World Journal of Vaccines
Vol. 1 No. 2 (2011) , Article ID: 4786 , 8 pages DOI:10.4236/wjv.2011.12004
Improving in Silico Prediction of Epitope Vaccine Candidates by Union and Intersection of Single Predictors
![]()
1School of Pharmacy, Medical University of Sofia, Sofia, Bulgaria; 2Life and Health Sciences, Aston University, Birmingham, United Kingdom.
Email: idoytchinova@pharmfac.net
Received February 16th, 2011; revised March 12th, 2011; accepted March 20th, 2011.
Keywords: MHC Class II Binders, T-cell Epitopes, HLA-DRB1 Alelles
ABSTRACT
The in silico prediction of peptide binding affinities to MHC proteins is a very important first step in the process of epitope-based vaccine design and development. Five MHC class II binding prediction servers were combined in different ways and the resulting performance of these combinations was evaluated using a test set, which consisted of 4540 known HLA-DRB1 binders. The five servers were: NetMHCIIpan, NetMHCII, ProPred, RANKPEP, and EpiTOP. The top 5% of the ranked predictions from each server were combined using union and intersection operators. The outputs were evaluated in terms of sensitivity and positive predictive value (PPV). The union operator showed high sensitivity (65% - 79%) and low PPVs (6% - 8%), while intersection outputs had low sensitivities (4% - 41%) yet significantly higher PPVs (14% - 31%). Thus there is a defining trade-off between sensitivity and PPV for each combination. The union of outputs from different servers brings more “noise” than “signal” to the resulting set of predicted binders. Conversely, selecting only commonly predicted binders increases the probability that an identified binder is a true binder.
1. Introduction
The epitope-based vaccines are a new generation of vaccines which are very well tolerated and have fewer side effects than the conventional vaccines. The in silico prediction of peptide binding affinities to MHC proteins is a very important first step in the process of vaccine design and development. Peptides which act as T-cell epitopes bind to MHC molecules; thus all T-cell epitopes are MHC binders but not all MHC binders are T-cell epitopes. Binding to a MHC protein is a necessary but not a sufficient condition for a peptide to be an epitope. Peptides presented by an MHC on the cell surface have either an intracellular or an extracellular origin. MHC class I molecules, present on most cell types, present peptides primarily from protein synthesized within the cell (endogenous processing pathway). MHC class II molecules, expressed on a restricted number of cell types, such as dendritic cells, B cells and macrophages, can present peptides derived from endocytosed extracellular proteins (exogenous processing pathway) [1]. A principal feature of MHC molecules is their allelic polymorphism: 3,411 human leukocyte antigens (HLA) class I and 1,222 HLA class II molecules were listed by the ImMunoGeneTics/HLA database in July 2010 [2].
Peptides binding to MHC class II proteins are typically between 10 and 20 residues in length. The complex comprising peptide and class II molecule is expressed on the cell surface and interacts exclusively with CD4+ T cells (helper T-cells, THC). TH cells help to trigger an appropriate immune response which may include localized inflammation and swelling due to recruitment of phagocytes or may lead to an antibody-mediated immune response via B-cell activation. X-ray data from peptide-MHC class II [3-6] and TCRpeptide-MHC class II complexes [7-9] indicate that nine amino acids are bound in an extended conformation within the peptide binding groove of the MHC. The MHC class II binding groove is formed by two separate protein chains: α and β [10]. A dozen hydrogen bonds are formed between the MHC and the peptide main-chain carbonyl and amide groups. There are five pockets in the binding groove: one deep pocket (denoted p1), and four shallow pockets (p4, p6, p7 and p9), that accept peptide side chains. Peptide side chains at p2, p3, p5 and p8 project outward toward the T-cell. Compared to MHC class I, the MHC class II peptide binding groove is open at both ends. The degree to which this allows many potential registers in which a peptide might bind remains a controversial issue [11,12].
Systematic mapping of peptides binding to MHC proteins involves the synthesis and testing of all overlapping peptides spanning the whole length of a target antigen: a costly and time-consuming task. Alternatively, computational methods which can predict the best binding peptides can be used before any experimental work, followed by synthesis and testing of a tiny subset of the potential peptides. There are now several servers for MHC class II binding prediction. A recent study indicates that only certain servers perform sufficiently well to be useful and useable [13].
Combining results from multiple prediction tools often increases overall accuracy. Such a consensus strategy was proposed by Mallios [14], who combined SYFPEITHI [15], ProPred [16] and the iterative stepwise discriminant analysis meta-algorithm [17]. MULTIPRED [18] integrates hidden Markov models (HMMs) and artificial neural networks (ANN). Six MHC class II predictors were combined by Karpenko et al. [19] basing its overall prediction on the probability distributions of the different scores. Wang et al. [20] applied a consensus method to calculate the median rank of the top three predictive methods for each MHC class II protein initially evaluated so as to rank all possible 8-, 9- and 10-mers from one protein. This rank was used then to select the top 1% of peptides within each protein.
Here, we explore the effectiveness of different strategies for combining five servers for MHC class II binding prediction: ProPred [16], RANKPEP [21], NetMHCIIpan [22], NetMHCII [23], and EpiTOP [24,25]. Previous work identified these servers as the best single predictors available [26]. Our aim here is to test their combined use, with the hope of generating more accurate and more reliable overall predictions than when used individually. The servers were used in union and intersection modes. Union output compiles the results of two or three servers, while the intersection output selects only commonly predicted binders.
2. Materials and Methods
2.1. Test Set
The test set comprised 4540 binders of different length, originating from 167 proteins. The data was extracted from the immune epitope database (December 2009) [27]. The study was performed on 12 DRB1 Alleles. The peptides bind the following alleles: DRB1*0101 (2051 Binders), DRB1*0301 (190 Binders), DRB1*0401 (392 Binders), DRB1 *0404 (159 Binders), DRB1*0405 (244 Binders), DRB1*0701 (336 Binders), DRB1*0802 (153 Binders), DRB1*0901 (160 Binders), DRB1*1101 (275 Binders), DRB1*1201 (24 Binders), DRB1*1302 (243 Binders) and DRB1* 1501 (313 Binders). Some of the servers do not predict binding to all DRB1 alleles used in the test set. Only servers NetMHCIIpan and EpiTOP make predictions for all 12 DRB1 alleles. Although many methods give quantitative predictions, in our evaluation they were used as classification methods. Each server was evaluated only on the alleles it predicts. The test set is available as supporting information.
2.2. Servers Used in the Study
The five best performing servers from our preliminary study were used here (Table 1) [26]. NetMHCIIpan and NetMHCII are ANN-based servers. ProPred predicts MHC class II binding peptides using quantitative matrices (QM) based on pocket profiles [28]. RANKPEP uses position-specific scoring matrices (PSSM) which represent the observed sequence-weighted frequency of all amino acids in every position of a sequence alignment. EpiTOP is a newly developed method for MHC class II binding prediction based on proteochemometrics [25]. It is a matrix-based method which considers both peptide and protein binding-site amino acids contributions.
2.3. Union Method
The complete sequence of each protein was submitted to each server and the results recorded. The top 5% of the ranked predicted binding nonamers was used as a threshold. Two-and three-server combinations were inspected. The top 5% of the best predicted binders from each

Table 1. Servers for MHC class II binders prediction used in the present study.
server were compiled into one set and compared to the set of known binders originating from the same protein. An identified binder was considered to be any nonamer sequence available within the tested binder peptide, which may be of arbitrary but longer length. Identified binders are shown as a percentage of all binders (sensitivity). Additionally, to test the precision of prediction, a positive predictive value (PPV) was calculated as a ratio of true binders to all predicted binders included in the top 5%.
In mathematical terms, the union method corresponds to applying the logical operator OR ( ). If the predicted top 5% best binders generated by server A form set a, and the top 5% of the best binders predicted by server B form set b, then the union set c = a
). If the predicted top 5% best binders generated by server A form set a, and the top 5% of the best binders predicted by server B form set b, then the union set c = a b.
b.
2.4. Intersection Method
The same sets of the top 5% of the best predicted binders generated by each server were used in the intersection method. Intersection sets contain only common nonamer binders identified from a particular protein, as predicted by different numbers of servers. Two-, three-, four-and five-server combinations were inspected. The common binders were compared to the set of known binders originating from the same protein. An identified binder was considered to be any nonamer sequence available within the tested binder peptide, which may be of arbitrary but longer length. The final sensitivity and PPV were assessed from the number of true binders identified by two, three, four, or five servers simultaneously.
In mathematical terms, the intersection method corresponds to the logical operator AND ( ). If the predicted top 5% best binders generated by server A form set a, and the top 5% of the best binders predicted by server B form set b, the intersection set c = a
). If the predicted top 5% best binders generated by server A form set a, and the top 5% of the best binders predicted by server B form set b, the intersection set c = a b.
b.
2.5. Performance Measures
Sensitivity is the proportion of experimentally determined binders that are predicted as binders. It is defined as true positives/(true positives + false negatives). Positive predictive value (PPV) is defined as true positives / (true positives + false positives). Server performance was assessed using the sensitivity and PPV at the top 5% of the best predicted binders.
3. Results
For this assessment, servers were selected on the basis of the following criteria: computational or machine-learning method-based, free web access, and the ability to predict binding to at least 9 of the 12 HLA-DRB1 alleles considered in this study. Given these criteria, previous studies indicated that the following were the best performing servers: NetMHCIIpan, NetMHCII, ProPred, RANKPEP, and EpiTOP.
3.1. Single Predictor Performance
The performance of single predictors is shown in Figure 1. At the top 5% threshold, NetMHCIIpan and NetMHCII perform best with 55% sensitivity. ProPred, EpiTOP and RANKPEP perform almost as well with sensitivities of 46%, 45% and 44%, respectively. PPVs range from 8% for RANKPEP and EpiTOP to 10% for NetMHCIIpan, NetMHCII and ProPred.
3.2. Union Method Performance
The results of combining two or three servers are shown in Figures 2 and 3. Any union combination performs better than single predictors. At the top 5% level, sensitivity ranges from 65% to 70% in two-server combinations and from 72% to 79% in three-server combinations. However, in terms of PPV, union outputs perform poorly. The highest PPV is 8%.
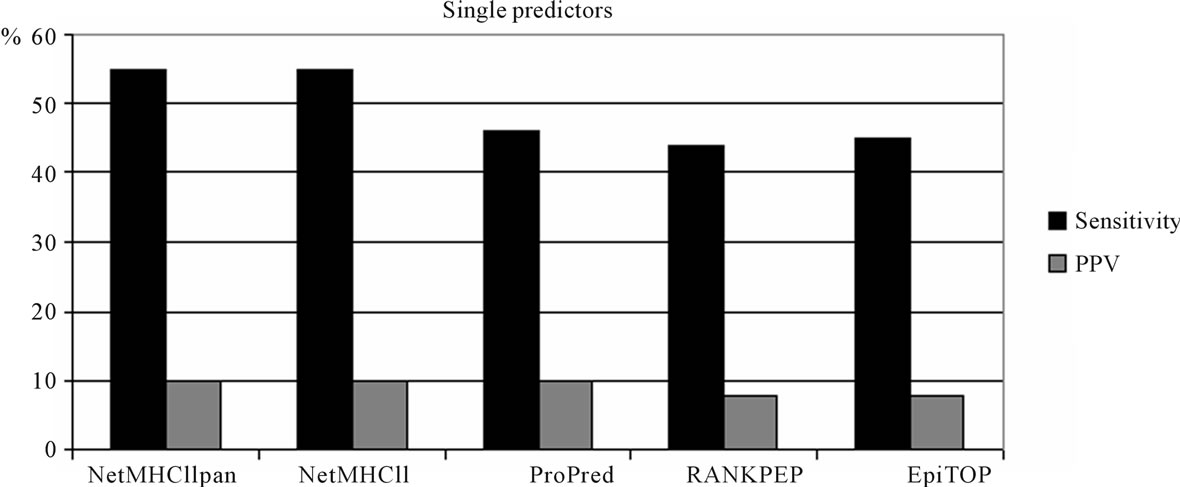
Figure 1. Single predictor performance.
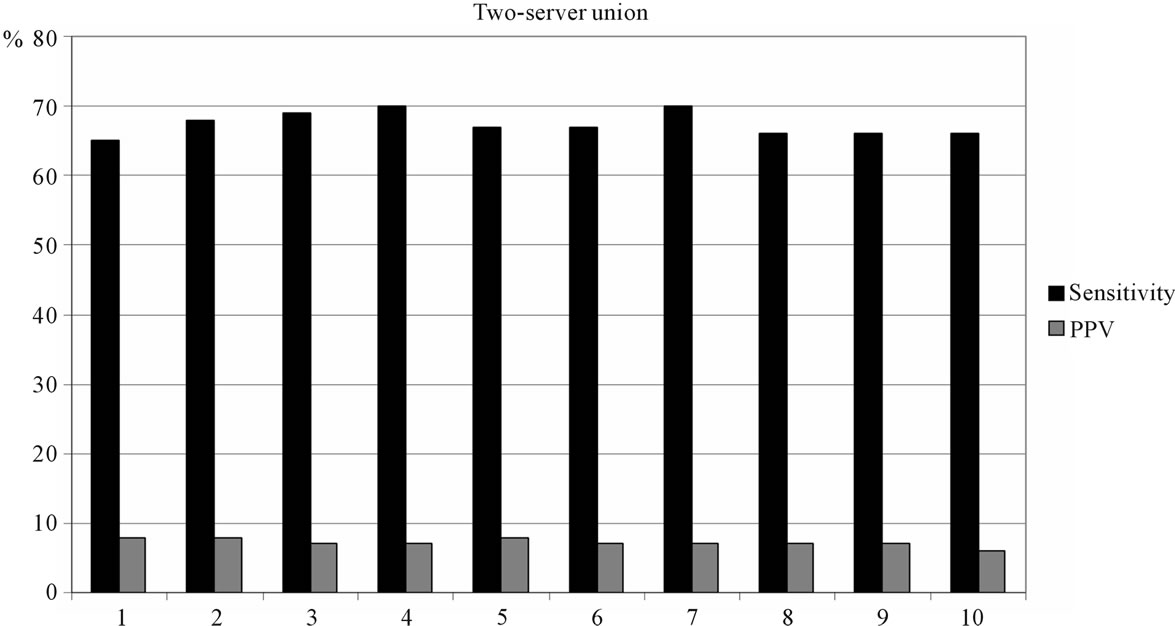
Figure 2. Two-server union method performance: 1 - NetMHCIIpan-+NetMHCII; 2 - NetMHCIIpan+ProPred; 3 - NetMHCIIpan + RANKPEP; 4 - NetMHCIIpan + EpiTOP; 5 - NetMHCII + ProPred; 6 - NetMHCII + RANKPEP; 7 - NetMHCII + EpiTOP; 8 - ProPred + RANKPEP; 9 - ProPred + EpiTOP; 10 - RANKPEP + EpiTOP.
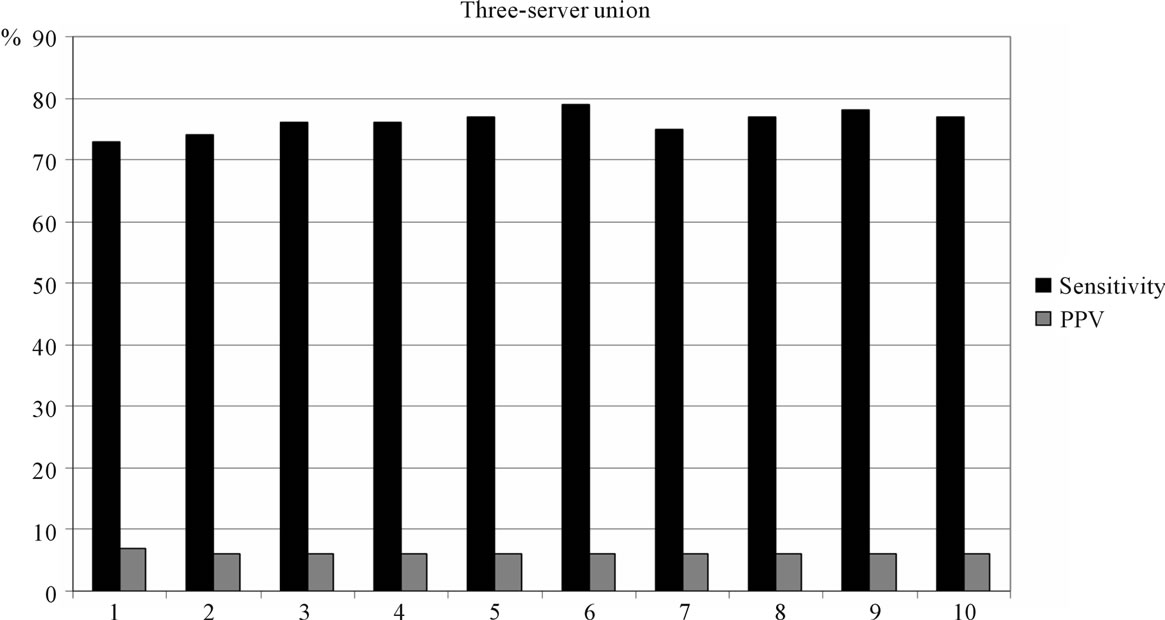
Figure 3. Three-server union method performance: 1 - NetMHCIIpan + NetMHCII + ProPred; 2 - NetMHCIIpan + NetMHCII + RANKPEP; 3 - NetMHCIIpan + NetMHCII + EpiTOP; 4 - NetMHCIIpan + ProPred + RANKPEP; 5 - NetMHCIIpan + ProPred + EpiTOP; 6 - NetMHCIIpan + RANKPEP + EpiTOP; 7 - NetMHCII + ProPred + RANKPEP; 8 - NetMHCII + ProPred + EpiTOP; 9 - NetMHCII + RANKPEP + EpiTOP; 10 - ProPred + RANKPEP + EpiTOP.
3.3. Intersection Method Performance
When servers are used in intersection mode, their sensitivities are typically poor (Figure 4). The common binders predicted by five servers identify only 4% of the known binders at the top 5% threshold. The four-server combinations recognize between 5% and 14%, threeserver combinations identify between 6% and 27%, and two-server combinations yield between 9% and 41% of the known binders. In contrast to the union mode, increasing the number of servers within the intersection decreases the sensitivity; yet the opposite is true for PPVs: increasing the number of servers increases the precision of the prediction, PPV reaches 31% for 5- server combination.
4. Discussions
In the present study, the impact on predictive accuracy of combining up to five servers for MHC class II binding
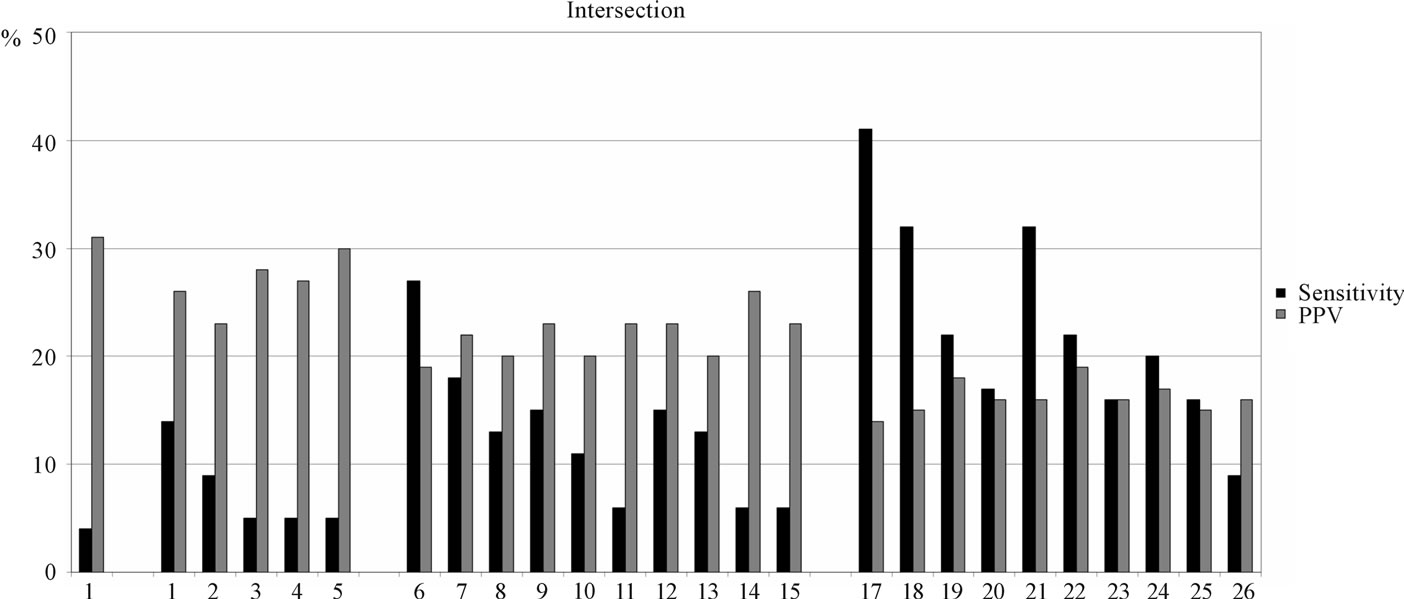
Figure 4. Intersection method performance: 1 - NetMHCIIpan + NetMHCII + ProPred + RANKPEP + EpiTOP; 2 - NetMHCIIpan + NetMHCII + ProPred + RANKPEP; 3 - NetMHCIIpan + NetMHCII + ProPred + EpiTOP; 4 - NetMHCIIpan + NetMHCII + RANKPEP + EpiTOP; 5 - NetMHCIIpan + ProPred + RANKPEP + EpiTOP; 6 - NetMHCII + ProPred + RANKPEP + EpiTOP; 7 - NetMHCIIpan + NetMHCII + ProPred; 8 - NetMHCIIpan + NetMHCII + RANKPEP; 9 - NetMHCIIpan + NetMHCII + EpiTOP; 10 - NetMHCIIpan + ProPred + RANKPEP; 11 - NetMHCIIpan + ProPred + EpiTOP; 12 - NetMHCIIpan + RANKPEP + EpiTOP; 13 - NetMHCII + ProPred + RANKPEP; 14 - NetMHCII + ProPred + EpiTOP; 15 - NetMHCII + RANKPEP + EpiTOP; 16 - ProPred + RANKPEP + EpiTOP; 17 - NetMHCIIpan + NetMHCII; 18 - NetMHCIIpan + ProPred; 19 - NetMHCIIpan + RANKPEP; 20 - NetMHCIIpan + EpiTOP; 21 - NetMHCII + ProPred; 22 - NetMHCII + RANKPEP; 23 - NetMHCII + EpiTOP; 24 - ProPred + RANKPEP; 25 - ProPred + EpiTOP; 26 - RANKPEP + EpiTOP.
prediction was evaluated. The top 5% of the best predicted binders for each server were combined using union and intersection operators. The outputs were evaluated in terms of sensitivity and positive predictive value. Union outputs showed high sensitivities (65% - 79%) and low PPVs (6% - 8%), while intersection outputs had low sensitivities (4% - 41%) but high PPVs (14% - 31%). The trade-off between sensitivity and PPV thus defines a combination. Uniting the outputs of different predictors brings more “noise” than “signal” to the resulting set of predicted binders. Conversely, selecting only the common predicted binders increases the probability of identifying true binders.
The predictive ability of any model depends strongly on the data used to derive it. Models work better interpolating between data than extrapolating from them. Generally, data used in MHC binding prediction methods fall into two categories: ligand-based and structure-based. Ligand-based data are focused on the structure of binding peptides, while structure-based data makes use of the 3D structures of target macromolecules and especially from structures of their binding sites. The nature of any analysis determines the choice of data and methods. Limitations in the quality and availability of data can have profound consequences for the efficiency and predictivity of resulting methods.
The aim of both ligand-based and structure-based MHC binding prediction is to identify viable biophores that interact with the great variety of binding sites implicit within the population of MHC molecules. In immunology, such biophores are typically called motifs. Ligand-based methods achieve this using sets of binding (and non-binding) peptides. Most of the known predictors are ligand-based, starting with motif-searching algorithms, progressing though different quantitative matrices, to more sophisticated machine-learning methods, such as ANN, HMM and SVM. Among the servers used in the present study NetMHCII and RANKPEP are ligandbased methods for MHC binding prediction.
In contrast, structure-based methods identify biophores using the structures of MHC binding sites. Sturniolo’s method based on MHC pocket profiles is an example of a structure-based method for MHC binding prediction [28]. Each MHC pocket on the binding site is determined by a set of amino acids; some are conserved, others are polymorphic. The interactions made by all natural amino acids with a given pocket establish the pocket profile. Pocket profiles are nearly independent of the remaining MHC binding site. QMs were generated for a large number of HLA class II alleles based on different combinations of pocket profiles. Among the servers used in the present study ProPred uses QMs based on Sturniolo’s pocket profiles. NetMHCIIpan and EpiTOP are mixed ligandand structure-based methods, because they consider information from both binding ligands and binding sites.
Apart from using different training sets, MHC class II binding predictors also differ in methodology. Each method can be evaluated in two ways: interpretative ability and predictive ability. The interpretation of models is a vital issue in immunoinformatics. In terms of interpretation, methods used for prediction could be classified into “easy to interpret” and “black boxes”. Motif-based search and QMs are “easy to interpret” methods, while machine-learning methods, like ANN, HMM and SVM, belong to the “black box” class. Because of many approximations, “easy to interpret” models often have moderate predictive ability, especially those using ligand -based training sets, while “black box” methods have a high capacity for identifying binders [29]. Thus there is often a trade-off between the interpretative and predictive abilities of MHC binding prediction methods. One must choose between easy to interpret but moderately predictive and highly predictive but uninterruptable methods. Using methods from the two groups in combination is a sensible compromise.
Peptides which bind to MHC proteins are extremely flexible molecules with very many low-energy conformations. Also, the binding site on class II proteins is open-ended which potentially allows a peptide to bind in several different registers [11,12]. To reduce this inherent uncertainty, MHC class II predictors use the “one binderone pattern” assumption. Another approximation used by QM methods is the additivity concept based on the hypothesis of independent binding of residues [30], which considers the binding affinity as a linear sum of the binding affinities at each peptide position. Of the servers tested, only EpiTOP avoids this assumption by including cross terms. Peptide binding to MHC molecules is neither single pattern-based, position-independent, or linear additive.
Because of its complexity, predicting MHC binding is seemingly beyond the power of a single predictor. We can seek to overcome such limitations by combining several predictors, each using different training sets and implementing different methods. In terms of the training sets used to develop the predictors, NetMHCII and RANKPEP are pure ligand-based, ProPred is pure structure-based, while NetMHCIIpan and EpiTOP are mixed methods. In terms of methodology used, ProPred, RANKPEP and EpiTOP use QMs, while NetMHCIIpan and NetMHCII use ANN. ProPred and RANKPEP apply the hypothesis of independent binding of peptide side chains, NetMHCIIpan and NetMHCII as ANN methods represent nonlinear relationships, while EpiTOP is a QM containing cross terms to capture the non-linearity of binding.
Considering all these differences between the predictors, it is not surprising that the intersection of the five servers has low sensitivity but high PPV. Single servers do not make good or bad predictions, they just make quite different predictions. Only 4% of the known binders in the test set are predicted by all five servers at the top 5% threshold. Four servers identify 14%, three servers find 27%, two servers recognize 41% of the known binders. At the same time, PPV increased with the number of servers: from 14% for two-server combinations to 31% for five-server combination. Thus, using the predictors in an intersection mode decreases the overall number of identified binders but increases the precision of prediction. Each prediction is much more likely to be correct, though many binders will be missed.
Results are quite different for the approach using data union. The sensitivity of all two-server combinations ranges from 65% to 70% at the top 5% threshold. The three-server combinations reach 79% sensitivity at the same level. Such sensitivity is currently beyond that of any single predictor. The best performing combination is NetMHCIIpan, RANKPEP and EpiTOP, followed closely by the NetMHCII, RANKPEP and EpiTOP combination with 78% sensitivity. Unfortunately, the precision of the predictions made by the union method is very low; the highest PPV is 8%. This means that increasing the number of predicted also increases false positives relative to the number of true positives, or more “noise” than “signal”.
Due to the high resource implications of experimental testing, when scanning a large proteome high numbers of false positives present a greater problem than high numbers of false negatives. Taking into account only the best predicted binders significantly reduces the number of false positives. With this in mind, three conclusions could be derived from the present study. First, combinations of different servers work better than single servers. Second, when the aim of the immunological study is to identify as many binders as possible, servers should be used in union mode. Third, when efficiency is a priority, and experimental work aims to pick out only a few, highly probable binders, server outputs should be combined using the intersection mode.
Meta-prediction is a now a well-established strategy within bioinformatic prediction [31-33]. This approach seeks to amalgamate the output of various predictors, typically internet servers, in an intelligent way so that the combined results possess greater accuracy than that of any individual predictor. Within Immunoinformatics, Trost et al. have used a heuristic method to address class I peptide-MHC binding [34], while Dai and co-workers have applied these methods to predicting peptides binding to class II MHCs [35]. Making use of such a tactic may in time prove of significant utility. In the current work, we have explored the optimality of combining server results, and largely verified the sagacity of this approach. In future work, the possibility remains to leverage the protocol we have developed in order to coalesce diverse server outputs in a similar reinforcing manner. Our work lays the solid foundation upon which to build future success. While limits exist to what computational vaccinology can achieve, it certainly offers tools and methods that can transform wider clinical and experimental endeavour. Immunoinformatic techniques are of true utilitarian value which can used by to foster and facilitate the design and discovery of vaccines, diagnostics, and reagents.
Supporting Information: The test set used in the study is given as supporting information.
5. Acknowledgements
The study was supported by a grant from the National Research Fund of Ministry of Education and Science, Bulgaria (Grant 02-1/2009).
REFERENCES
- O. Rötzschke and K. Falk, “Origin, Structure and Motifs of Naturally Processed MHC Class II Ligands,” Current Opinion in Immunology, Vol. 6, No. 1, February 1994, pp. 45-51. doi:10.1016/0952-7915(94)90032-9
- J. Robinson, M. J. Waller, P. Parham, N. De Groot, R. Bontrop, L. J. Kennedy, P. Stoehr and S. G. E. Marsh, “IMGT/HLA and IMGT/MHC: Sequence Databases for the Study of the Major Histocompatibility Complex,” Nucleic Acids Research, Vol. 31, No. 1, 2003, pp. 311- 314. doi:10.1093/nar/gkg070
- A. Dessen, C. M. Lawrence, S. Cupo, D. M. Zaller and D. C. Wiley, “X-Ray Crystal Structure of HLA-DR4 (DRA* 0101, DRB1*0401) Complexed with a Peptide from Human Collagen II,” Immunity, Vol. 7, No. 4, October 1997, pp. 473-481. doi:10.1016/S1074-7613(00)80369-6
- Z. Zavala-Ruiz, E. J. Sundberg, J. D. Stone, D. B. DeOliveira, I. C. Chan, J. Svendsen, R. A. Mariuzza and L. J. Stern, “Exploration of the P6/P7 Region of the Peptidebinding Site of the Human Class II Major Histocompatability Complex Protein HLA-DR1,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 44904-44912. doi:10.1074/jbc.M307652200
- Z. Zavala-Ruiz, I. Strug, B. D. Walker, P. J. Norris and L. J. Stern, “A Hairpin Turn in a Class II MHC-Bound Peptide Orients Residues outside the Binding Groove for T Cell Recognition,” Proceeding of National Acadademy of Sciences USA, Vol. 101, No. 36, 2004, pp. 13279-13284. doi:10.1073/pnas.0403371101
- M. M. Fernandez, R. Guan, C. P. Swaminathan, E. L. Malchiodi and R. A. Mariuzza, “Crystal Structure of Staphylococcal Enterotoxin I (SEI) in Complex with a Human Major Histocompatibility Complex Class II Molecule,” Journal of Biological Chemistry, Vol. 281, 2006, pp. 25356-25364. doi:10.1074/jbc.M603969200
- J. Hennecke and D. C. Wiley, “Structure of a Complex of the Human a/b T Cell Receptor (TCR) HA1.7, Influenza Hemagglutinin Peptide, and Major Histocompatibility Complex Class II Molecule, HLA-DR4 (DRA*0101 and DRB1*0401): Insight into TCR Cross-Restriction and Alloreactivity,” Journal of Experimental Medicine, Vol. 195, 2002, pp. 571-581. doi:10.1084/jem.20011194
- L. Deng, R. J. Langley, P. H. Brown, G. Xu, L. Teng, Q. Wang, M. I. Gonzales, G. G. Callender, M. I. Nishimura, S. L. Topalian and R. A. Mariuzza, “Structural Basis for the Recognition of Mutant Self by a Tumor-Specific, MHC Class II-Restricted T Cell Receptor,” Nature Immunology, Vol. 8, 2007, pp. 398-408. doi:10.1038/ni1447
- L. Wang, Y. Zhao, Z. Li, Y. Guo, L. L. Jones, D. M. Kranz, W. Mourad and H. Li, “Crystal Structure of a Complete Ternary Complex of TCR, Superantigen and Peptide-MHC,” Nature Structural & Molecular Biology, Vol. 14, 2007, pp. 169-171. doi:10.1038/nsmb1193
- C. A. Janeway, P. Travers, M. Walport and J. D. Capra, “Immunobiology: The Immune System in Health and Disease,” Elsevier Science Ltd., Amsterdam, 1999.
- I. A. Doytchinova and D. R. Flower, “Towards the in Silico Identification of Class II Restricted T-Cell Epitopes: a Partial Least Squares Iterative Self-Consistent Algorithm for Affinity Prediction,” Bioinformatics, Vol. 19, No. 17, 2003, pp. 2263-2270. doi:10.1093/bioinformatics/btg312
- J. C. Tong, G. L. Zhang, T. W. Tan, J. T. August, V. Brusic and S. Ranganathan, “Prediction of HLA-DQ3.2β Ligands: Evidence of Multiple Registers in Class II Binding Peptides,” Bioinformatics, Vol. 22, No. 10, 2006, pp. 1232-1238. doi:10.1093/bioinformatics/btl071
- H. H. Lin, G. L. Zhang, S. Tongchusak, E. L. Reinherz and V. Brusic, “Evaluation of MHC-II Peptide Binding Prediction Servers: Applications for Vaccine Research,” BMC Bioinformatics, Vol. 9, Supplement 12, 2008, p. S22.
- R. R. Mallios, “A Consensus Strategy for Combining HLA-DR Binding Algorithms,” Human Immunology, Vol. 64, No. 9, September 2003, pp. 852-856. doi:10.1016/S0198-8859(03)00142-3
- H. Rammensee, J. Bachmann, N. P. Emmerich, O. A. Bachor and S. Stevanovic, “SYFPEITHI: Database for MHC Ligands and Peptide Motifs,” Immunogenetics, Vol. 50, No. 3-4, 1999, pp. 213-219. doi:10.1007/s002510050595
- H. Singh and G. P. Raghava, “ProPred: Prediction of HLA-DR Binding Sites,” Bioinformatics, Vol. 17, No. 12, 2001, pp. 1236-1237. doi:10.1093/bioinformatics/17.12.1236
- R. R. Mallios, “Predicting Class II MHC/Peptide MultiLevel Binding with an Iterative Stepwise Discriminant Analysis Meta-Algorithm,” Bioinformatics, Vol. 17, No. 10, 2001, pp. 942-948. doi:10.1093/bioinformatics/17.10.942
- G. L. Zhang, A. M. Khan, K. N. Srinivasan, J. T. August and V. Brusic, “Neural Models for Predicting Viral Vaccine Targets,” Journal of Bioiformatics and Computational Biology, Vol. 3, No. 5, 2005, pp. 1207-1225. doi:10.1142/S0219720005001466
- O. Karpenko, L. Huang and Y. Dai, “A Probabilistic Meta-Predictor for the MHC Class II Binding Peptides,” Immunogenetics, Vol. 60, No. 1, 2008, pp. 25-36. doi:10.1007/s00251-007-0266-y
- P. Wang, J. Sidney, C. Dow, B. Mothe, A. Sette and B. Peters, “A Systematic Assessment of MHC Class II Peptide Binding Predictions and Evaluation of a Consensus Approach,” PLoS Computational Biology, Vol. 4, 2008, p. e1000048. doi:10.1371/journal.pcbi.1000048
- P. A. Reche, J. P. Glutting and E. L. Reinherz, “Enhancement to the RANKPEP Resource for the Prediction of Peptide Binding to MHC Molecules Using Profiles,” Immunogenetics, Vol. 56, No. 6, 2004, pp. 405-419. doi:10.1007/s00251-004-0709-7
- M. Nielsen, C. Lundegaard, T. Blicher, B. Peters, A. Sette, S. Justesen, S. Buus and O. Lund, “Quantitative Predictions of Peptide Binding to Any HLA-DR Molecule of Known Sequence: NetMHCIIpan,” PLoS Computational Biology, Vol. 4, 2008, p. e1000107.
- M. Nielsen and O. Lund, “NN-Align: An Artificial Neural Network-Based Alignment Algorithm for MHC Class II Peptide Binding Prediction,” BMC Bioinformatics, Vol. 10, 2009, p. 296. doi:10.1186/1471-2105-10-296
- I. Dimitrov, P. Garnev, D. R. Flower and I. Doytchinova, “Peptide Binding to the HLA-DRB1 Supertype: A Proteochemometrics Analysis,” European Journal of Medicinal Chemistry, Vol. 45, No. 1, January 2010, pp. 236- 243. doi:10.1016/j.ejmech.2009.09.049
- I. Dimitrov, P. Garnev, D. R. Flower and I. Doytchinova, “EpiTOP—A Proteochemometric Tool for MHC Class II Binding Prediction,” Bioinformatics, Vol. 26, No. 16, 2010, pp. 2066-2068. doi:10.1093/bioinformatics/btq324
- I. Dimitrov, P. Garnev, D. R. Flower and I. Doytchinova, “MHC Class II Binding Prediction: A Little Help from a Friend,” Journal of Biomedicine and Biotechnology, Vol. 2010, Special Issue Vaccine Informatics, 2010, Article ID705821.
- B. Peters and A. Sette, “Integrating Epitope Data into the Emerging Web of Biomedical Knowledge Resources,” Nature Reviews Immunology, Vol. 7, June 2007, pp. 485- 490. doi:10.1038/nri2092
- T. Sturniolo, E. Bono, J. Ding, L. Raddrizzani, O. Tuereci, U. Sahin, M. Braxenthaler, F. Gallazzi, M. P. Protti, F. Sinigaglia and J. Hammer, “Generation of Tissue-Specific and Promiscuous HLA Ligand Databases Using DNA Microarrays and Virtual HLA Class II Matrices,” Nature Biotechnology, Vol. 17, June 1999, pp. 555-561. doi:10.1038/9858
- D. R. Flower, “Vaccines: Data Driven Prediction of Binders, Epitopes and Immunogenicity. Bioinformatics for Vaccinology,” Wiley-Blackwell, Hoboken, 2008.
- K. C. Parker, M. A. Bednarek and J. E. Coligan, “Scheme for Ranking Potential HLA-A2 Binding Peptides Based on Independent Binding of Individual Peptide SideChains,” Journal of Immunology, Vol. 152, No. 1, 1994, pp. 163-175.
- M. Pawlowski, M. J. Gajda, R. Matlak and J. M. Bujnicki, “MetaMQAP: A Meta-Server for the Quality Assessment of Protein Models,” BMC Bioinformatics, Vol. 9, 2008, p. 403. doi:10.1186/1471-2105-9-403
- B. Wallner, P. Larsson and A. Elofsson, “Pcons.Net: Protein Structure Prediction Meta Server,” Nucleic Acids Research, Vol. 35 (Web Server Issue), 2007, pp. W369-374.
- I. Friedberg, T. Harder and A. Godzik, “JAFA: A Protein Function Annotation Meta-Server,” Nucleic Acids Research, Vol. 34 (Web Server Issue), 2006, pp. W379-381.
- O. Karpenko, L. Huang and Y. Dai, “A Probabilistic Meta-Predictor for the MHC Class II Binding Peptides,” Immunogenetics, Vol. 60, No. 1, 2008, pp. 25-36. doi:10.1007/s00251-007-0266-y
- B. Trost, M. Bickis and A. Kusalik, “Strength in Numbers: Achieving Greater Accuracy in MHC-I Binding Prediction by Combining the Results from Multiple Prediction Tools,” Immunome Research, Vol. 3, 2007, p. 5. doi:10.1186/1745-7580-3-5