meet the Challenges of Drug Development. Nature Reviews Chemistry, 2, 409-421. https://doi.org/10.1038/s41570-018-0055-1
- 5. Kogure, K., Hama, S., Kisaki, M., Takemasa, H., Tokumura, A. and Suzuki, I. (2004) Structural Characteristic of Terminal Dicarboxylic Moiety Required for Apoptogenic Activity of Alpha-Tocopheryl Esters. Biochimica et Biophysica Acta,1672, 93-99.
- 6. Neuzil, J. and Massa, H. (2005) Hepatic Processing Determines Dual Activity of α-Tocopheryl Succinate: A Novel Paradigm for a Shift in Biological Activity Due to Pro-Vitamin-to-Vitamin Conversion. Biochemical and Biophysical Research Communications, 327, 1024-1027, https://doi.org/10.1016/j.bbrc.2004.12.115
- 7. Chen, Y.-C. (2017) Evaluation of Greenhouse Gas Emissions from Waste Management Approaches in the Islands. Waste Management & Research, 35, 691. https://doi.org/10.1177/0734242X17707573
- 8. Yang, Y.L, Ramaswamy, S.G. and Jakoby, W.B. (1998) Enzymatic Hydrolysis of Organic Cyclic Carbonates. The Journal of Biological Chemistry, 273, 7814-7817. https://doi.org/10.1074/jbc.273.14.7814
- 9. Rauwerdink, A. and Kazlauskas, R.J. (2015) How the Same Core Catalytic Machinery Catalyzes 17 Different Reactions: The Serine-Histidine-Aspartate Catalytic Triad of α/β-Hydrolase Fold Enzymes. ACS Catalysis, 5, 6153-6176. https://doi.org/10.1021/acscatal.5b01539
- 10. Zhou, Y., Wang, S. and Zhang, Y. (2010) Catalytic Reaction Mechanism of Acetylcholinesterase Determined by Born-Oppenheimer Ab Initio QM/MM Molecular Dynamics Simulations. The Journal of Physical Chemistry B, 114, 8817-8825. https://doi.org/10.1021/jp104258d
- 11. Hedstrom, L. (2002) Serine Protease Mechanism and Specificity. Chemical Reviews, 102, 4501-4524. https://doi.org/10.1021/cr000033x
- 12. Neitzel, J.J. (2010) Enzyme Catalysis: The Serine Proteases. Nature Education, 3, 21.
- 13. Kobayashi, K., Kimura, S., Togawa, E. and Wada, M. (2013) Thermal Expansion Behavior of Hydrate Paramylon in the Low-Temperature Region. Carbohydrate Polymers, 91, 543-548. https://doi.org/10.1016/j.carbpol.2012.08.067
- 14. Karr-Lilienthal, L.K., Kadzere, C.T., Grieshop, C.M. and Fahey Jr., G.C. (2005) Chemical and Nutritional Properties of Soybean Carbohydrates as Related to Nonruminants: A Review. Livestock Production Science, 97, 1-12. https://doi.org/10.1016/j.livprodsci.2005.01.015
- 15. Bode, L. (2009) Human Milk Oligosaccharides: Prebiotics and beyond Lars Bode. Nutrition Reviews, 67, S183-S191. https://doi.org/10.1111/j.1753-4887.2009.00239.x
- 16. Cummings, J.H. and Stephen, A.M. (2007) Carbohydrate Terminology and Classification. European Journal of Clinical Nutrition, 61, S5-S18. https://doi.org/10.1038/sj.ejcn.1602936
- 17. Englyst, K.N., Liu, S. and Englyst, H.N. (2007) Nutritional Characterization and Measurement of Dietary Carbohydrates. European Journal of Clinical Nutrition, 61, S19-S39. https://doi.org/10.1038/sj.ejcn.1602937
- 18. Mata, Y., Diéguez, M., Pàmies, O. and Claver, C. (2005) New Carbohydrate-Based Phosphite-Oxazoline Ligands as Highly Versatile Ligands for Palladium-Catalyzed Allylic Substitution Reactions. Advanced Synthesis & Catalysis, 347, 1943-1947. https://doi.org/10.1002/adsc.200505192
- 19. Goodman, C. (2014) A Family of Starch-Active Polysaccharide Monooxygenases. Proceedings of the National Academy of Sciences, 111, 13822-13827. https://doi.org/10.1073/pnas.1408090111
- 20. Miao, M., Jiang, B., Jin, Z.Y. and BeMiller, J.N. (2018) Microbial Starch-Converting Enzymes: Recent Insights and Perspectives. Comprehensive Reviews in Food Science and Food Safety, 17, 1238-1259. https://doi.org/10.1111/1541-4337.12381
- 21. Hayek, M. and Shriner, R.L. (1944) Hydrolysis of Starch by Sulfurous Acid. Industrial & Engineering Chemistry Research, 36, 1001-1003. https://doi.org/10.1021/ie50419a007
- 22. Oates, C.G. (1997) Towards an Understanding of Starch Granule Structure and Hydrolysis. Trends in Food Science & Technology, 8, 375-382.
- 23. Ring, S.G., Gee, J.M., Whittam, M., Orford, P. and Johnson, I.T. (1988) Resistant Starch: Its Chemical Form in Foodstuffs and Effect on Digestibility in Vitro. Food Chemistry, 28, 97-109. https://doi.org/10.1016/0308-8146(88)90139-2
- 24. Azevedo, H.S., Gama, F.M. and Reis, R.L. (2003) In Vitro Assessment of the Enzymatic Degradation of Several Starch Based Biomaterials. Biomacromolecules, 4, 1703-1712. https://doi.org/10.1021/bm0300397
- 25. Nielsen, J.E., Beier, L., Otzen, D., Borchert, T.V., Frantzen, H.B., Andersen, K.V. and Svendsen, A. (1999) Electrostatics in the Active Site of an α-Amylase. European Journal of Biochemistry, 264, 816-824.
- 26. Briggs, A.J., Evans, C.M., Glenn, R. and Kirby, A.J.J. (1983) Stereoelectronic Effects at Oxygen. A Very Large Effect on the Hydrolysis of a Conformationally Locked Acetal: Implications for β-Glycosidase Mechanisms. Journal of the Chemical Society. Perkin Transactions 2, 1637-1640. https://doi.org/10.1039/P29830001637
- 27. Spano, L.A., Medeiros, J. and Mandels, M. (1976) Enzymatic Hydrolysis of Cellulosic Wastes to Glucose. Resource Recovery and Conservation, 1, 279-294. https://doi.org/10.1016/0304-3967(76)90039-1
- 28. Zhu, J.Y. and Pan. X.J. (2010) Woody Biomass Pretreatment for Cellulosic Ethanol Production: Technology and Energy Consumption Evaluation. Bioresource Technology, 101, 4992-5002. https://doi.org/10.1016/j.biortech.2009.11.007
- 29. Assam, A.M.J. (1989) Pretreatment of Cane Bagasse with Alkaline Hydrogen Peroxide for Enzymatic Hydrolysis of Cellulose and Ethanol Fermentation. Journal of Environmental Science and Health, 24, 421-433. https://doi.org/10.1080/03601238909372658
- 30. Bose, S., Armstrong, D.W. and Petrich, J.W.J. (2010) Enzyme-Catalyzed Hydrolysis of Cellulose in Ionic Liquids: A Green Approach toward the Production of Biofuels. The Journal of Physical Chemistry B, 114, 8221-8227. https://doi.org/10.1021/jp9120518
- 31. Saharay, M., Guo, H. and Smith, J.C. (2010) Catalytic Mechanism of Cellulose Degradation by a Cellobiohydrolase, CelS. PLoS ONE, 5, e12947. https://doi.org/10.1371/journal.pone.0012947
- 32. Deslongchamps, P. and Moreau, C. (1971) Ozonolysis of Acetals. (1) Ester Synthesis, (2) THP Ether Cleavage, (3) Selective Oxidation of β-Glycoside, (4) Oxidative Removal of Benzylidene and Ethylidene Protecting Groups. Canadian Journal of Chemistry, 49, 2465-2467. https://doi.org/10.1139/v71-405
- 33. Deslongchamps, P., Moreau, C., Fréhel, D. and Atlani, P. (1972) The Importance of Conformation in the Ozonolysis of Acetals. Canadian Journal of Chemistry, 50, 3402-3404. https://doi.org/10.1139/v72-548
- 34. Deslongchamps, P. (1993) Intramolecular Strategies and Stereoelectronic Effects. Glycosides Hydrolysis Revisited. Pure and Applied Chemistry, 65, 1161-1178. https://doi.org/10.1351/pac199365061161
- 35. Reddy, A. and Maley, F.J. (1996) Studies on Identifying the Catalytic Role of Glu-204 in the Active Site of Yeast Invertase. Biological Chemistry, 271, 13953-13958. https://doi.org/10.1351/pac199365061161
- 36. Davidson, J.M., Linforth, R.S.T., Hollowood, T.A. and Taylor, A.J.J. (1999) Effect of Sucrose on the Perceived Flavor Intensity of Chewing Gum. Journal of Agricultural and Food Chemistry, 47, 4336-4340. https://doi.org/10.1021/jf9901082
- 37. Farine, S., Versluis, C., Bonnici, P.J., Heck, A., Peschet, J.L., Puigserver, A. and Biagini, A. (2001) Separation and Identification of Enzymatic Sucrose Hydrolysis Products by High-Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection. Journal of Chromatography A, 920, 299-308. https://doi.org/10.1016/S0021-9673(01)00612-4
- 38. Klostergaard, H.J. (1976) Inversion of Sucrose and Fructose Structure. Journal of Chemical Education, 53, 298. https://doi.org/10.1021/ed053p298
- 39. Dordick, R.S. and Clarke, G.A.J. (1979) Salt Effects on the Hydrolysis of Sucrose. Journal of Chemical Education, 56, 352. https://doi.org/10.1021/ed056p352
- 40. Toufeili, I.A., Dziedzic, S.Z. and Rathbone, E.B. (1986) C-Methylation of Sucrose: Synthesis of 6- and 6’-C-Methylsucrose. Carbohydrate Research, 148, 279-285. https://doi.org/10.1016/S0008-6215(00)90395-1
- 41. Kehlbeck, J.D., Slack, C.C., Turnbull, M.T. and Kohler, S.J.J. (2014) Exploring the Hydrolysis of Sucrose by Invertase Using Nuclear Magnetic Resonance Spectroscopy: A Flexible Package of Kinetic Experiments. Journal of Chemical Education, 91, 734-738. https://doi.org/10.1021/ed300889s
- 42. Heinzerling, P., Schrader, F. and Schanze, S.J. (2012) Measurement of Enzyme Kinetics by Use of a Blood Glucometer: Hydrolysis of Sucrose and Lactose. Journal of Chemical Education, 89, 1582-1586. https://doi.org/10.1021/ed200735f
- 43. Mendes, A.A. and De Castro, H.F. (2005) Effect on the Enzymatic Hydrolysis of Lipids from Dairy Wastewater by Replacing Gum Arabic Emulsifier for Sodium Chloride. Brazilian Archives of Biology and Technology, 48, 135.
- 44. Mala, J.G.S. and Takeuchi, S. (2008) Understanding Structural Features of Microbial Lipases—An Overview. Analytical Chemistry Insights, 3, 9.
- 45. Krishna, S.H. and Karanth, N.G. (2002) Lipases and Lipase-Catalyzed Esterification Reactions in Nonaqueous Media. Catalysis Reviews—Science and Engineering, 44, 499-591. https://doi.org/10.1081/CR-120015481
- 46. Duan, G., Ching, C.B., Lim, E. and Ang, C.H. (1997) Kinetic Study of Enantioselective Esterification of Ketoprofen with n-Propanol Catalysed by an Lipase in an Organic Medium. Biotechnology Letters, 19, 1051-1055. https://doi.org/10.1023/A:1018420022398
- 47. Tundo, P., Selva, M. and Bomben, A. (2004) Org. Synth. Coll., 10, 640.
- 48. Philippidis, G.P., Smith, T.K. and Wyman, C.E. (1993) Study of the Enzymatic Hydrolysis of Cellulose for Production of Fuel Ethanol by the Simultaneous Saccharification and Fermentation Process. Biotechnology and Bioengineering, 41, 846-853. https://doi.org/10.1002/bit.260410903
- 49. Mustafa, M.Y., Calay, R.K. and Román, E. (2016) Biogas from Organic Waste—A Case Study. Procedia Engineering, 146, 310-317. https://doi.org/10.1016/j.proeng.2016.06.397
- 50. Zhang, R., Pan, Z. and Zheng, Y. (2009) International Journal of Agricultural and Biological Engineering, 2, 51.
- 51. Parmar, I. and Rupasinghe, H.P.V. (2013) Bio-Conversion of Apple Pomace into Ethanol and Acetic Acid: Enzymatic Hydrolysis and Fermentation. Bioresource Technology, 130, 613-620. https://doi.org/10.1016/j.biortech.2012.12.084
- 52. Muzio, G., Maggiora, M., Paiuzzi, E., Oraldi, M. and Canuto, R.A. (2012) Aldehyde Dehydrogenases and Cell Proliferation. Free Radical Biology & Medicine, 52, 735-746. https://doi.org/10.1016/j.freeradbiomed.2011.11.033
- 53. Liu, Z.-J., Sun, Y.-J., Rose, J., Chung, Y.-J., Hsiao, C.-D., Chang, W.-R., Kuo, I., Perozich, J., Lindahl, R., Hempel, J. and Wang, B.-C. (1997) The First Structure of an Aldehyde Dehydrogenase Reveals Novel Interactions between NAD and the Rossmann Fold. Nature Structural & Molecular Biology, 4, 317-326.
- 54. Marchitti, S.A., Brocker, C., Stagos, D. and Vasiliou, V. (2008) Non-P450 Aldehyde Oxidizing Enzymes: The Aldehyde Dehydrogenase Superfamily. Expert Opinion on Drug Metabolism & Toxicology, 4, 697-720. https://doi.org/10.1517/17425255.4.6.697
- 55. Bélafi-Bako, K., Kabiri Badr, A., Nemestothy, N., Ehrenstein, U. and Gubicza, L. (2003) Kinetics of Ethyl Acetate Formation by Lipase in Organic Solvent and Solvent-Free System. Chemical Papers, 57, 278-281.
- 56. Hwang, S.O. and Park, Y.H. (1994) Ethyl Acetate Production in the Gas Phase. Biotechnology Letters, 16, 379-384.
- 57. Morval, L., Mihaltz, P. and Hollo, J. (1992) Short Chain Flavour Esters Synthesis by Microbial Lipases. Applied Microbiology and Biotechnology, 36, 581-586.
- 58. Richard, J.P. (2011) Enzymatic Catalysis of Proton Transfer and Decarboxylation Reactions. Pure and Applied Chemistry, 83, 1555. https://doi.org/10.1351/PAC-CON-11-02-05
- 59. Hollenstein, M. (2013) Deoxynucleoside Triphosphates Bearing Histamine, Carboxylic Acid, and Hydroxyl Residues—Synthesis and Biochemical Characterization. Organic & Biomolecular Chemistry, 11, 5162-5172. https://doi.org/10.1039/c3ob40842f
- 60. Lee, Y.-S. and Cho, Y.-D. (2001) Enzymatic Catalysis of Proton Transfer and Decarboxylation Reactions. Biochemical Journal, 360, 657-665. https://doi.org/10.1042/bj3600657