World Journal of Vaccines
Vol. 1 No. 4 (2011) , Article ID: 8641 , 7 pages DOI:10.4236/wjv.2011.14013
JJo, a Recombinant Dimer of Conformationally Restricted Peptide Elicits Protective Response against Group A Streptococcus (GAS) Isolates from a GAS-Endemic Region
![]()
1Department of Microbiology and Cell Biology, Indian Institute of Science, Bangalore, India; 2School of Biological Sciences, University of Wollongong, Wollongong, Australia; 3Queensland Institute of Medical Research, Brisbane, Australia; 4The National Centre for Immunisation Research and Surveillance, Children’s Hospital at Westmead, Westmead, Australia.
Email: *sri.sriprakash@qimr.edu.au
Received August 5th, 2011; revised September 12th, 2011; accepted September 23rd, 2011.
Keywords: Streptococcus pyogenes, vaccine development, rheumatic fever
ABSTRACT
A peptide (J14) containing conformationally restricted epitopes from the M protein of group A streptococcus (GAS) is capable of eliciting protective immune response against GAS infection. However, the protective response may be lost possibly due to its weak secondary-structure when the antigen is fused with other antigens in a recombinant polyepitope vaccine construct. We previously showed that JJo, a conformationally stabilized derivative of dimeric J14, overcomes this problem. We now show that anti JJo antibodies react with diverse GAS isolates found in the Indian sub-continent and that these antibodies are opsonic for GAS. The GAS strains used in this study were isolated from throat and skin swabs from Mumbai, Chennai and Vellore. Sera from mice immunized with recombinant JJo peptide were tested by ELISA, immunofluorescence, flow-cytometry, indirect bactericidal assay and mouse challenge assays to determine specific immunogeni-city, opsonic functions and protection against an Indian isolate. We propose that JJo is a robust antigen suitable for inclusion in recombinant multi-epitope vaccines which are potentially affordable option for the pediatric population of developing countries.
1. Introduction
The human pathogen Streptococcus pyogenes, commonly referred to as group A streptococcus (GAS), causes a wide variety of diseases. These include mild upper respiratory and skin infections, the incidences of which are very high among school going children, as well as severe invasive diseases such as necrotizing fasciitis and toxic shock syndrome and the post-infectious sequelae-rheumatic fever (RF) and rheumatic heart disease (RHD) [1]. Although the incidence of RF and RHD has declined in many parts of the world, they continue to be a major cause of cardiovascular morbidity and mortality in developing countries [2,3]. This is particularly evident in India, where it is estimated that 600,000 children <15 years of age suffer from RHD [3]. RF and RHD are autoimmune diseases and the M protein, a major streptococcal virulence factor, has been implicated in their pathogenesis. The M protein is also responsible for typespecific immunity against GAS [4]. Therefore, this protein has been a major target for vaccine design.
For most parts, M protein is a coiled coil protein which contains a number of internal repeat sequences (A, B and C repeats) [5]. The amino terminus is highly polymorphic and is the basis for typing while the carboxyl terminus including the C-repeat region (CRR) is highly conserved among GAS isolates [5]. Comprehensive dissection of the CRR has resulted in the identification of protective opsonic B cell epitopes that are void of cross reactive Band T-cell epitopes; hence these B cell epitopes are regarded as safe vaccine candidates [6]. These peptides however, are conformationally restricted. To help maintain their alpha helical conformation, an attribute necessary for inducing a protective immune response, they are embedded within a short heterologous coil-promoting amino acid sequence. One such resulting chimeric peptide antigen is called J14 [7].
When conjugated to diphtheria toxoid, J14 elicits an immune response that reacts against multiple M types [8]. However, to date the efficacy of J14 has largely been shown against commonly circulating strains in developed countries [9]. In order to target vaccines to populations with high GAS disease burden and a large diversity of circulating GAS strains, such as populations of the Indian subcontinent, it is necessary to demonstrate that a vaccine is effective in controlling local strains. In addition to broad cross reactivity, a vaccine that would make the most impact in such communities would also need to be affordable. In an attempt to address this, we have expressed J14 as a recombinant protein [10].
When designing recombinant polyepitope vaccines containing J14 as one of the antigens, we and others [10,11] found that the immune responses to J14 moiety in the recombinant fusion proteins was poor while the other antigens elicited expected responses. We reasoned that the molecular forces required to present J14 in coiled coil conformation may be weak, and hence could be sensitive to the context of its fusion partner(s) in recombinant polyepitope constructs. To overcome this limitation we designed JJo, a dimer of J14 with continuous heptad repeat to strengthen its coil-promoting propensity [10]. JJo is immunogenic in the mouse model and can induce antibody response that recognizes the original vaccine antigen peptide J14 as well as the native M protein [10]. However, JJo has never been tested for the ability to stimulate an immune response that reacts against diverse GAS strains. Since binding of antibodies to the surface of GAS is a prerequisite for opsonisation, in the present work we used immunofluorescence and flow cytometry to screen a number of GAS isolates belonging to major Indian M types for their ability to bind antibodies raised against JJo as a first step toward vaccine efficacy test. We further show that the JJo antibodies are functional and can kill GAS in an opsonophagocytosis assay and offer protection against systemic GAS infection in a mouse model.
2. Materials and Methods
2.1. GAS Isolates and Microbiological Methods
The GAS strains used in this study were isolated from throat or skin swabs as part of community survey in Mumbai, Chennai and Vellore. The strains were maintained in Todd-Hewitt broth (THB) supplemented with 10% glycerol (v/v) at −80˚C until used. All GAS isolates were typed using standard emm-sequencing protocols [12]. GAS C28 and Mu3 represented emm 63.3 and 81.2 two major emm types among our collection of GAS isolates used in this study. PL1 an Australian isolate [13] and JRS145, an M-negative strain [14] were used for comparisons where appropriate.
2.2. Protein Expression and Purification
Recombinant JJo was expressed and purified as described before [10]. Briefly, recombinant JJo was purified from E. coli cellular fractions by Nickel ion affinity chromatography. JJo expression was induced with 0.5 mM IPTG and subsequently purified under native conditions. The E. coli cell pellet was incubated for 30 minutes in lysis buffer (10mM Imidazole) containing 1 mM phenyl-methylsulphonyl fluoride (PMSF) and lysozyme (1 mg/ml) to promote cell lysis. The clarified cell lysate was passed through Ni-NTA resin pre-equilibriated with ice-cold lysis buffer, the column was washed five times with wash buffer (50 mM imidazole) and JJo protein was subsequently eluted with elution buffer (500 mM imidazole). Fractions containing JJo were pooled and dialyzed against 2 L of phosphate buffer saline at 4˚C for 20 h with two buffer changes.
2.3. Murine Immunization and Challenge Model
To assess whether the recombinant vaccine candidate JJo could stimulate an antibody response against the parent peptide (J14) in mice of different genetic backgrounds, we determined the immunogenicity of purified JJo in Balb/c, C3HeJ and B10BR mice following methods previously described [8]. In addition, antibodies were also raised in Quackenbush mice to assess functionality in an out bred strain. All animal experiments were carried out in accordance with Indian Institute of Science (IISc) Animal Ethics Committee. On day 0, six week old female BALB/c, B10Br, C3HeJ or Quackenbush mice (n = 10) as detailed in the results received 30 μg of antigen emulsified with complete Freund’s adjuvant (CFA), at the base of the tail. Mice received boosts with 15 μg of antigen in PBS, on days 21, 28 and 35 unless otherwise specified. Blood samples (10 μl) were taken before each immunisation via a snip at the end of the tail, on days 0, 20, 27 and 34. A full tail bleed (~200 μl) was taken 7 days after the final boost on day 42. The antibody responses in individual mice of the same genetic background were quantified by ELISA as described earlier [8]. Whole cell ELISAs were also carried out with sera from Balb/c and C3HeJ immunised mice [15]. Endpoint antibody titre was determined as the lowest dilution that had an absorbance higher than that of the average plus three standard deviations of the sham control serum. The sera from mice of the same genetic background were subsequently pooled for further studies.
On day 50, C3HeJ mice were challenged intraperitoneally with a pre-determined minimal lethal dose, following previously described methods [16]. To enhance virulence for these experiments, GAS isolates were serially passaged several times in mice [17]. However, among the major Indian GAS strains in our collection, only Mu3 showed increased virulence in our model. Following challenge with Mu3 (400 μl of 108 cfu/ml), mice were observed daily for 10 days to monitor survival. Appropriate sham controls (mice immunized with CFA in PBS only) were included.
2.4. Immunofluorescence and Flow-Cytometry
For immunofluorescence, GAS strains grown overnight in 5 ml of THB/1% neopeptone at 37˚C, were collected and resuspended in PBS to an OD600 of 0.1. Twenty microlitres of bacterial suspension was smeared and fixed on poly-lysine microscopic slides with 3% paraformaldehyde. Fixed bacterial cells were blocked with 5% skim milk in PBS-Tween 20 for 1 h in a humid chamber at 37˚C, then rinsed in PBS. Any cell-surface Fc receptors were subsequently blocked with purified nonspecific polyclonal human IgG (Sigma, 1:200) for 1 h at 37˚C. Blocked slides were incubated with anti-JJo or sham control (CFA in PBS vaccinated) mouse serum (1:50 dilution; Balb/C). The bound antibody was detected with fluorescein isothiocyanate (FITC)-conjugated secondary antibody (anti mouse-IgG) (Sigma, USA, 1:200). JRS145, an M-negative mutant was used as a control. Slides were visualized at 520 nm under a confocal microscope (LSM-510 meta, Zeiss). The cell suspensions prepared for immunofluorescence were also processed in parallel for flow cytometric analysis as described earlier [8] with slight modifications. Cells were blocked with 3% bovine serum albumin (BSA) and human IgG prior to incubation with pooled anti-JJo or sham control serum (1:50 dilution). Cells with bound antibodies, were detected with anti-mouse IgG-FITC conjugated secondary antibody (Sigma, USA,) and subsequently counted using FACS Calibur flow cytometer (B&D, USA). CellQuest program was used to analyze the data.
2.5. Indirect Bactericidal Assay
Bactericidal assays were performed as described previously [16], GAS isolates were cultured overnight in Todd-Hewitt broth at 37˚C. Fifty μl of the diluted culture (105) were mixed with 50 μl of serum (normal or immune serum from B10BR, Quackenbush and C3HeJ mice) and then the mixture was incubated for 3 hrs at 37˚C with end-over-end mixing with 400 μl of non-opsonic human blood which supports growths of GAS by > 32 times [17]. Subsequently 50 μl aliquots of this mixture were plated in duplicate using the pour plate method. Based on our previous calibrations GAS culture of a 105 dilution consistently gave 50 - 100 cfu in 50 μl. CFUs were counted after 24 h of incubation at 37˚C. Fifty microliters of the bacterial dilution were also plated likewise to confirm the size of the inoculum. Bactericidal activity of immune sera (% reduction in mean CFU) was calculated as: 1 – [mean CFUs in the presence of immune mouse sera/ mean CFUs in the presence of sera from PBS immunised mice] × 100. Each sample was tested in duplicate.
3. Results
3.1. Characterization of Antibody Response to JJo
Earlier studies [16] showed that J14, a conformational restricted peptide antigen which is genetically restricted, offered protection against GAS infection when conjugated to diphtheria toxoid. However, J14 lost its immunogenicity when it was expressed with a fusion partner in a recombinant construct [10]. We attributed this to the loss of secondary conformation of the peptide in the context of its fusion partner. To solve this problem, we redesigned the antigen to contain two moieties of J14 [10]. In the present study, this new vaccine candidate, called JJo, is shown to stimulate a JJo-specific humoral response in mice representing different genetic backgrounds (Balb/c, C3HeJ and B10BR). Titres against JJo in all the three genetic background are similar (mean titre 25600; Figure 1(a)).
In a separate experiment, sera from these mice reacted with the parent peptide J14 (data not shown). These results confirm our earlier results obtained with Balb/c [10]. Taken together, our results show that the immune response of the recombinant JJo is not genetically restricted and hence unlike short B cell epitopes which need help with heterologous T cell epitopes (such as diphtheria toxoid), recombinant JJo on its own is immunogenic without additional T-cell help.
3.2. Binding Characteristics of JJo Antibodies to GAS Isolates
In order to test that JJo antibodies bind to M protein on the GAS cell surface, we used several independent approaches. Firstly, the immune sera reacted with whole cells of two representative strains representatives of prevalent Indian M-types (C28 and Mu3) in a whole-cell
 (a)
(a)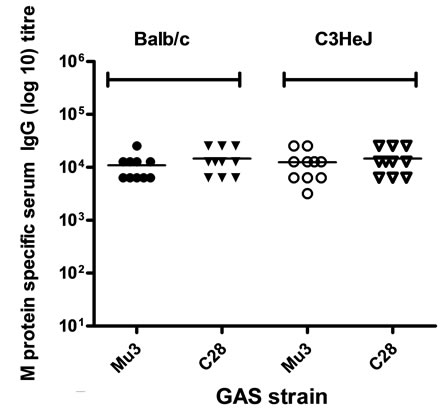 (b)
(b)
Figure 1. Comparison of immunogenicity of JJo in mice with different genetic backgrounds. (a) Microtiter plates were coated with recombinant JJo (500 ng/well); (b) Microtiter plates coated with Mu3 or C28 GAS isolates. For whole cell ELISA, the GAS isolates were grown overnight in THB media at 37˚C. The streptococci cells washed and resuspended in PBS to an OD600 of 0.4. Diluted cells (100 μl) were aliquoted to each well of the microtitre plate and incubated at 37˚C for 30 min. Non-adherent bacteria were removed by washing and standard ELISA protocol was followed.
ELISA experiment (Figure 1(b)) suggesting that the cell surface antigen in its native form is recognized by JJo antibodies. The binding characteristics in both these strains are similar. We then extended this study by probing a broader set of Indian strains with IFA (Figure 2(a)) and flow cytometry (FACS) (Figure 2(b)). The Results of both IFA and FACS (Table 1) confirmed binding of JJo specific antibodies to the surface of GAS cells of diverse M types. All GAS strains typically contained within their last C-terminal repeat M-specific sequences corresponding to either J14 or J14.1 [18]. Anti-J14 antisera cross react with J14 and J14.1 and vice versa [19]. Therefore it is not surprising that anti-JJo cross-reacted
 (a)
(a)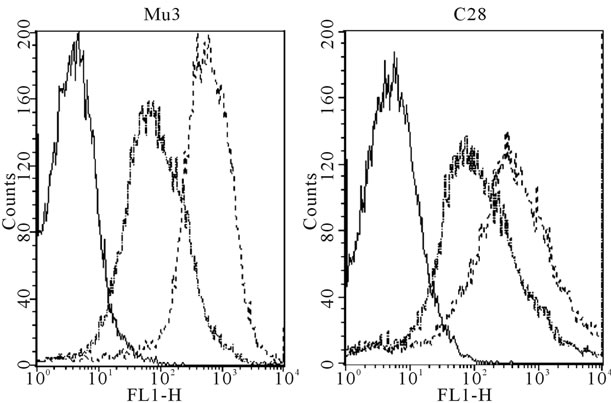 (b)
(b)
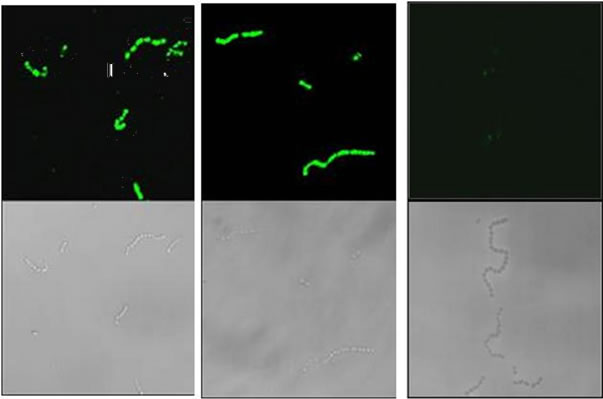
Figure 2. Immunofluorescent microscopy and flow cytometric analysis of representative Indian GAS isolates. (a) Immunofluorescent micrographs and corresponding bright field images of GAS strains Mu3 (left panel), C28 (middle panel) and JRS145 (right panel) probed with JJo specific antibodies. JJo antibodies bound strongly to Indian GAS isolates Mu3 and C28, however no binding was observed to M-negative JRS145. Sham control sera did not bind to either GAS strain (data not shown); (b) Flow cytometric analysis of GAS isolates Mu3 (left graph) and C28 (right graph) probed with JJo or sham control antisera. A small shift to the right was observed when cells were incubated with sham control sera indicating a low level of non-specific binding. However, a large shift to the right was observed following incubation with anti-JJo antibody. The fluorescent peak for the bacterial cell control was at far right, indicative of no antibody binding. For each sample, data for 10,000 events were analyzed.
with all the GAS isolates tested.
While JJo antibodies bound to all of the GAS strains tested the levels of binding as measured by the signal intensity did vary somewhat from strain to strain. In addition, for some isolates there was poor correlation between the fluorescence intensity observed in IFA and the mean fluorescence intensity (MFI) as determined
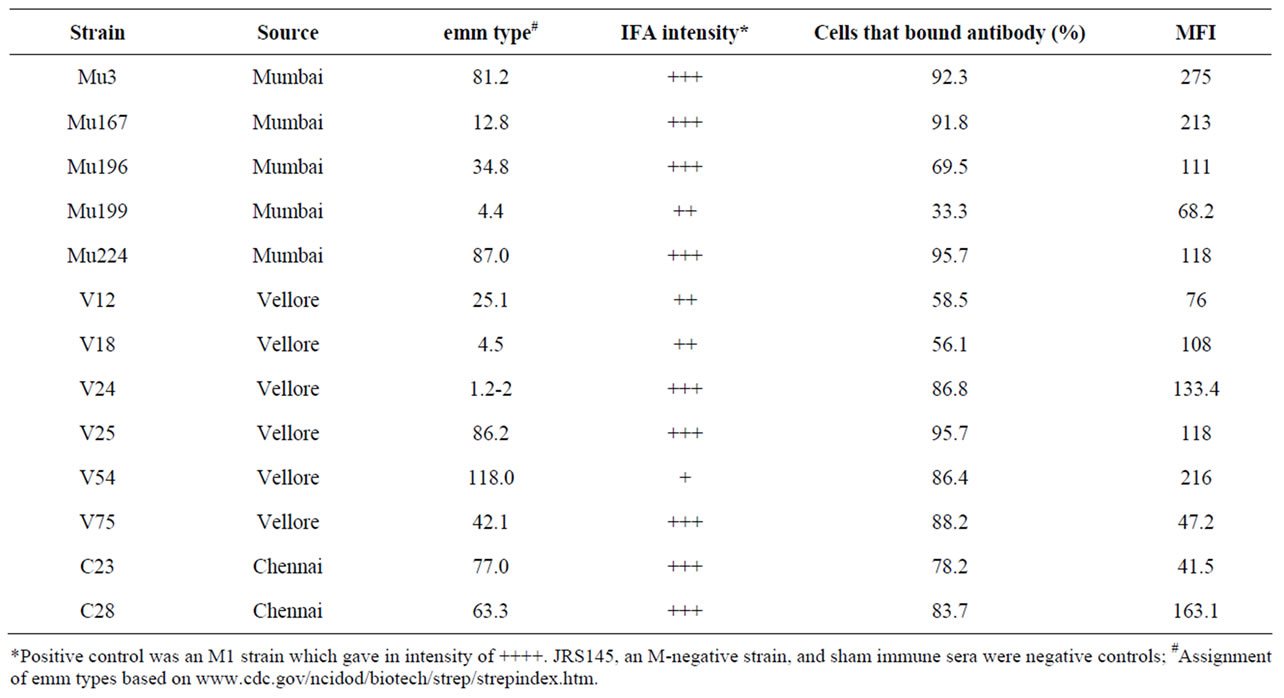
Table 1. Summary of the strains tested in this study and their reactivity of JJo antibodies as determined by immunofluorescence and flow cytometry.
by FACS. Taken together it is clear that JJo antibodies react with M proteins from all the GAS strains tested. However, native M protein on the surface of cells may not be equally accessible for binding by the JJo antibodies. This is not unexpected as differences in capsular thickness [20], the number of CRRs [21], the level of expression of M protein or loss of the M protein on surface [22] have been described and could account for this observed inequality. In the IFA, JJo antiserum did not react with M-negative GAS strain JRS145 (Figure 2(a)), which suggests that the anti-JJo antibody specifically reacts with M protein on the cell surface.
3.3. JJo Antibodies Opsonize GAS
Anti-phagocytic function is the main biological attribute of the M protein. To test whether JJo antibodies, like antibodies to the parent J14 molecule, are capable of opsonizing GAS strain C28 or PL1 was incubated with human blood in an indirect bactericidal assay. We opted to use Australian blood donors because we found it difficult to identify suitable donors (whose blood supports growth of GAS) in our cohort of volunteers from a region highly endemic for GAS infection (probably because of natural antibodies to GAS). We also did not find suitable donors for bactericidal assays for Mu3 which was the strain passaged for GAS challenge model. As shown in Table 2, antibodies raised in Quackenbush and B10BR mice to recombinant JJo or peptide J14 (conjugated to diphtheria toxoid) are opsonic to PL1 whereas sham immunized mice sera aren’t. The results were also reproducible with C28 when tested against a suitable donor blood from Australia (Table 2).
3.4. JJo Antibodies Protect Mice against GAS Infection
In the mouse challenge model for protection against systemic GAS infection, it is necessary initially to passage GAS strains through mice for several cycles to increase their virulence in this model [23]. We succeeded in doing this for one Indian GAS strain, Mu3. If indeed the reactive epitope for JJo antiserum is abundantly accessible in a strain, as for instance in Mu3, we expected that the antiserum to be protective against the strain in a mousechallenge experiment. To test this, we immunized C3HeJ mice with JJo. The immunized mice were challenged with minimal lethal dose of mouse-passaged Mu3. As predicted, the JJo immunized mice survived the challenge significantly better than sham-immunized mice (p = 0.019; Log-ranked Mantel-Cox test; Figure 3). These results confirm that JJo antibodies are protective against GAS infection with an endemic Indian isolate.
4. Discussion
Our present study clearly suggests broad reactivity of JJo
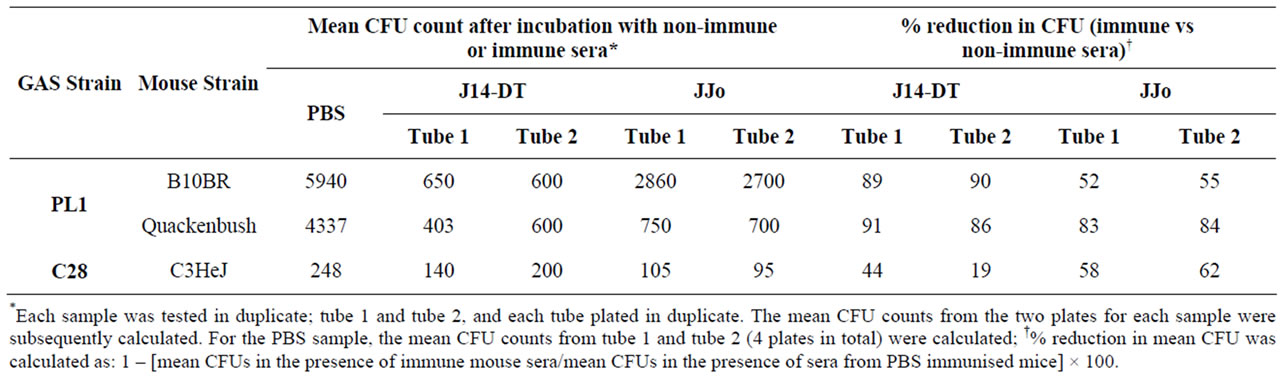
Table 2. In vitro bactericidal activity of antibodies raised against JJo in mice with different genetic backgrounds.
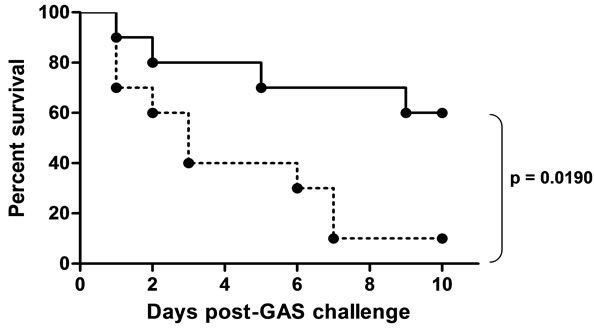
Figure 3. Survival curves for JJo immunized C3HeJ mice following intraperitoneal challenge with mouse-passaged Mu3 GAS strain. The percentage survival determined up to 10 days following challenge. Mice immunized with JJo (solid line) survived better than control mice (dashed line; p = 0.0190). Ten mice were included in each group.
antibodies against variety of GAS strains, and the functionality of these antibodies tested against a sub-set of these isolates using in-direct bactericidal assay and a mouse model of systemic GAS infection. While our study shows the overall coverage of strains for JJo-based vaccine is good, to avoid possible escape of strains from JJo antibody mediated clearance due to poor accessibility, there is strong argument for a polyepitope vaccine approach for GAS. In a similar attempt to make recombinant vaccine construct with broad coverage, Penfound et al. [11] recently combined J14 with other N-terminal epitopes from 6 M types. However, they consistently found that J14 was not immunogenic in such polyepitope constructs. Earlier we too demonstrated [10] conformational stabilization of J14 was required for appropriate immune response in a recombinant construct. We showed that the JJo derivative of J14 when expressed as a recombinant fusion protein elicited J14-specific immune response. These experiments confirmed earlier work [24] suggesting requirement for conformational stability for protective immune response. Our current work advocates that JJo replace J14 in all future recombinant polyepitope vaccine designs.
5. Acknowledgements
The authors thank Drs KN Bramhadatan and T Menon for Vellore and Chennai strains, Dr SG Ramachandra, central animal facility, IISc for facilitating the mouse work, Dr Omana Joy, FACS facility for FACS analysis and Ms Aakriti Anand for her enthusiastic assistance.
The funding for this work is from Australia India Strategic Research Fund, The Department of Biotechnology, India and the National Health and Medical Research Council, Australia.
REFERENCES
- A. L. Bisno, “Group A Streptococcal Infections and Acute Rheumatic Fever,” New England Journal of Medicine, Vol. 325, No. 11, 1991, pp. 783-793. doi:10.1056/NEJM199109123251106
- S. Padmavati, “Rheumatic Fever and Rheumatic Heart Disease in India at the Turn of the Century,” Indian Heart Journal, Vol. 53, 2001, pp. 35-37.
- A. Shet and E. Kaplan, “Addressing the Burden of Group A Streptococcal Disease in India,” Indian Journal of Paediatrics, Vol. 71, No. 1, 2004, pp. 41-48. doi:10.1007/BF02725655
- J. H. Robinson and M. A. Kehoe, “Group A Streptococcal M Proteins: Virulence Factors and Protective Antigens,” Immunology Today, Vol. 13, No. 9, 1992, pp. 362-367. doi:10.1016/0167-5699(92)90173-5
- V. A. Fischetti, “Streptococcal M Protein,” Scientific American, Vol. 264, No. 6, 1991, pp. 58-65. doi:10.1038/scientificamerican0691-58
- W. A. Hayman, E. R. Brandt, W. A. Relf, J. Cooper, A. Saul and M. F. Good, “Mapping the Minimal Murine T cell and B Cell Epitopes within a Peptide Vaccine Candidate from the Conserved Region of the M Protein of Group A Streptococcus,” International Immunology, Vol. 9, No. 11, 1997, pp. 1723-1733. doi:10.1093/intimm/9.11.1723
- S. Pruksakorn, A. Galbraith, R. A. Hougusten and M. F. Good, “Conserved T and B Cell Epitopes on the M Protein of Group A Streptococci. Induction of Bactericidal Antibodies,” The Journal of Immunology, Vol. 149, 1992, pp. 2729-2735.
- M. R. Batzloff, W. A. Hayman,M. R. Davies, M. Zeng, S.Pruksakorn, E. R. Brandt, M. F. Good, “Protection against Group A Streptococcus by Immunization with J8-Diphtheria Toxoid: Contribution of J8- and Diphtheria Toxoid-Specific Antibodies to Protection,” The Journal of Infectious Diseases, Vol. 187, No. 10, 2003, pp. 1598- 1608. doi:10.1086/374800
- H. Vohra, N. Dey, S. Gupta, A. K. Sharma, R. Kumar, D. J. McMillan and M. F. Good, “M Protein Conserved Region Antibodies Opsonise Multiple Strains of Streptococcus Pyogenes with Sequence Variations in C-Repeats,” Research in Microbiology, Vol. 156, No. 4, 2005, pp. 575-582. doi:10.1016/j.resmic.2004.12.009
- M. M. Georgousakis, A. Hofmann, M. R. Batzloff, D. J. McMillan and K. S. Sriprakash, “Structural Optimisation of a Conformational Epitope Improves Antigenicity When Expressed as a Recombinant Fusion Protein,” Vaccine, Vol. 27, No. 48, 2009, pp. 6799-6806. doi:10.1016/j.vaccine.2009.08.049
- T. A. Penfound, E. Y. Chiang, E. A. Ahmed and J. B. Dale, “Protective Efficacy of Group A Streptococcal Vaccines Containing Type-Specific and Conserved M Protein Epitopes,” Vaccine, Vol. 28, No. 31, 2010, pp. 5017-5022. doi:10.1016/j.vaccine.2010.05.018
- B. Beall, R. Facklam and T. Thompson, “Sequencing Emm-Specific PCR Products for Routine and Accurate Typing of Group A Streptococci,” Journal of Clinical Microbiology, Vol. 34, 1996, pp. 953-958.
- E. R. Brandit, K. S. Sriprakash, R. I. Hobb, W. A. Hayman, W. Zeng, M. R. Batzloff, D. C. Jackson and M. F. Good, “New Multi-Determinant Strategy for a Group A Streptococcal Vaccine Designed for the Australian Aboriginal Population,” Nature Medicine, Vol. 6, No. 4, 2000, pp. 455-459.
- M. G. Caparon, R. T. Geist, J. Perez-Casal and J. R. Scott, “Environmental Regulation of Virulence in Group A Streptococci: Transcription of the Gene Encoding M Protein Is Stimulated by Carbon Dioxide,” Journal of Bacteriology, Vol. 174, 1992, pp. 5693-5701.
- H. S. Courtney, O. I. Penfound, T. V. Nizet, M. A. Pence, B. Kreikemeyer, A. Podbielski, D. L. Hasty and J. B. Dale, “Relationship between Expression of the Family of M Proteins and Lipoteichoic Acid to Hydrophobicity and Biofilm Formation in Streptococcus Pyogenes,” PLoS One, Vol. 4, No. 1, 2009, p. e4166. doi:10.1371/journal.pone.0004166
- M. R. Batzloff, H. Yan, M. R. Davies, J. Hartas, G. H. Lowell, G. White, D. S. Burt, T. Leanderson and M. F. Good, “Toward the Development of an Antidisease, Transmission-Blocking Intranasal Vaccine for Group A Streptococcus,” The Journal of Infectious Diseases, Vol. 192, No. 8, 2005, pp. 1450-1455. doi:10.1086/466528
- G. Stollerman, F. Kantor and B. Gordon, “Accessory Plasma Factors Involved in the Bactericidal Test for Type-Specific Antibody to Group A Streptococci. I. Atypical Behavior of Some Human and Animal Bloods,” Journal of Experimental Medicine, Vol. 108, 1958, pp. 475-491. doi:10.1084/jem.108.4.475
- A. C. Steer, G. Magor, A. W. J. Jenney, J. Kado, M. F. Good, D. J. McMillan, M. Batzloff and J. R. Carapetis “Emm and C-Repeat Region Molecular Typing of BetaHemolytic Streptococci in a Tropical Country: Implications for Vaccine Development,” Journal of Clinical, Microbiology, Vol. 47, No. 8, 2009, pp. 2502-2509. doi:10.1128/JCM.00312-09
- N. Yoonim, C. Olive, C. Pruksachatkunakorn and S. Pruksakorn, “Bactericidal Activity of M Protein Conserved Region Antibodies against Group A Streptococcal Isolates from the Northern Thai Population,” BMC Microbiology, Vol. 6, 2006, p. 71.
- J. C. Levin and M. R. Wessels, “Identification of csrR/ csrS, a Genetic Locus That Regulates Hylarunic Acid Capsule Synthesis in Group A Streptococcus,” Molecular Microbiology, Vol. 30, No. 1, 1998, pp. 209-219.
- P. R. Smeesters, D. J. McMillan and K. S. Sriprakash, “The Streptococcal M Protein: A Highly Versatile Molecule,” Trends in Microbiology, Vol. 18, No. 6, 2010, pp. 275-282. doi:10.1016/j.tim.2010.02.007
- W. J. Simpson and P. P. Cleary, “Expression of M Type 12 Protein by a Group A Streptococcus Exhibits Phaselike Variation: Evidence for Coregulation of Colony Opacity Determinants and M Protein,” Infection and Immunity, Vol. 55, No. 10, 1987, pp. 2448-2455.
- S. P. O’Connor and P. P. Cleary, “In Vivo Streptococcus Pyogenes C5a Peptidase Activity: Analysis Using Transposonand Nitrosoguanidine-Induced Mutants,” The Journal of Infectious Diseases, Vol. 156, No. 3, 1987, pp. 495-504. doi:10.1093/infdis/156.3.495
- W. A. Relf, J. Cooper, E. R. Brandt, W. A. Hayman, R. F. Anders, S. Pruksakorn, B. Currie, A. Saul and M. F. Good, “Mapping a Conserved Conformational Epitope from the M Protein of Group A Streptococci,” Peptide Research, Vol. 9, 1996, pp. 12-20.