Green and Sustainable Chemistry
Vol.2 No.3(2012), Article ID:21740,15 pages DOI:10.4236/gsc.2012.23015
Ultrasonically Assisted Regioselective Nitration of Aromatic Compounds in Presence of Certain Group V and VI Metal Salts
Department of Chemistry, Osmania University, Hyderabad, India
Email: *kcrajannaou@yahoo.com
Received May 25, 2012; revised June 30, 2012; accepted July 19, 2012
Keywords: Ultrasonically Assisted; Regioselective Nitration; Group V and VI Metal Salts; Reduction in Reaction Times
ABSTRACT
Ultrasonically assisted nitration reactions (USANR) with anilides, moderately activated and non-activated aromatic compounds underwent smoothly and afforded good yields of products with high regio selectivity. Observed longer reaction times (6 - 8 hrs.) in metal catalyzed reactions reduced to (1 - 2 hrs.) under sonication. When ortho position is blocked para derivatives are obtained, and ortho nitro products are obtained when para position is blocked. In case of USANR of aromatic carbonyl and related compounds the effect of sonication is much more effective. The reactions could be completed only in few minutes.
1. Introduction
Aromatic compounds such as benzene afford a variety of derivatives when they are allowed to react with other compounds under different conditions. The π-Electron cloud, located above and below the plane of benzene ring is more involved in holding carbon nuclei together through resonance than the π-electrons of a carbon-carbon double bond in an aliphatic unsaturated hydrocarbon. Yet, these π-electrons are much more loosely held in comparison with π-electrons and are easily available to a reagent that is seeking electrons. In its typical reactions benzene ring serves as a source of electrons. Benzene reacts with electron deficient molecules or species (electrophiles) in substitution reactions to afford benzene derivatives. A reaction is referred to as SE when a substitution reaction involves the attack by electrophiles [1-3].
Electrophilic nitration reactions are carried out by acid catalyzed reactions of HNO3 and its derivatives. The nitronium ion (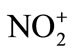 ) is established as the reactive nitrating species by Ingold in 1940’s following an early suggestion by Euler. Nitrating agents possess general formula NO2-X which serve as sources of the
) is established as the reactive nitrating species by Ingold in 1940’s following an early suggestion by Euler. Nitrating agents possess general formula NO2-X which serve as sources of the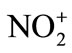 , nitronium ion. The ease of X-elimination from NO2-X, Euler derived a relative sequence of nitrating activity of certain nitrating agents: nitroniumion (
, nitronium ion. The ease of X-elimination from NO2-X, Euler derived a relative sequence of nitrating activity of certain nitrating agents: nitroniumion (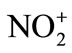 ) > nitracidiumion (
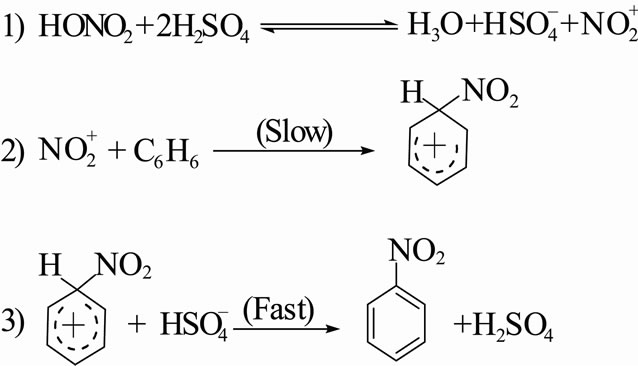
) > nitracidiumion (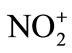 -OH2) > nitryl chloride (NO2-Cl) > nitrogen pentoxide (N2O5) > methyl nitrate (NO2-O-CH3). However, now the range of nitrating agents is much wider. The reactivity of the nitrating agent is also affected by the solvent used. A decreasing activity is observed in the series of solvents of increasing nucleophilicity: Sulfuric acid > nitric acid > nitro methane (NO2CH3) > acetic acid > 1,4-dioxane > water. Nitration reaction proceeds through the formation of
-OH2) > nitryl chloride (NO2-Cl) > nitrogen pentoxide (N2O5) > methyl nitrate (NO2-O-CH3). However, now the range of nitrating agents is much wider. The reactivity of the nitrating agent is also affected by the solvent used. A decreasing activity is observed in the series of solvents of increasing nucleophilicity: Sulfuric acid > nitric acid > nitro methane (NO2CH3) > acetic acid > 1,4-dioxane > water. Nitration reaction proceeds through the formation of 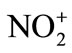 electrophile in the first step, followed by the formation of nitro derivative of carbocation in the second step. The carbocation thus formed supplements a proton to bisulphite ion to give H2SO4 by affording nitro benzene.
electrophile in the first step, followed by the formation of nitro derivative of carbocation in the second step. The carbocation thus formed supplements a proton to bisulphite ion to give H2SO4 by affording nitro benzene.
This reaction is simply an acid-base equilibrium in which sulfuric acid serves as the acid and the much weaker nitric acid serves as a base. The very strong acid, sulfuric acid, causes nitric acid to ionize in the sense HO-…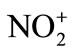 , rather than in the usual way of H+… -ONO2. The nitronium ion is well known, existing in salts such as nitronium perchlorate [
, rather than in the usual way of H+… -ONO2. The nitronium ion is well known, existing in salts such as nitronium perchlorate [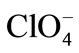
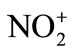 ], and nitronium fluoroborate [
], and nitronium fluoroborate [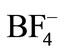
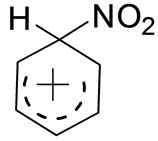
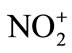 ]. Needing electrons, the nitronium ion finds them particularly available in the π-electron cloud of the Benzene ring and so in step [3-8], attaches itself to one of the carbon atoms by a covalent bond. This forms the carbocation; it’s a resonance hybrid of the three structures, represented as:
]. Needing electrons, the nitronium ion finds them particularly available in the π-electron cloud of the Benzene ring and so in step [3-8], attaches itself to one of the carbon atoms by a covalent bond. This forms the carbocation; it’s a resonance hybrid of the three structures, represented as:
Apart from therapeutic and diagnostic applications, i.e., medical ultra sonography and teeth cleaning; ultrasound also has many applications in chemical and biochemical sciences. In the recent past the study of ultrasonically assisted reactions has been the subject of interest to synthetic and physical organic chemists probably due to a dramatic reduction in the reaction times followed by astonishingly high increase in the yields of reaction products [9,10]. The first example in which ultrasound induced a divergent pathway relative to thermal conditions was reported by Ando and co-workers more than 30 years ago [9-13]. Ever since the concept of chemical ultrasonics is introduced in 1927, a number of chemical reactions have been observed to occur under sonicated conditions [11-14]. One of the most fascinating areas of organic sonochemistry is sonochemical switching. In some cases, application of ultrasound may completely change the distribution of products or even cause the formation of different substances. Sonochemistry shares some aims with green chemistry, as it also uses smaller quantities of non-hazardous chemicals and solvents, reduces energy consumption, and increases product selectivity. The use of ultrasound in chemical reactions provides specific activation based on a physical phenomenon, known as acoustic cavitation. Recently, ultrasound has been utilized to accelerate a wide number of synthetically useful organic reactions [15-23]. In a recent review [20], David Bremner excellently elaborated some of the more modern uses of ultrasound in organic synthesis in different sections such as oxidations, carbon-nitrogen bond formation, carbon-carbon bond formation, cycloadditions, bond cleavage reactions, biological systems and sonochemical switching.
Since more than one decade our group is also actively working on exploiting the use of a variety of eco-friendly materials such as metal ions and surfactants as catalysts and non-conventional energy sources (such as microwave and ultra sound) to assist various organic transformations [24,25]. These reactions generally did not proceed at room temperature and is too sluggish even at elevated temperatures. However, under sonicated conditions these reactions exhibited astonishingly high yields of the products and dramatic rate enhancements. The reaction times were reduced from several hours (in thermal reactions) to few minutes in ultrasonic irradiated reactions (the reaction flask was clamped in an ultrasonic bath of 2 MHz frequency at 40˚C for the purpose of irradiations). Encouraged by these results we embarked on the use ultrasonics as a non-conventional energy source to assist nitration reactions of the present work.
2. Experimental Details
2.1. General Procedure
To a solution of 4-Hydroxy benzaldehyde 1b (0.099 ml, 1 mmol) in dichloromethane (DCM) or dichloro ethane (DCE) ammonium molybdate (1.235 gms, 1 mmol), and 70% HNO3 (0.063 ml, 1 mmol) were added and the contents refluxed for 1hr. Progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was filtered and washed with water. The organic layer was separated and dried over Na2SO4 and evaporated under vacuum. The crude product was purified by column chromatography using ethyl ace-tatehexane (3:7) as eluent to get pure product 4-Hydroxy- 3-nitro benzaldehyde as Yellow powder (mp 138˚C - 140˚C) in 85% yield. The nitrated products were characterized by comparing their spectral (IR, 1H-NMR, mass, TLC) and physical data with authentic samples [26-31]. Certain representative spectra are given as supplementary data.
2.2. Spectroscopic Data
4-Nitrophenol: Yellow powder; mp 112˚C - 114˚C (lit. [26] 114˚C); IR (KBr) (νmax/cm–1): 3331, 1614, 1592, 1500, 1346; 1H NMR (250 MHz, CDCl3): δ 8.18 (d, 2H, J = 5.50 Hz, Ar-H), 7.00 (d, 2H, J = 5.49 Hz, Ar-H), 6.35 (s, 1H, -OH); GC-Ms/EI: m/z (%) = 139 (M+, 14), 109 (50), 81 (33), 65 (84), 53 (31), 46 (22), 39 (100), 30 (77), 28 (41).
2-Nitrophenol: Yellow powder; mp 42˚C - 44˚C (lit. [26] 45˚C); IR (KBr) (νmax/cm–1): 3241, 1623, 1606, 1593, 1538, 1479; 1H NMR (250 MHz, CDCl3): δ 10.5 (s, 1H, -OH), 8.10 (d, 1H, J = 8.50 Hz, Ar-H), 7.59 (m, 1H, Ar-H), 7.16 (d, 1H, J = 8.4 Hz, Ar-H), 7.00 (m, 1H, Ar-H); GC-Ms/ EI: m/z (%) = 139 (M+, 40), 109 (35), 81 (55), 65 (60), 53 (38), 39 (100), 30 (66).
4-Chloro-2-nitrophenol: Yellow powder; mp 84˚C - 86˚C (lit [27] 85˚C - 89˚C); IR (KBr) (νmax/cm–1): 3490, 1631, 1531, 1469, 1384, 1346, 1238; 1H NMR (250 MHz, DMSO): 11.33 (brs, 1H, -OH), 7.90 (s, 1H, Ar-H), 7.54 (d, 1H, J = 8.9 Hz), 7.14 (d, 1H, J = 8.9 Hz, Ar-H); GC-Ms/ EI: m/z (%) = 173 (M+, 16), 156 (7), 143 (26), 128 (10), 115 (41), 99 (39), 73 (29), 63 (100), 51 (20), 37 (13), 30 (26).
4-Chloro-3-nitrobenzaldehyde: Yellow powder; mp 140˚C - 147˚C (lit. [31] 148˚C - 150˚C); IR (KBr) (νmax/ cm–1): 3101, 2873, 2715, 2596, 1701, 1593, 1431, 1330; 1H NMR (250 MHz, DMSO): δ 10.1 (s, 1H, -CHO), 8.22 (s, 1H, Ar-H), 7.90 (d, 1H, J = 3.1 Hz, Ar-H), 7.55 (d, 1H, J = 3.1 Hz, Ar-H); GC-Ms/ EI: m/z (%) = 185 (M+, 9), 167 (10), 149 (22), 129 (15), 97 (19), 73 (45), 57 (100).
2,4-Dinitrophenol: Yellow powder; mp 107˚C - 110˚C (lit. [31] 108˚C - 112˚C); IR (KBr) (νmax/cm–1): 3587, 3529, 1608, 1569, 1531, 1523, 1438, 1357, 1280; 1H NMR (250 MHz, DMSO): δ 11.02 (brs, 1H, -OH), 8.65 (s, 1H, Ar-H), 8.26 (d, 1H, J = 9.3 Hz), 7.22 (d, 1H, J = 9.2 Hz, Ar-H); GC-Ms/ EI: m/z (%) = 184 (M+, 4), 167 (10), 129 (10), 111 (12), 85 (22), 69 (100).
4-Hydroxy-3-nitrobenzaldehyde: Yellow powder; mp 138˚C - 140˚C (lit [31] 140˚C - 141˚C); IR (KBr) (νmax/cm–1): 3236, 2816, 1693, 1620, 1562, 1423, 1334, 1269, 1126; 1H NMR (250 MHz, CDCl3): δ 11.01 (brs, 1H, -OH), 9.94 (s, 1H, -CHO), 8.63 (s, 1H, Ar-H), 8.14 (d, 1H, J = 3.5 Hz), 7.33 (d, 1H, J = 3.5 Hz, Ar-H); GC-Ms/ EI: m/z (%) = 167 (M+, 10), 149 (25), 129 (11), 111 (11), 85 (23), 69 (100).
2-Hydroxy-5-nitrobenzaldehyde: Yellow powder; mp 127˚C - 129˚C (lit. [27] 129˚C - 130˚C); IR (KBr) (νmax/ cm–1): 3460, 3074, 2935, 1666, 1620, 1477, 1334, 1323; 1H NMR (250 MHz, DMSO): δ 11.5 (brs, 1H, -OH), 9.91 (s, 1H, -CHO), 8.49 (s, 1H, Ar-H), 8.32 (d, 1H, J = 9.1 Hz, Ar-H), 7.05 (d, 1H, J = 9.1 Hz, Ar-H); GC-Ms/EI: m/z (%) = 167 (M+, 11), 149 (13), 129 (7), 111 (12), 85 (24), 69 (100).
4-Nitroaniline: Yellow powder; mp (146˚C - 148˚C (lit. [27] 146˚C - 149˚C); IR (KBr) (νmax/cm–1): 3475, 3363, 1601, 1530, 1348; 1H NMR (250 MHz, CDCl3): δ 7.94 (d, 2H, J = 5.50 Hz, Ar-H), 6.76 (d, 2H, J = 5.49 Hz, Ar-H), 4.25 (brs, 2H, -NH2); GC-Ms/ EI: m/z (%) = 138 (M+, 45).
3-Hydroxy-4-nitro Acetophenone: δ 10.68 (s 1H, OH) 2.38 (s 3H, COMe) 7.62 (d, 1H, J = 8.5Hz), 8.10 (m 1H, J = 8.5 Hz, J = 3.5Hz) 7.52 (d 1H, J = 3.5Hz); m/z = 181.
4-Amino-2-nitro Phenol: δ 4.45 (s 2H, NH2) 9.92 (s 1H, OH) 6.76 (d, 1H, J = 8Hz) 6.82 (d 1H, J = 8Hz), 7.24 (s 1H); m/z = 154.
3. Results & Discussion
3.1. Nitration Reactions under Conventional (Thermal) Conditions
Nitration of non-active and moderately active aromatic compounds were catalyzed by Bismuth nitrate (BN), sodium Bismuthate (SB) and Group VI B metal salts such as potassium chromate (PCR), ammonium molybdate (AMB) and sodium tungstate (STG). To substantiate the generality, the reaction is conducted with an array of non-activated, moderately activated and activated organic compounds. General reaction conditions are shown in Scheme 1.
The data presented in Tables 1-5 show that the reactions with aromatic compounds such as phenols (entries 1 to 4) anilides (entries 7 to 14) underwent smoothly with
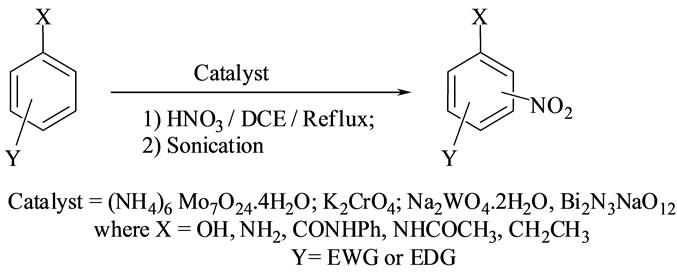
Scheme 1. Nitration of aromatic compounds.
moderate to long reaction times (6 to 8 hours) and afforded good yields of products with good regioselectivity. It is of interest to note that para substituted compounds (entries 2 to 4, 8, 9, 11 to 14 in Tables 1-5) underwent nitration only at ortho position and afforded excellent yield of products, while ortho substituted compound (entry 7) gave very good yield of para derivative. Under similar conditions aniline, chloro benzene, toluene and ethyl benzene (entries 5, 19, 20, 21) have undergone smooth nitration with high regioselectivity.
Even though, benzene failed to undergo nitration; it is also of interest to note that aromatic hydrocarbons such as anthracene, naphthalene, α-naphthol, β-naphthol, 2-amino anthracene and 1-naphthalde-hyde underwent smooth nitration with quantitative yields (80% to 90%). When the reaction protocol is extended to activated-aromatic compounds such as carbonyl compounds, interestingly the reaction times are drastically reduced from several hours to about one hour. Related data are given in Tables 6- 10.
Nitration of aromatic carbonyl and related compounds underwent rapidly affording high yields of the corresponding mono nitro derivatives with high regioselectivity. The reactions were clean, no attack being observed on the alkyl portion of the ketones. The present nitration produces a remarkably high yield of para nitro compounds (entries 1a, 1c, 1g, 1h, 1j, 1k, 1l, 1m, 1o, 1p & 1q). To check the regioselectivity of the reaction, the reaction is carried out with different para substituted aromatic carbonyl and related compounds (entries 1b, 1d, 1e, 1f, 1i, 1r) which afforded only meta nitrated product in good yield. General reaction conditions are given in Scheme 2. In the absence of metal salts, the nitration did not proceed. The results of the present work are summarized in Tables 6-10.
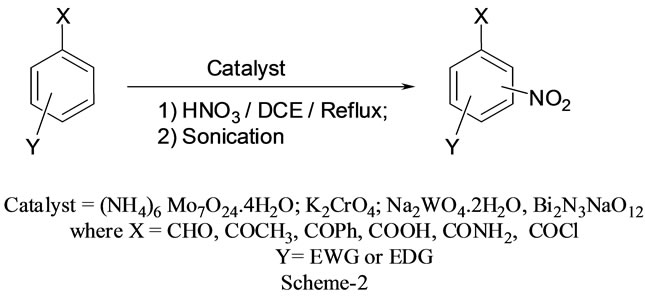
Scheme 2. Nitration of aromatic aldehydes.
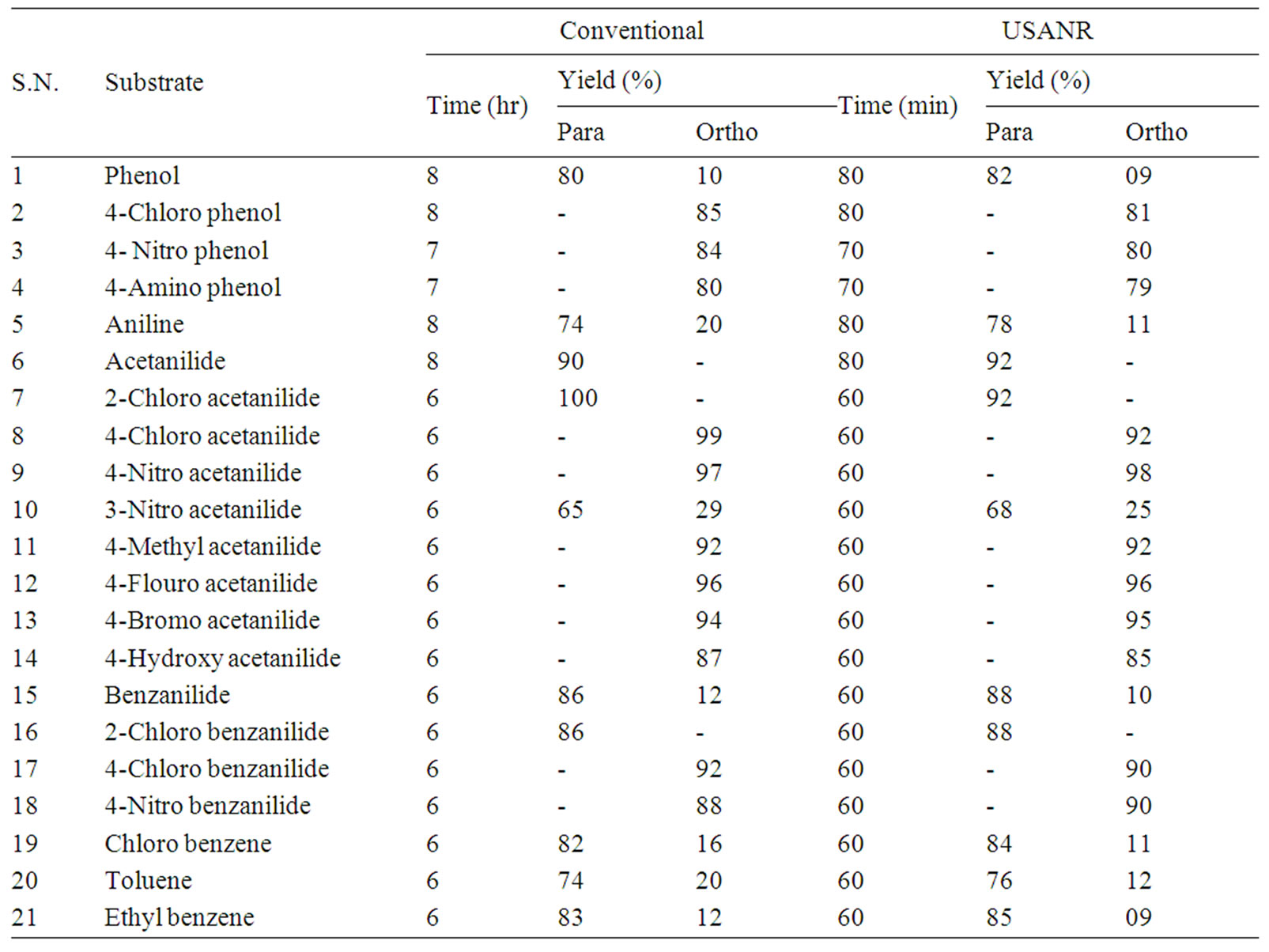
Table 1. Ammonium molybdate as catalyst for regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
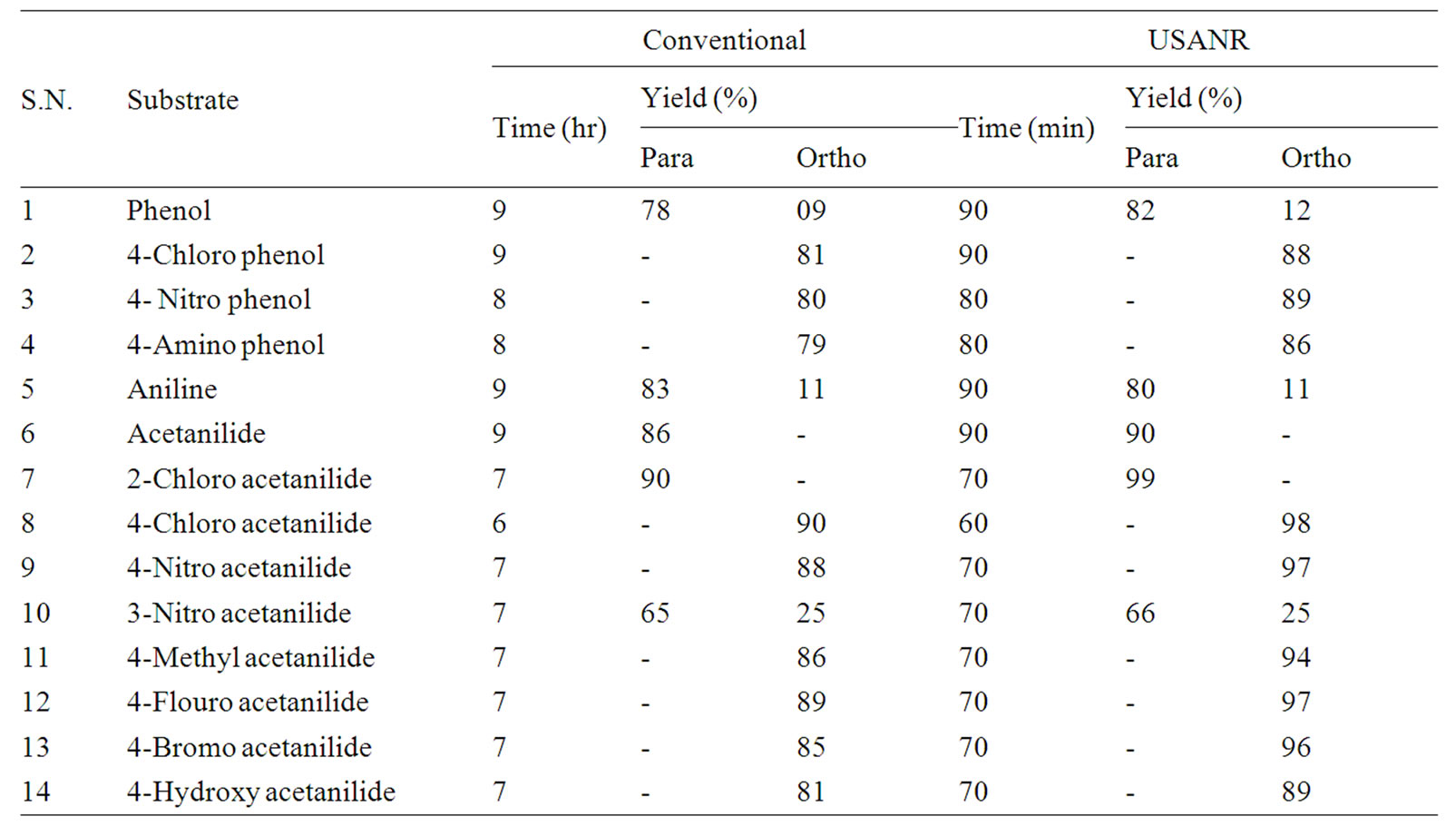
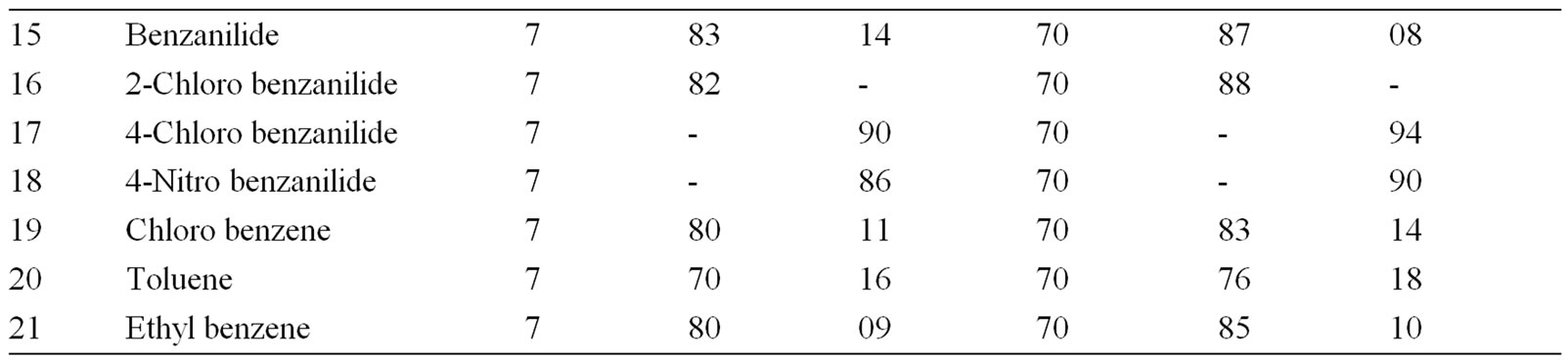
Table 2. Ultrasonically assisted potassium chromate catalyzed regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
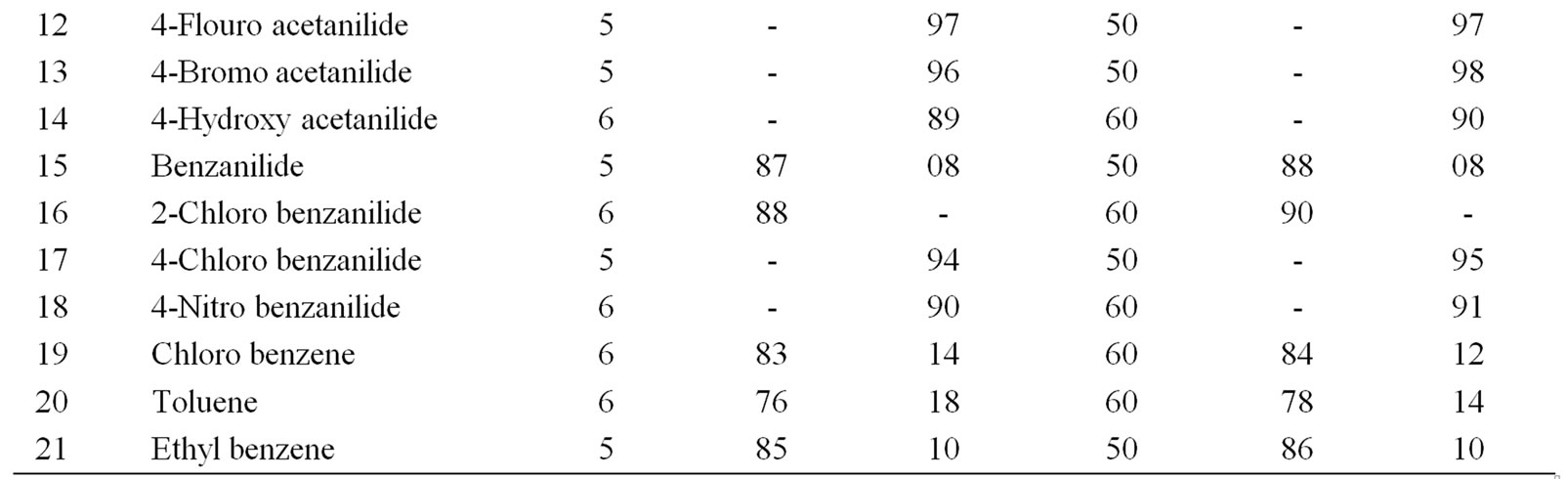
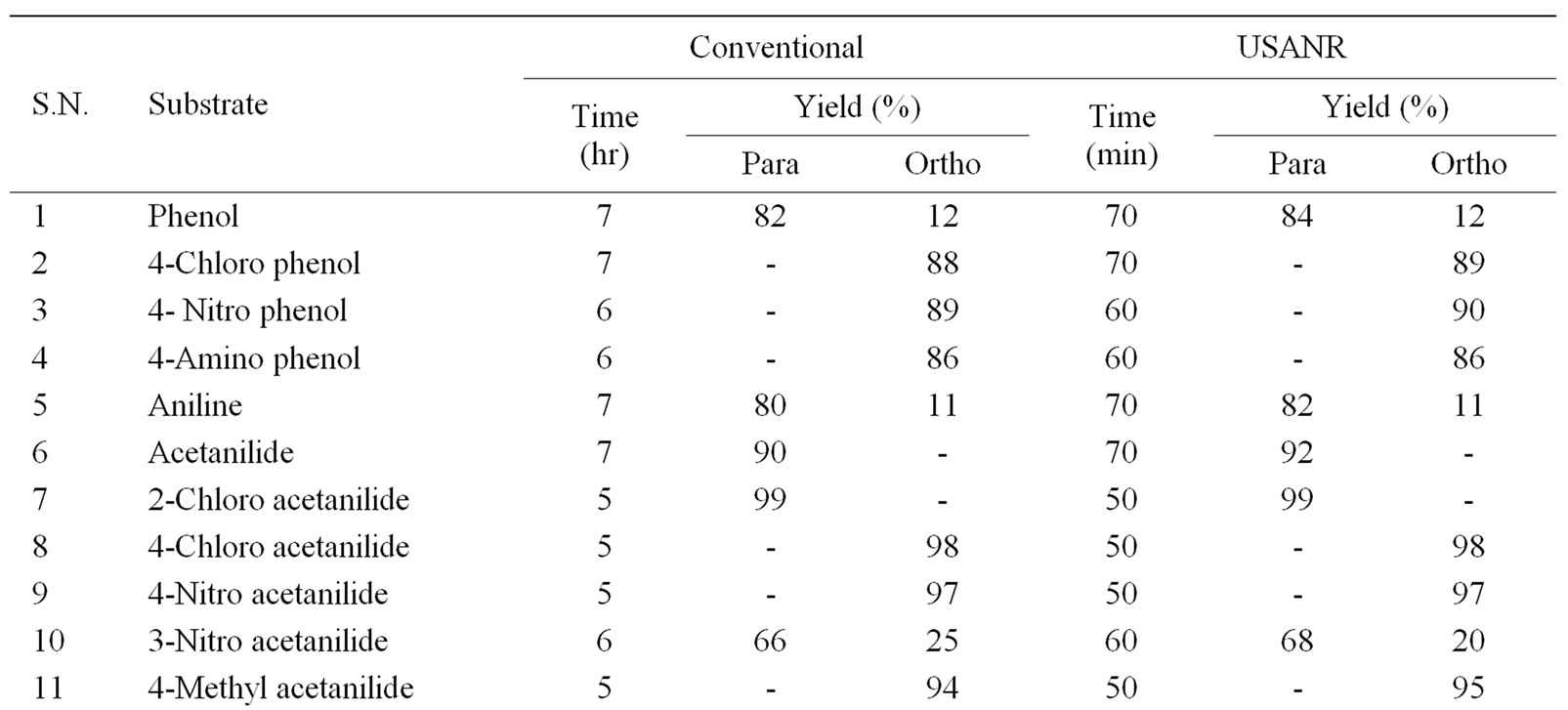
Table 3. Ultrasonically assisted sodium tungstate catalyzed regio selective nitration of anilides, non-activated and moderately activated organic compounds under mild acid conditions.
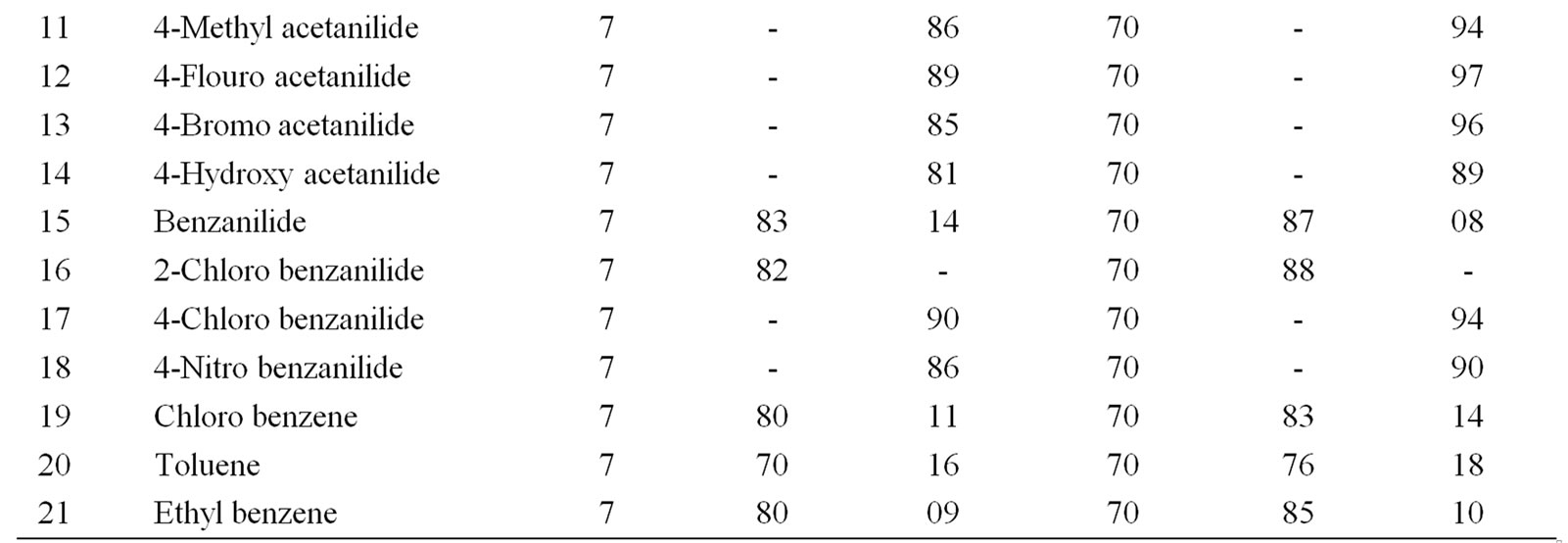
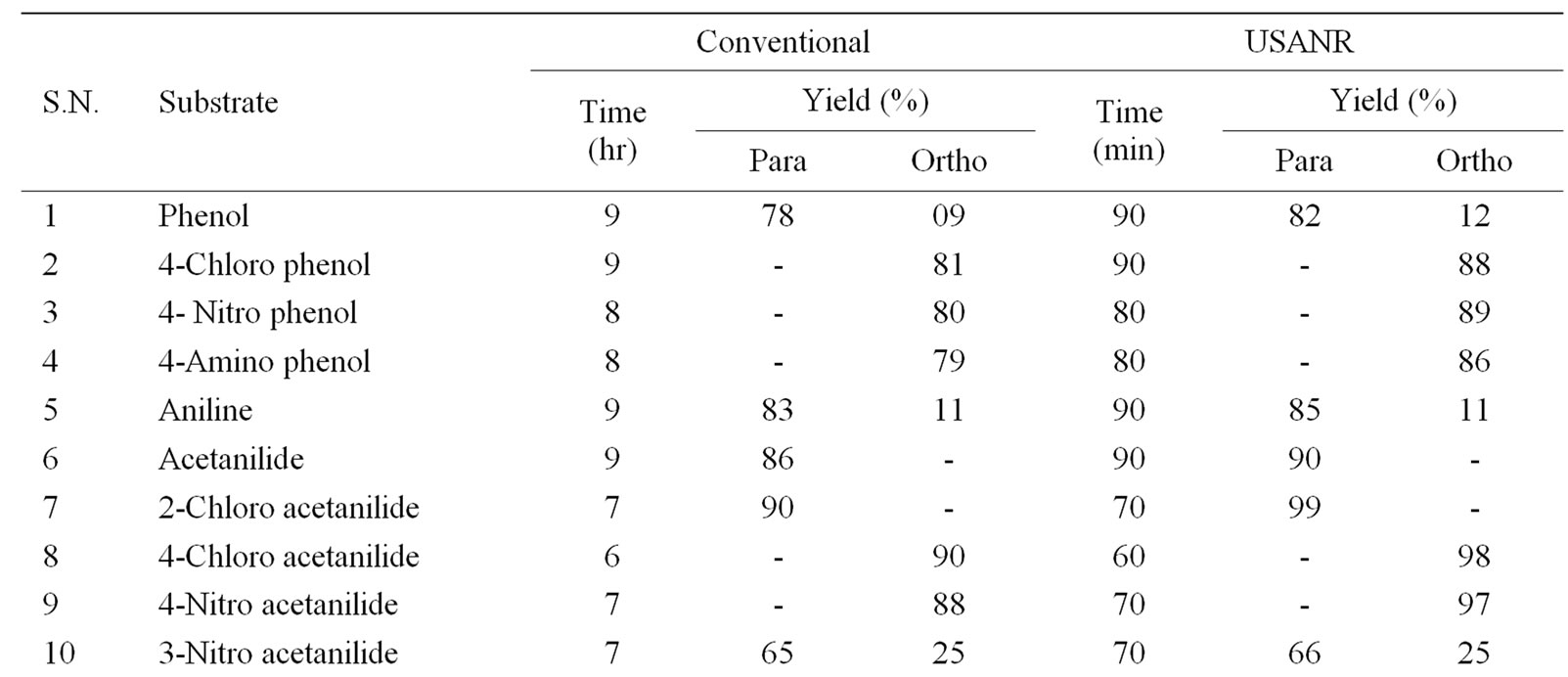
Table 4. Ultrasonically assisted bismuth nitrate catalyzed nitration of non-active and moderately active aromatic organic compounds under mild acid conditions.
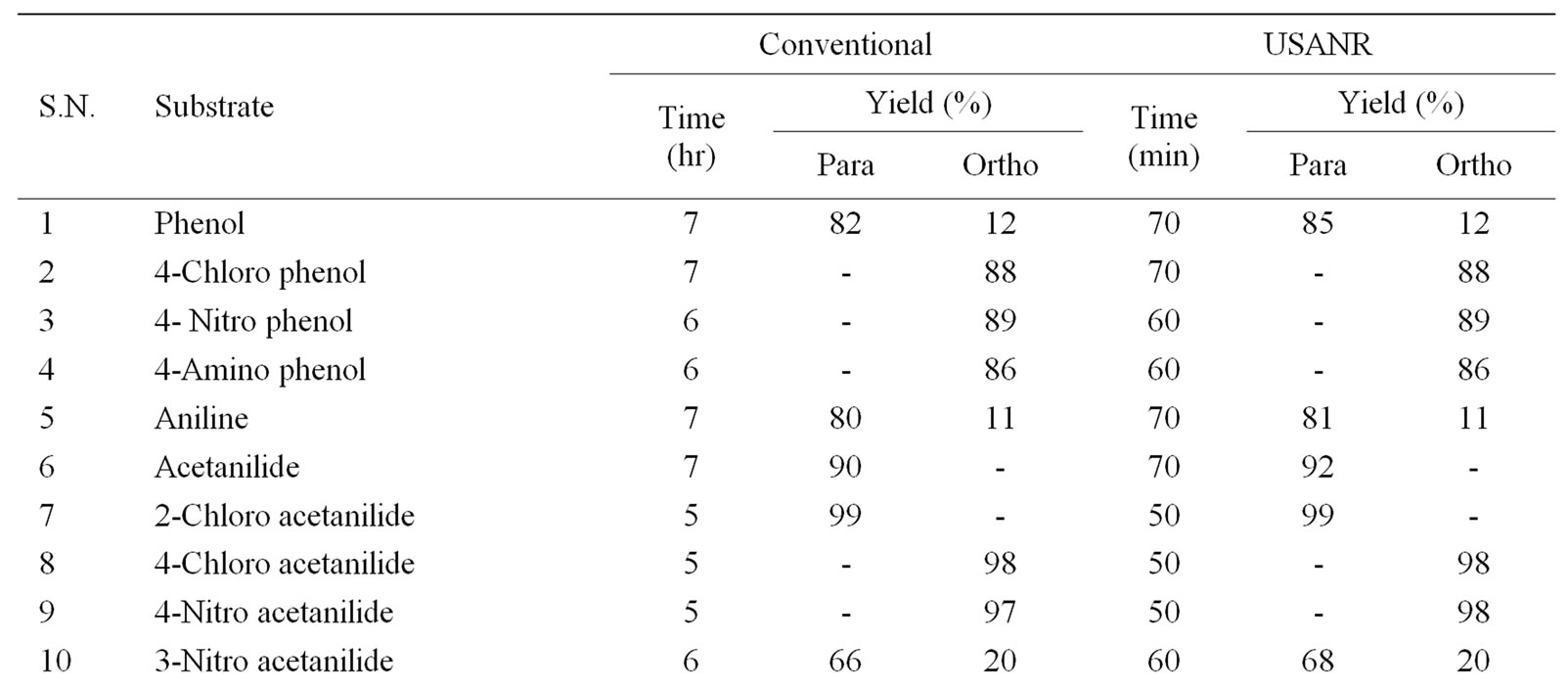
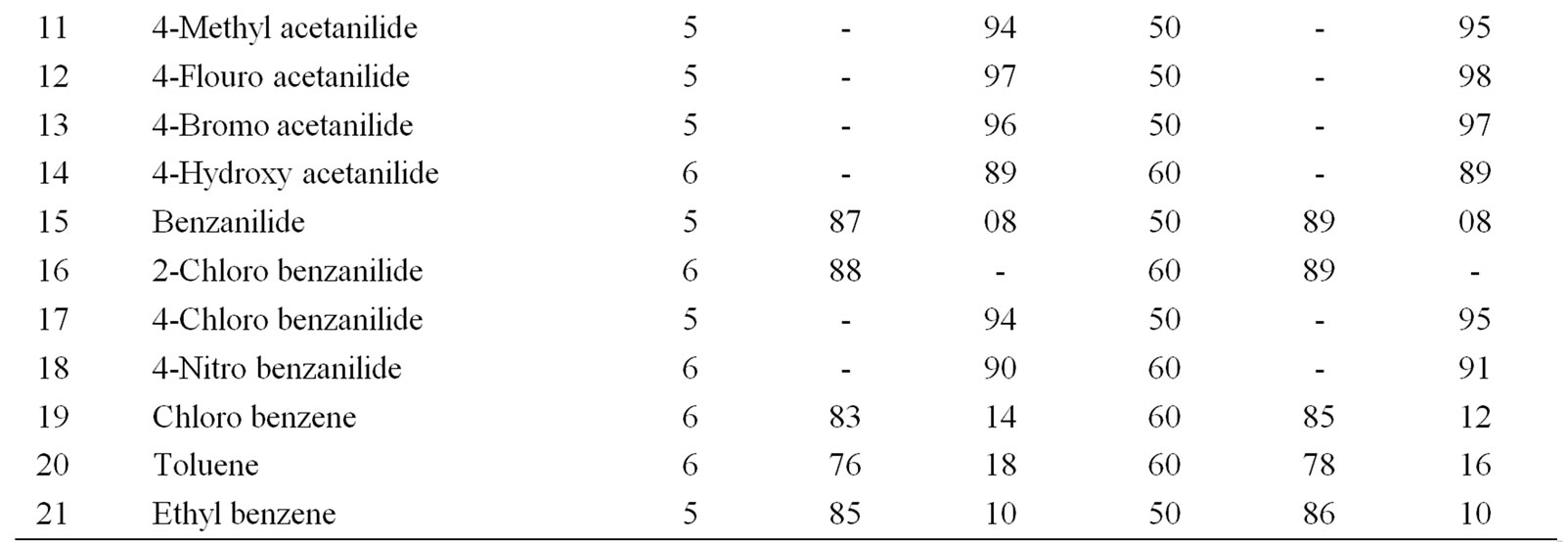
Table 5. Ultrasonically assisted sodium bismuthate catalyzed nitration of non-active and moderately active aromatic organic compounds under mild acid conditions.
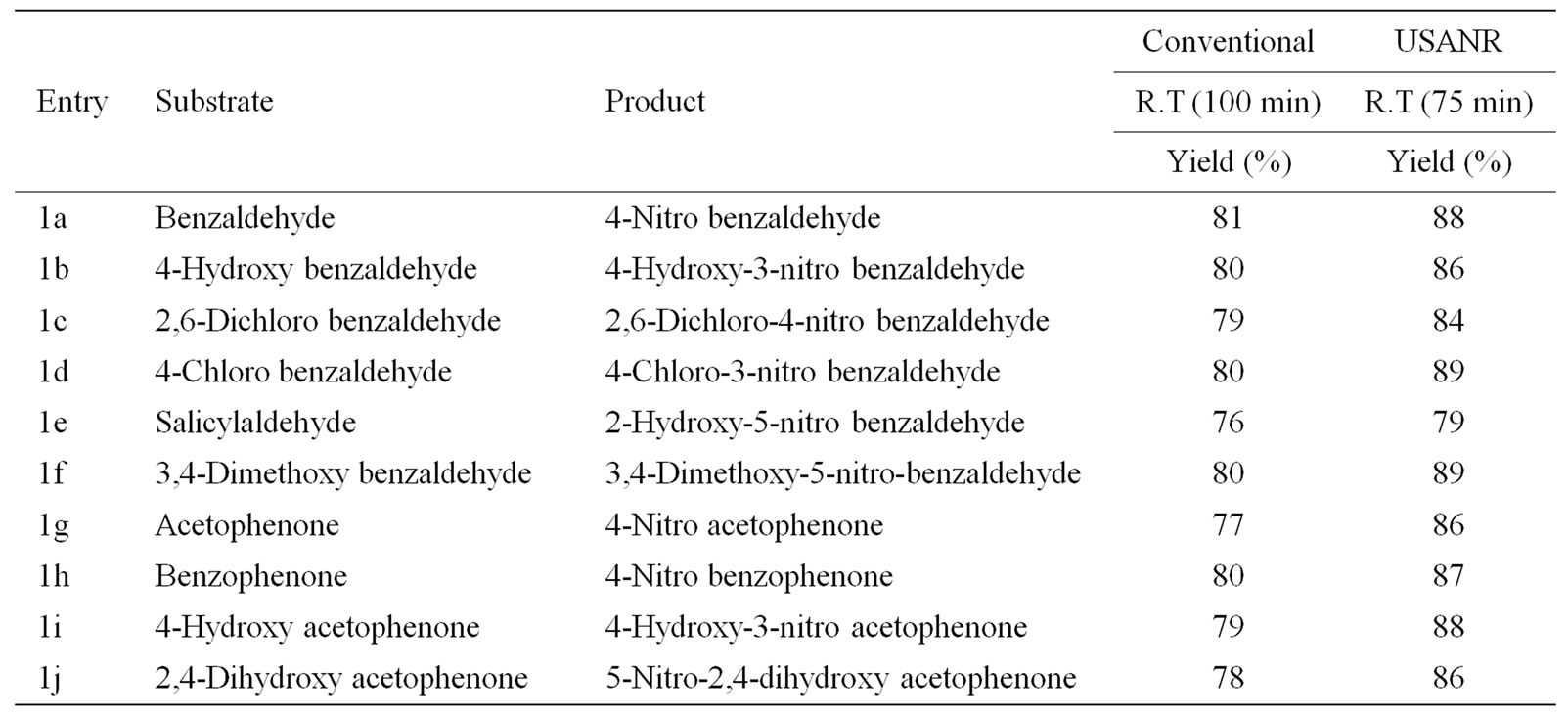

Table 6. Ultrasonically assisted potassium chromate catalyzed nitration of carbonyl and related compounds under mild acid conditions.
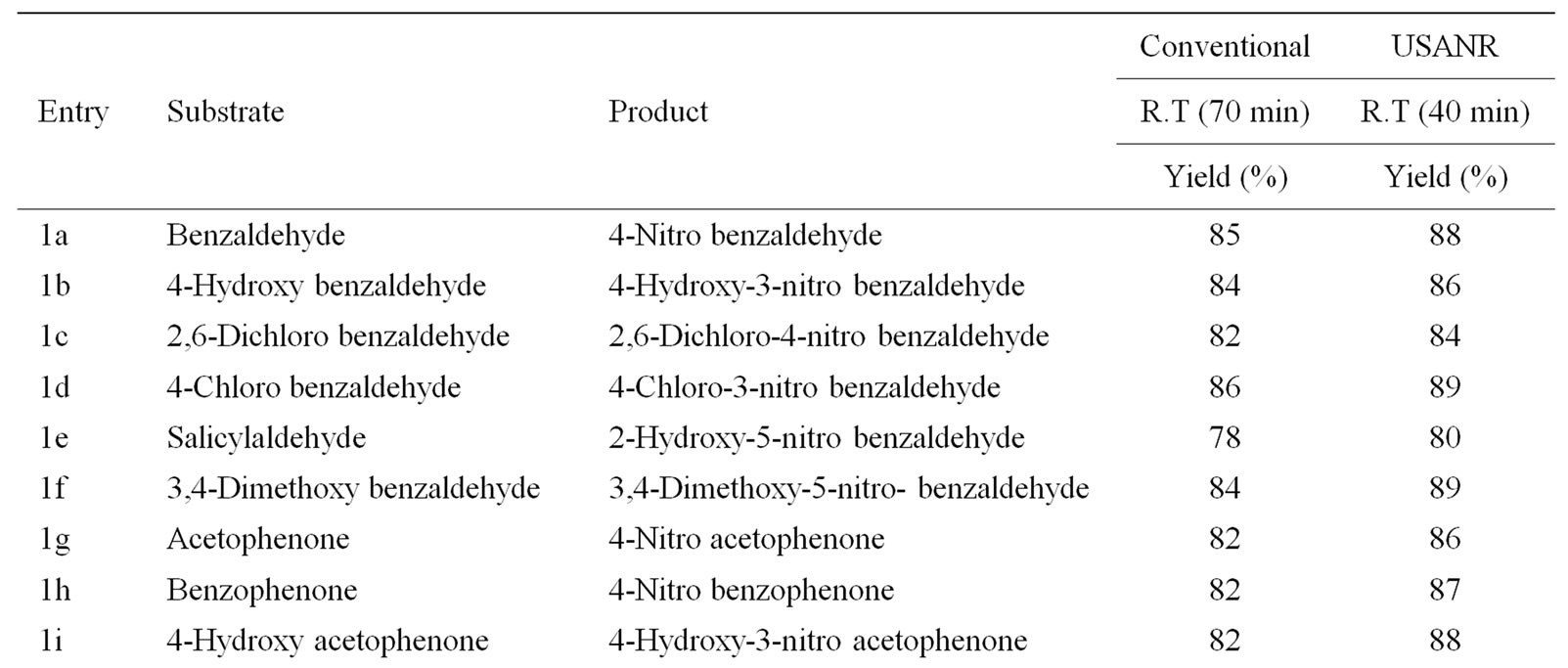

Table 7. Ultrasonically assisted ammonium molybdate catalyzed nitration of carbonyl and related compounds under mild acid conditions.
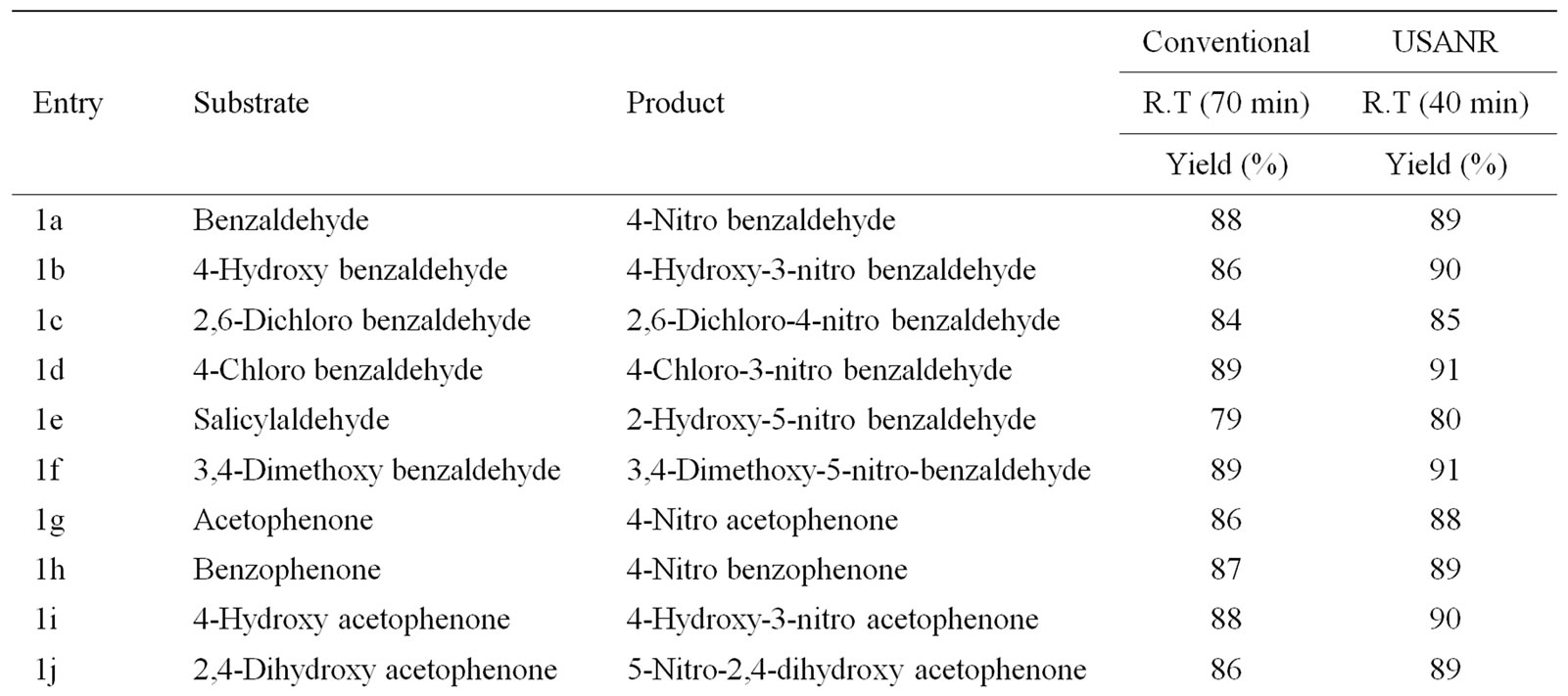

Table 8. Ultrasonically assisted sodium tungstate catalyzed nitration of carbonyl and related compounds under mild acid conditions.


Table 9. Ultrasonically assisted sodium bismuthate catalyzed nitration of carbonyl and related compounds under mild acid conditions.

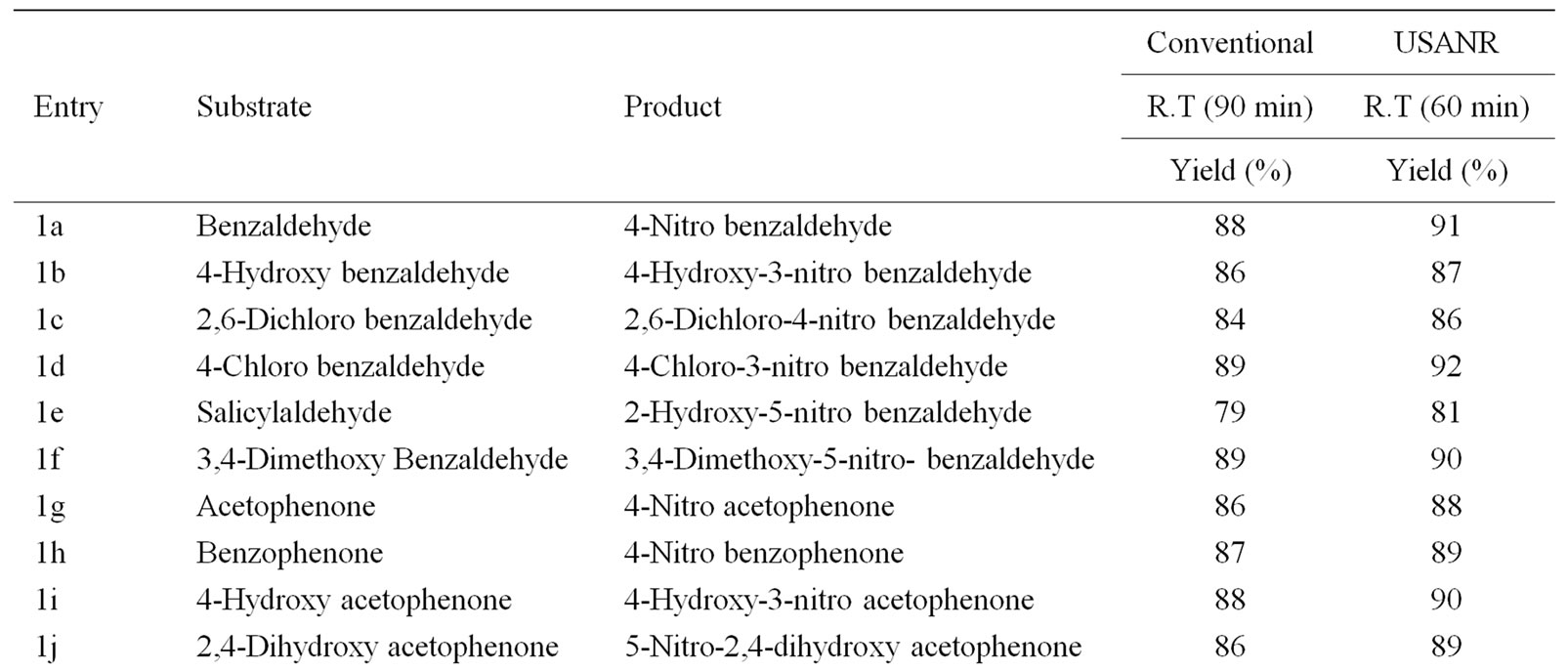
Table 10. Ultrasonically assisted bismuth nitrate catalyzed nitration of carbonyl and related compounds under mild acid conditions.
The observed para selectivity could be explained due to the reversible nucleophilic hydration of carbonyl group by utilizing the water molecules present in the molybdenum species. Hydration of the carbonyl group makes it a non-electron withdrawing species, which in turn facilitates para nitration in the subsequent steps. Further, it is of great interest to note that benzoyl chloride (entry 10) also underwent para nitration without loss of chloride. It is also found that in no case under these conditions oxidation of aldehydic group to corresponding carboxylic group has been observed. The results clearly indicate that the direct nitration of aromatic carbonyl and related compounds can be achieved successfully by employing ammonium molybdate and nitric acid as nitrating agent with high para-regioselectivity.
3.2. Ultrasonically Assisted Nitration Reactions
Certain recent non-conventional activation techniques, such as ultrasonically assisted methods have become stimulus to physical and organic chemists because of their use to enhance and promote organic synthesis. Ultrasonically assisted organic reactions (USAR) afforded dramatic rate enhancements followed by achieving the selectivity, ease of experimental manipulation. The use of this non-traditional tool has been found to of great help in overcoming many of the difficulties associated with conventional reactions, and offered both processes related and environmental advantages.
In the present study we have conducted all the nitration reactions under sonication. All the reactions in the present study underwent with highly remarkable rate and yield enhancements followed by a reduction in the reaction times as could be seen form the data presented in Tables 1-10.
The spectrum of ultrasound spreads over a wide range of frequencies, (10 Hz - 10 MHz) as shown in Figure 1. A higher frequency range (1 MHz -10 MHz) is used for medical and therapeutic purposes, medium frequency range (20 kHz - 100 kHz) is used for sonochemistry and low frequency range is used for human handling (Figure 1).
Literature reports have demonstrated that ultrasonic irradiations not only accelerate chemical reactions, but also promote new systems and also reduce the number of
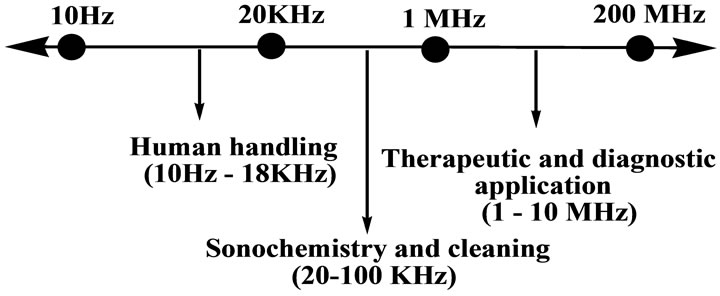
Figure 1. Ultrasound range diagram.
steps which are generally required for normal reactions. It is also well documented in literature that the ultrasonically assisted reactions occur through a transient state of high temperature and pressure and the chemical effects of ultrasound are due to the phenomenon of acoustic cavitation [12,13], as shown in Figure 2. When the reaction takes under ultrasonic conditions, sound waves pass through the solution containing reaction mixture and induce molecular motion through a series of compression and rarefaction cycles. At sufficiently high powers, the rarefaction cycle is able to pull molecules apart and create voids that are effectively bubbles containing small quantities of solvent vapour. At ultrasonic frequencies the succeeding compression wave does not fully collapse the bubbles and they grow in size through subsequent cycles by a process known as rectified diffusion. Eventually they become unstable and undergo violent collapse in a process known as acoustic cavitation, as shown in Figure 2 [14,15]. Depending on the frequency, extreme temperatures and pressures can be generated within the bubble and at the point of bubble collapse. This phenomenon provides a source of energy which can be used to enhance the rate of a chemical process. Each cavitation bubble acts itself as a localized micro reactor which causes rate accelerations in the ultrasonically assisted nitration reactions (USNR). Cavitation induces very high local temperatures and the liquid and enhanced mass transfer. Figure 2 represents the development and collapse of bubbles due to cavitation.
3.3. Recyclisation of the Catalyst
For large scale application of the reaction, it would be desirable to recycle the catalyst. The reaction with 0.25 equivalents or 0.5 equivalents of ammonium molybdate is very slow. Therefore the reaction is carried out at 1:1:1 molar ratio. Furthermore, the catalyst is readily recycled by a simple filtration after ensuring that it is free from nitrate ions by washing with water. The recovered catalyst was used as such and found to be active for further nitrations
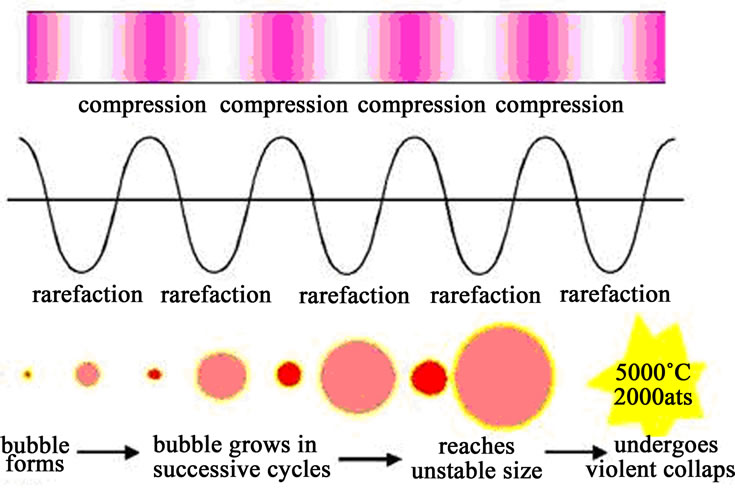
Figure 2. The process of acoustic cavitation.
with no reduction in yield and with no change in selectivity. Benzaldehyde, benzamide and benzoic acid were selected as the substrates to examine the catalyst recycling. To ascertain how long the efficacy of the catalyst would last, the reaction was repeated minimum three to four times using the same batch of catalyst. The results were reproducible with good accuracy.
4. Conclusion
In conclusion we have demonstrated that ultrasonically assisted nitration reactions (USANR) underwent smoothly with good yields and high regioselectivity is observed for anilides, moderately and non-activated aromatic compounds. Longer reaction times (6 - 8 hrs) of metal catalyzed reactions reduced to (1 - 2 hrs) under sonication. When ortho position is blocked para derivatives are obtained while ortho nitro products are obtained when para position is blocked. In case of USANR of aromatic carbonyl and related compounds the effect of sonication is highly significant; this could be seen from the observed reaction times ranging in few minutes.
5. Acknowledgements
The authors are thankful to Professor P. K. Saiprakash for constant encouragement and Heads of the Chemistry Department at Osmania University and Nizam College, Hyderabad for facilities.
REFERENCES
- G. A. Olah, R. Malhotra and S. C. Narang, “Nitration: Methods and Mechanism,” VCH, New York, 1989.
- C. K. Ingold, “Structure and Mechanism in Organic Chemistry,” 2nd Edition, Cornell University Press, Ithaca, 1969.
- K. Schofield, “Aromatic Nitrations,” Cambridge University Press, Cambridge, 1980.
- P. B. D. Dela mare and J. H. Ridd, “Aromatic Substitution, Nitration and Halogenation,” Bufferworths, London, 1959.
- G. A. Olah, “Mechanism of Electrophilic Aromatic Substitutions,” Accounts of Chemical Research, Vol. 4, No. 7, 1971, pp. 240-248. doi:10.1021/ar50043a002
- J. H. Ridd, “Mechanism of Aromatic Nitration,” Accounts of Chemical Research, Vol. 4, No. 7, 1971, pp. 248-253. doi:10.1021/ar50043a003
- H. Fener and A. T. Nielson, “Nitro Compounds: Recent Advances in Synthesis and Chemistry,” VCH Publishers, New York, 1990.
- N. C. Marziano, C. Tortato and M. Sampali, “Thermodynamic Nitration Rates of Aromatic Compounds. Part 3. Nitration of Aromatic Compounds in Concentrated Aqueous Trifluoromethanesulphonic Acid,” Journal of the Chemical Society, Perkin Transactions, Vol. 2, 1991, pp. 423-429. doi:10.1039/p29910000423
- S. V. Ley and C. M. R. Low, “Ultrasound in Synthesis,” Springer, Berlin, 1989. doi:10.1007/978-3-642-74672-7
- W. T. Richards and A. L. Loomi, “The Chemical Effects of High Frequency Sound Waves I. A Preliminary Survey,” Journal of the American Chemical Society, Vol. 49, No. 12, 1927, pp. 3086-3100. doi:10.1021/ja01411a015
- T. J. Mason and J. P. Lorimer, “Sonochemistry: Theory, Applications and Uses of Ultrasound in Chemistry,” John Wiley and Sons, New York, 1988.
- T. J. Mason, “Ultrasound in Synthetic Organic Chemistry,” Chemical Society Reviews, Vol. 26, No. 6, 1997, pp. 443-451. doi:10.1039/cs9972600443
- V. Singh, K. P. Kaur, A. Khurana and G. L. Kad, “Ultrasound: A Boon in Synthesis of Organic Compounds,” Resonance, Vol. 3, No. 9, 1998, p. 56-60.
- T. J. Mason, “Chemistry with Ultrasound,” Elsevier Science Publishers Ltd., London, 1990.
- T. J. Mason and D. Peters, “Practical Sonochemistry: Power Ultrasound Uses and Applications,” 2nd Edition, Hoorwood Publishing, Chichester, 2003.
- K. S. Suslick, “Ultrasound: Its Chemical, Physical and Biological Effects,” VCH, New York, 1988.
- M. A. Margulis, “Advances in Sonochemistry,” Vol. 1, JAI Press, London, 1990, p. 49.
- J. Alvarez-Builla and J. Ezquerra, “Phase Transfer Catalysis under Ultrasound. Alkylation of Isoquinoline Reissert Compound,” Journal of the Chemical Society, Chemical Communications, No. 1, 1984, pp. 54-55. doi:10.1039/c39840000054
- T. Ando, S. Sumi, T. Kawate, J. Ichihara and T. Hanafusa; “Sonochemical Switching of Reaction Pathways in SlidLiquid Two-Phase Reactions,” Journal of the Chemical Society, Chemical Communications, No. 7, 1984, pp. 439- 440. doi:10.1039/c39840000439
- H. Bremner, “Recent Advances in Organic Synthesis Utilizing Ultrasound,” Ultrasonics Sonochemistry, Vol. 1, No. 2, 1994, pp. 119-124.
- G. Cravotto and P. Cintas; “Power Ultrasound in Organic Synthesis: Moving Cavitational Chemistry Fromacademia to Innovative and Large-Scale Applications,” Chemical Society Reviews, Vol. 35, No. 2, 2006, pp 180-196. doi:10.1039/b503848k
- H. Fillion and J. L. Luche, “Ch.9: Selected Experiments,” In: J. L. Luche, Ed., Synthetic Organic Sonochemistry, Plenum, New York, 1998.
- R. Cella and H. A. Stefani, “Ultrasound in Heterocycles Chemistry,” Tetrahedron, Vol. 65, No. 13, 2009, pp 2619- 2649. doi:10.1016/j.tet.2008.12.027
- K. C. Rajanna, M. M. Ali, S. Sana, Tasneem and P. K. Saiprakash, “Ultrasonically Accelerated Vilsmeier Haack Cyclisation and Formylation Reactions,” Synthetic Communications, Vol. 32, No. 13, 2002, pp. 1351-1356, doi:10.1081/SCC-120003631
- K. C. Rajanna, M. Satish Kumar, P. Venkanna, S. Ramgopal and M. Venkateswarlu, “Vilsmeier Haack Adducts as Effective Reagents for Regioselective Nitration of Aromatic Compounds under Conventional and Non-Conventional Conditions,” International Journal of Organic Chemistry, Vol. 1, No. 4, 2011, pp. 250-256. doi:10.4236/ijoc.2011.14036
- K. C. Rajanna, M. M. Ali, S. Sana and P. K. Saiprakash, “Mild, Efficient and Selective Nitration of Anilides, NonActivated and Moderately Activated Aromatic Compounds with Ammonium Molybdate and Nitric Acid as a New Nitrating Agent,” Chemistry Letters, Vol. 29, No. 1, 2000, pp. 48-49. doi:10.1246/cl.2000.48
- A. I. Vogel, A. R. Tatchell, B. S. Furnis and A. J. Hannaford, “Vogel’s Text Book of Practical Organic Chemistry,” 4th Edition, Longman, London & New York, 1986.
- http://www.sigmaaldrich.com
- http://www.chemexper.com
- http://www.rdchemicals.com
- D. R. Lide and H. P. R. Frederikse, “CRC Handbook of Chemistry and Physics,” 83rd Edition, CRC Press, Boca Raton, 2002-2003.
Supplementary Data
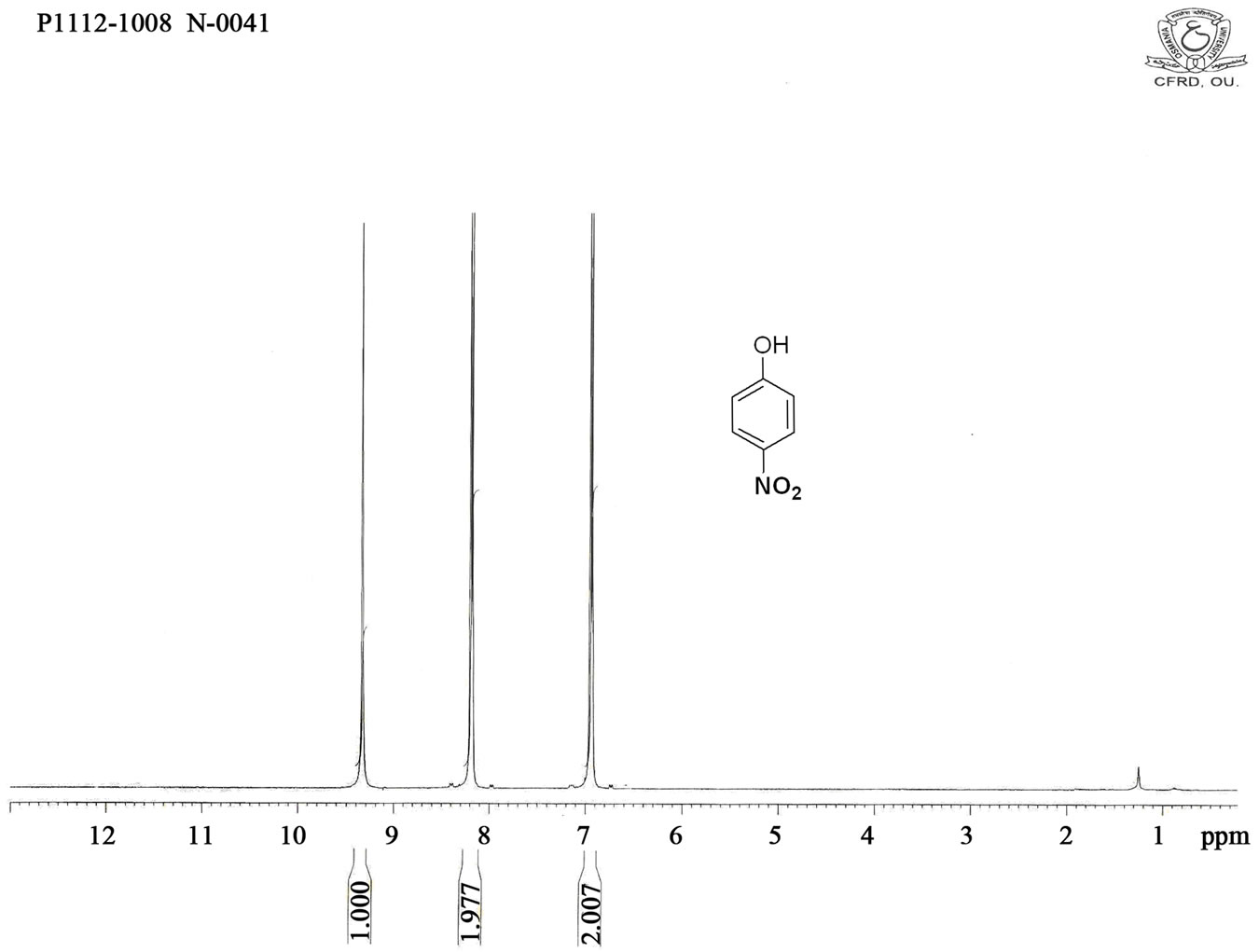
Figure S1. HNMR spectrum of 4-nitro phenol.
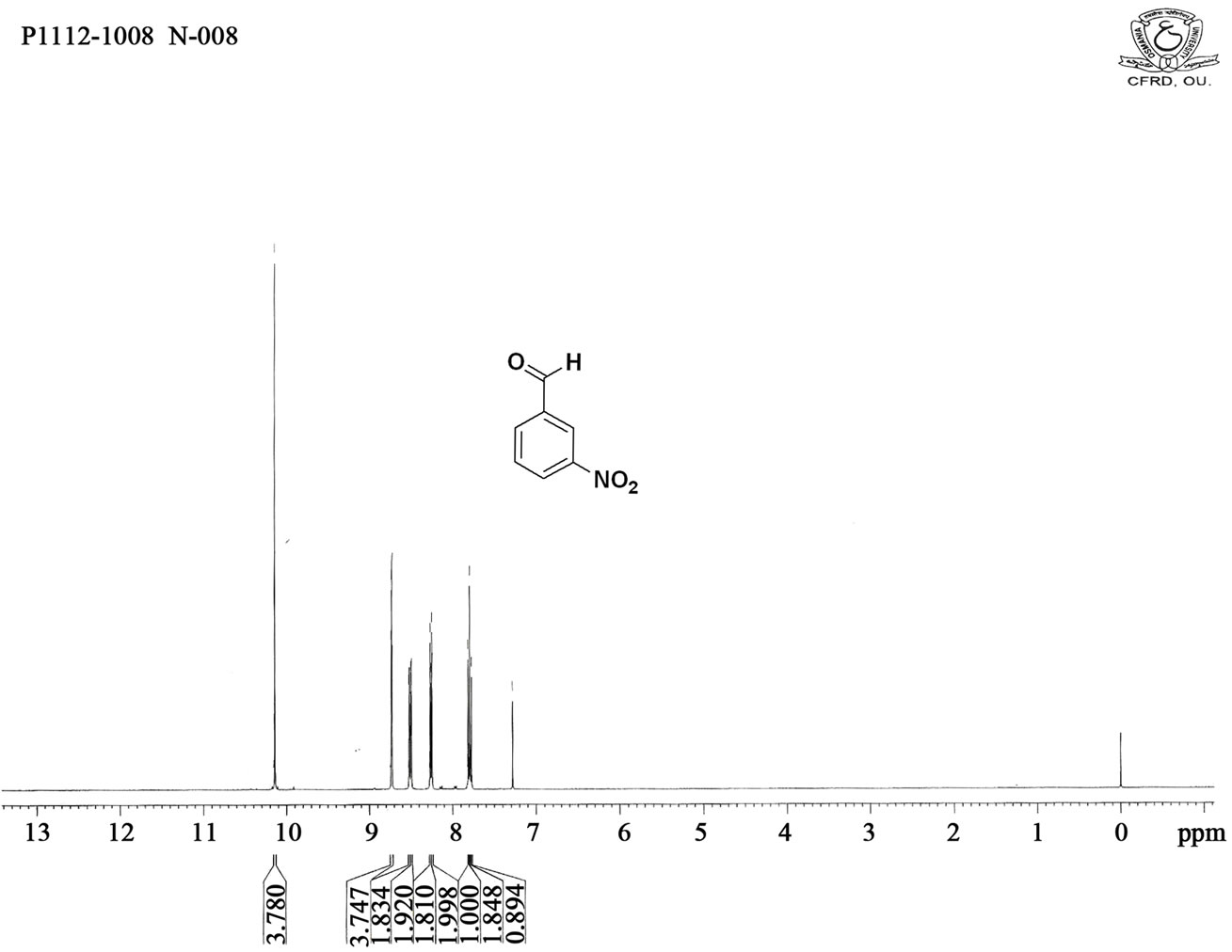
Figure S2. HNMR spectrum of 3-nitro benzaldehyde.
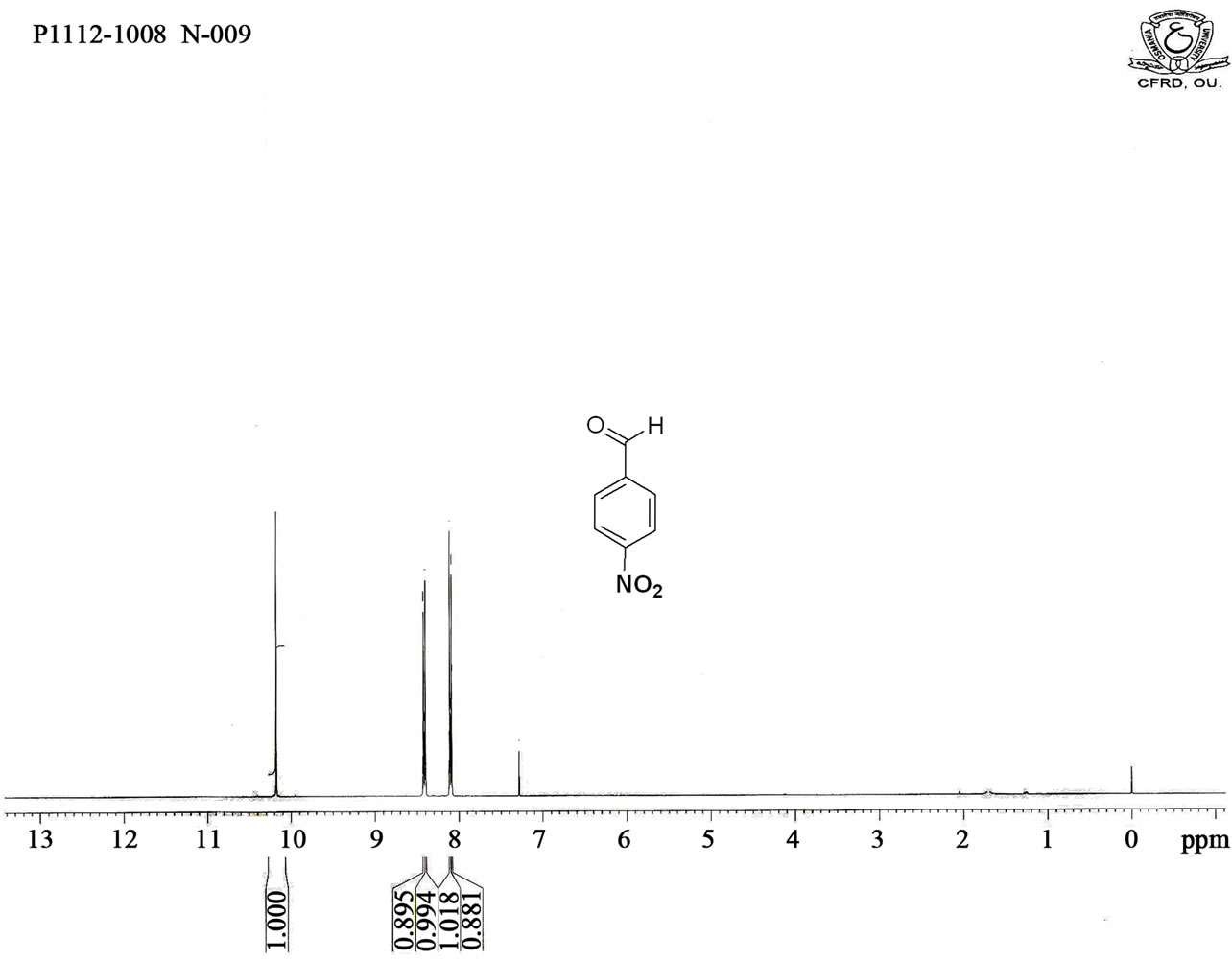
Figure S3. HNMR spectrum of 4-nitro benzaldehyde.
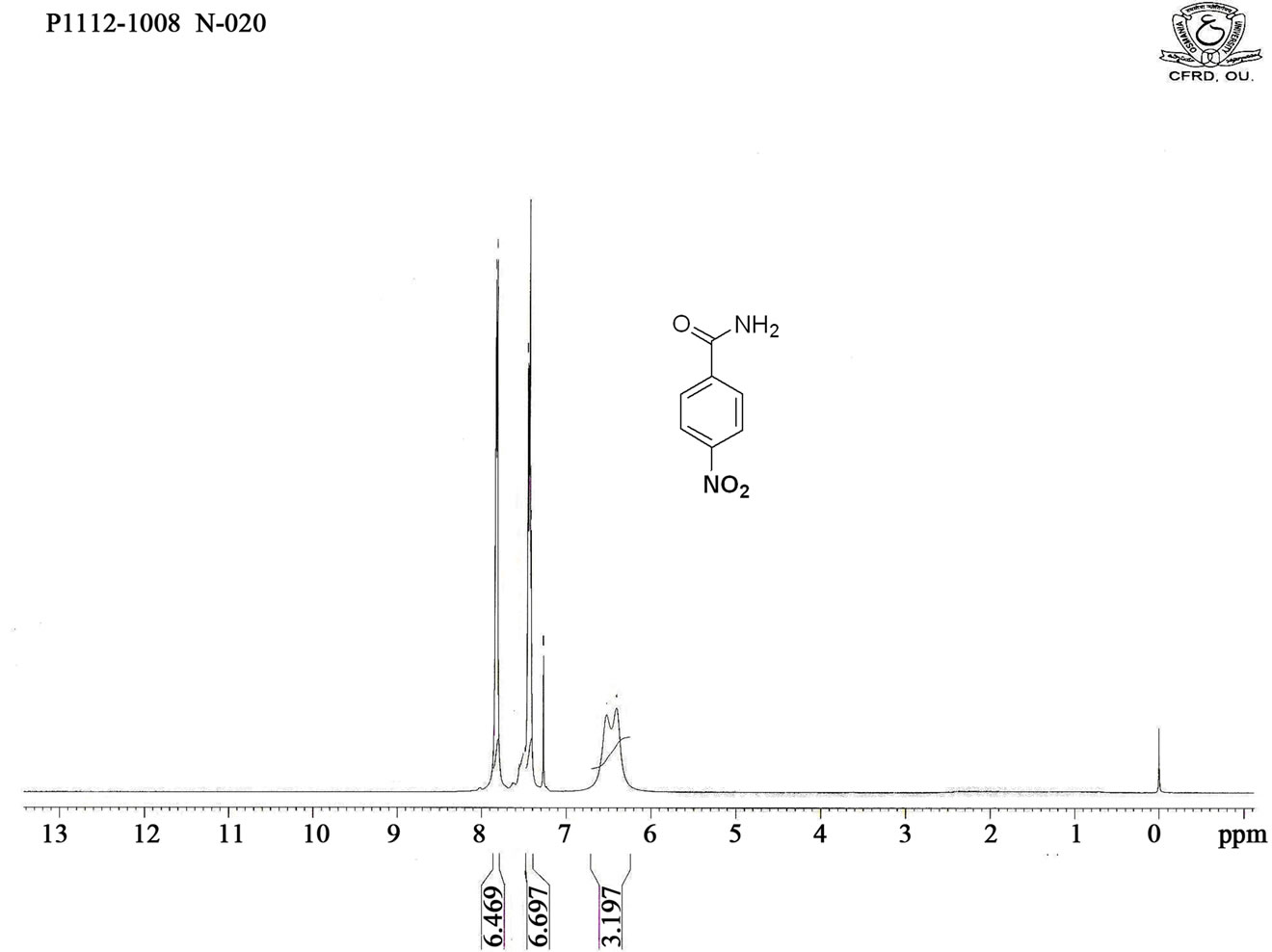
Figure S4. HNMR spectrum of 4-nitro benzamide.
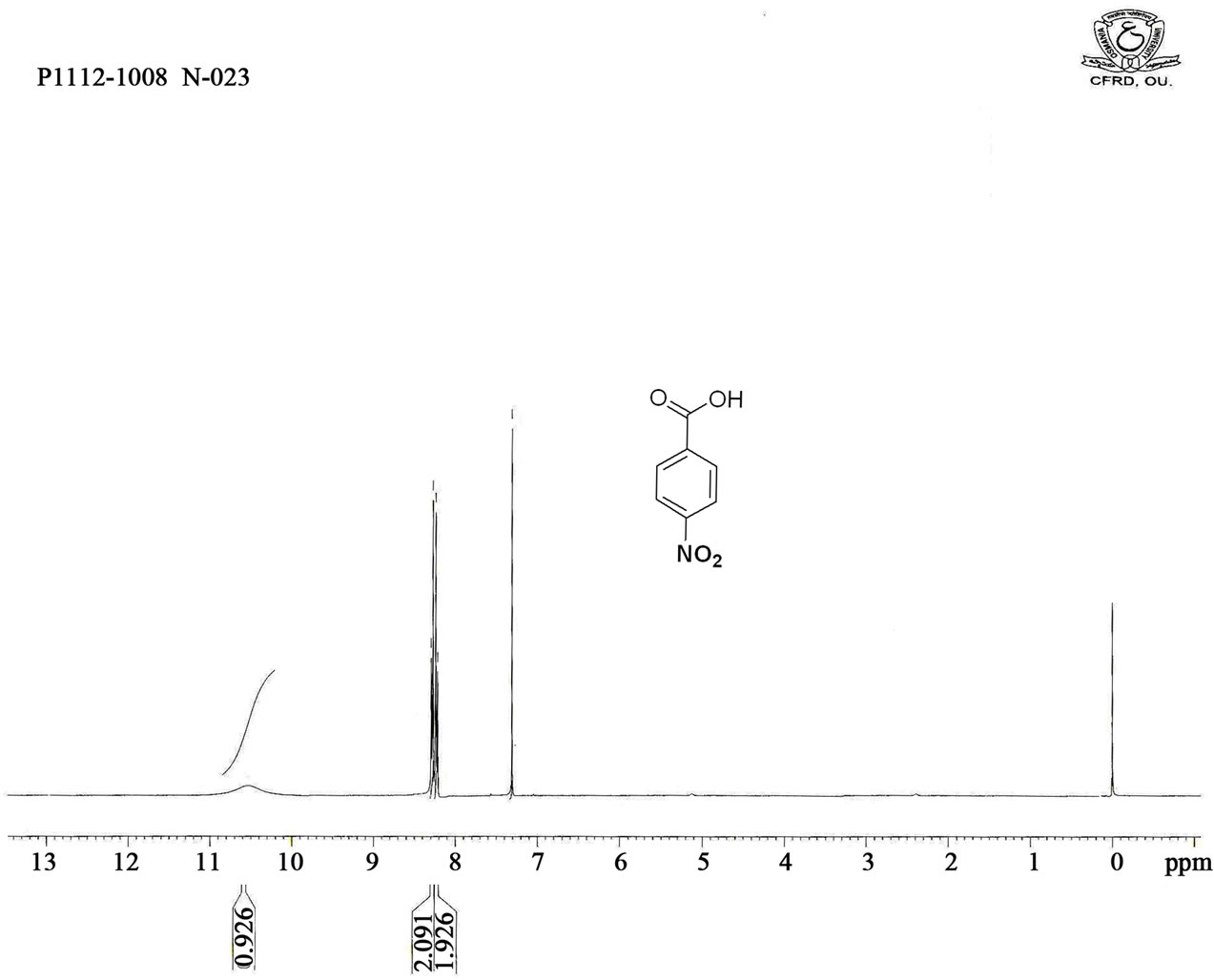
Figure S5. HNMR spectrum of 4-nitro benzoic acid.
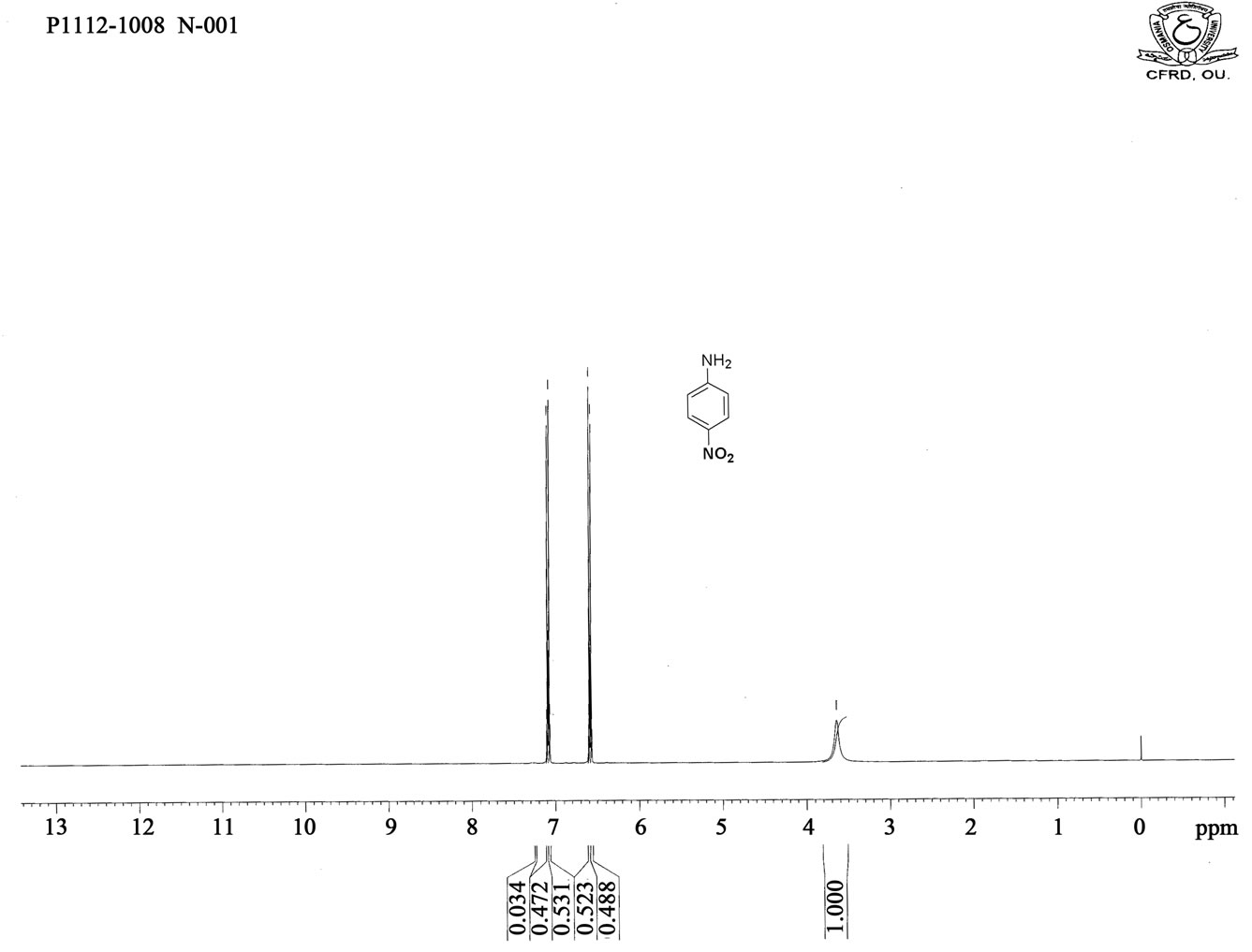
Figure S6. HNMR spectrum of 4-nitro aniline.
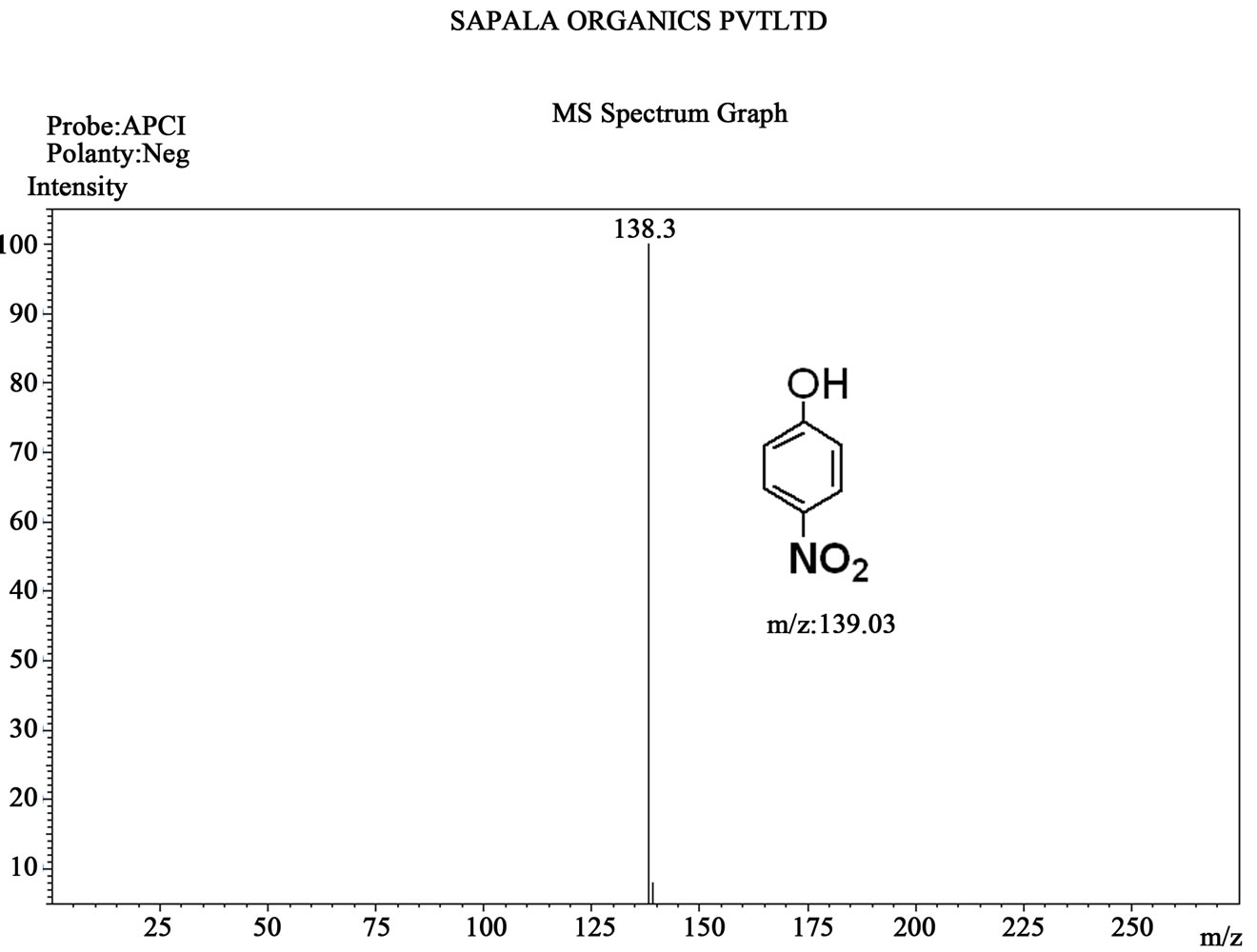
Figure S7. Mass spectrum of 4-nitro phenol.
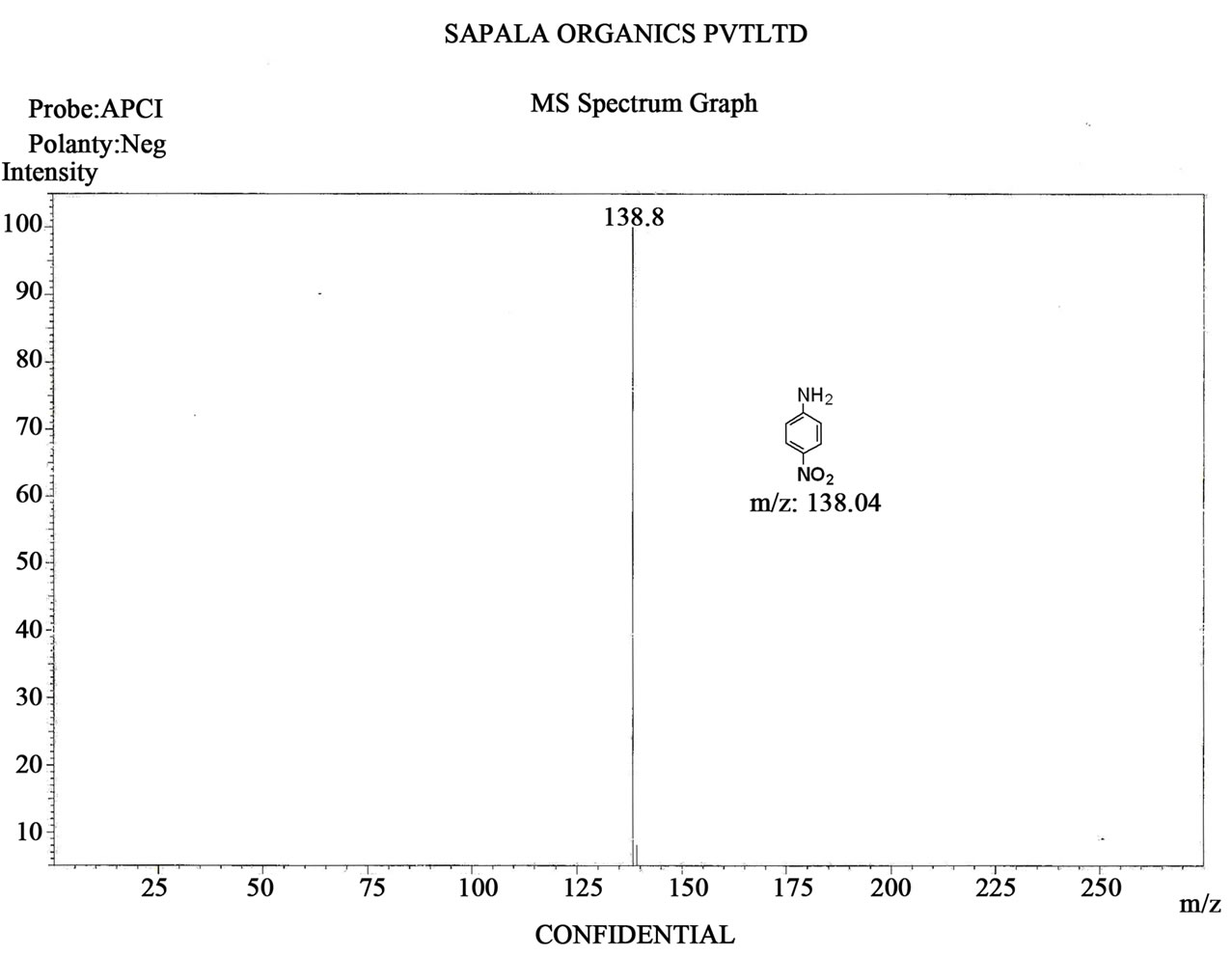
Figure S8. Mass spectrum of 4-nitro aniline.
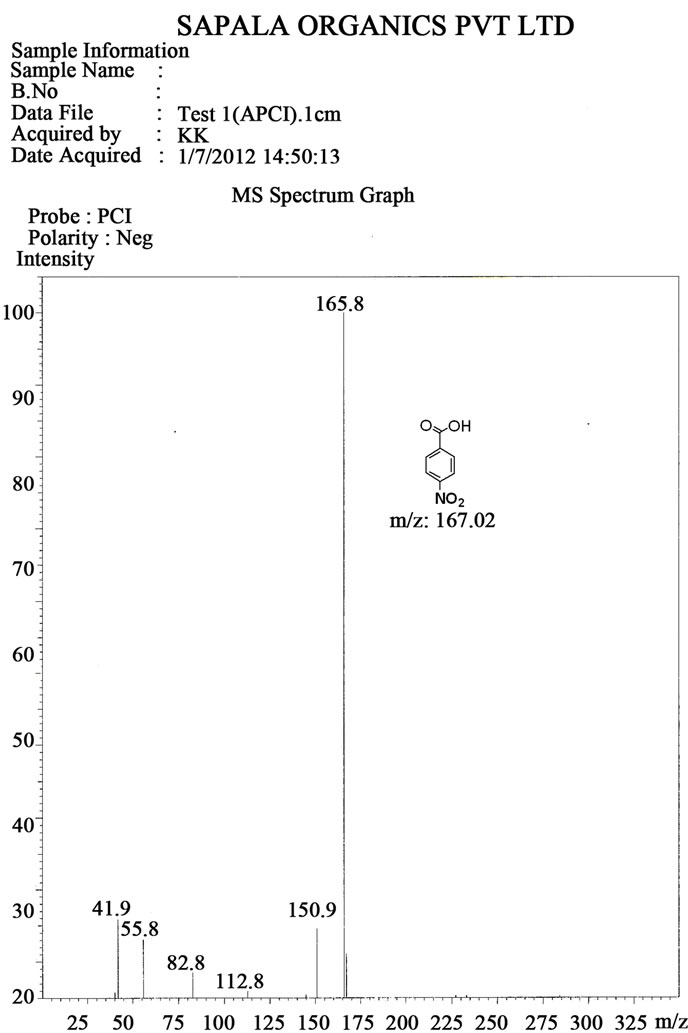
Figure S9. Mass spectrum of 4-nitro benzoic acid
NOTES
*Corresponding author.