Atmospheric and Climate Sciences
Vol. 2 No. 3 (2012) , Article ID: 21376 , 36 pages DOI:10.4236/acs.2012.23025
Application, Evaluation, and Process Analysis of the US EPA’s 2002 Multiple-Pollutant Air Quality Modeling Platform
Department of Marine, Earth, and Atmospheric Sciences, North Carolina State University, Raleigh, USA
Email: *yzhang9@ncsu.edu
Received May 7, 2012; revised June 1, 2012; accepted June 9, 2012
Keywords: Multi-Pollutant; Air Toxics; Model Evaluation; Process Analysis
ABSTRACT
A multiple-pollutant version of CMAQ v4.6 (i.e., CMAQ-MP) has been applied by the US EPA over continental US in 2002 to demonstrate the model’s capability in reproducing the long-term trends of ambient criteria and hazardous air pollutants (CAPs and HAPs, respectively) in support of regulatory analysis for air quality management. In this study, a comprehensive model performance evaluation for the full year of 2002 is performed for the first time for CMAQ-MP using the surface networks and satellite measurements. CMAQ-MP shows a comparable and improved performance for most CAPs species as compared to an older version of CMAQ that did not treat HAPs and used older versions of national emission inventories. CMAQ-MP generally gives better performance for CAPs than for HAPs. Max 8-h ozone (O3) mixing ratios are well reproduced in the O3 season. The seasonal-mean performance is fairly good for fine particulate matter (PM2.5), sulfate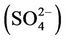 , and mercury (Hg) wet deposition and worse for other CAPs and HAPs species. The reasons for the model biases may be attributed to uncertainties in emissions for some species (e.g., ammonia (NH3), elemental carbon (EC), primary organic aerosol (POA), HAPs), gas/aerosol chemistry treatments (e.g., secondary organic aerosol formation, meteorology (e.g., overestimate in summer precipitation), measurements (e.g.,
, and mercury (Hg) wet deposition and worse for other CAPs and HAPs species. The reasons for the model biases may be attributed to uncertainties in emissions for some species (e.g., ammonia (NH3), elemental carbon (EC), primary organic aerosol (POA), HAPs), gas/aerosol chemistry treatments (e.g., secondary organic aerosol formation, meteorology (e.g., overestimate in summer precipitation), measurements (e.g.,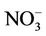 ), and the use of a coarse grid resolution. CMAQ cannot well reproduce spatial and seasonal variations of column variables except for nitrogen dioxide (NO2) and the ratio of column mass of HCHO/NO2. Possible reasons include inaccurate seasonal allocation or underestimation of emissions, inaccurate BCONs at higher altitudes, lack of model treatments such as mineral dust or plume-in-grid process, and limitations and errors in satellite data retrievals. The process analysis results show that in addition to transport, gas chemistry or aerosol/emissions play the most important roles for O3 or PM2.5, respectively. For most HAPs, emissions are important sources and cloud processes are a major sink. Simulated
), and the use of a coarse grid resolution. CMAQ cannot well reproduce spatial and seasonal variations of column variables except for nitrogen dioxide (NO2) and the ratio of column mass of HCHO/NO2. Possible reasons include inaccurate seasonal allocation or underestimation of emissions, inaccurate BCONs at higher altitudes, lack of model treatments such as mineral dust or plume-in-grid process, and limitations and errors in satellite data retrievals. The process analysis results show that in addition to transport, gas chemistry or aerosol/emissions play the most important roles for O3 or PM2.5, respectively. For most HAPs, emissions are important sources and cloud processes are a major sink. Simulated 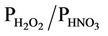 and HCHO/NO2 indicate VOC-limited chemistry in major urban areas throughout the year and in other non-urban areas in winter, but NOX-limited chemistry in most areas in summer.
and HCHO/NO2 indicate VOC-limited chemistry in major urban areas throughout the year and in other non-urban areas in winter, but NOX-limited chemistry in most areas in summer.
1. Introduction
Hazardous air pollutants (HAPs) or air toxics are the pollutants known to cause serious effects on human health, such as cardiovascular, neurological, and other organ system problems and adverse environmental issues. 188 air toxics are identified and regulated under the 1990 Clean Air Act. HAPs are emitted from a variety of sources, including large manufacturing facilities, combustion facilities, small commercial, and both onroad and nonroad mobile sources [1]. In contrast with criteria air pollutants CAPs such as O3 and PM2.5, HAPs are normally controlled by state or local air toxics monitoring programs rather than the National Ambient Air Quality Standards (NAAQS) [2]. In recent years, the US Environmental Protection Agency (EPA) has launched several programs (e.g., National Air Toxics Assessment), in order to gain a better understanding of the impacts of air toxics emissions on public health and environment and eventually strengthen the nation’s air quality management system [3]. One of the major activities as part of those programs is the development and evaluation of the 2002 multiscale multiple pollutants (MP) air quality modeling platform to integrate across the complex chemical and physical processes for MPs in a single modeling framework in support of scientific research and regulatory analysis.
The US EPA’s Models-3 Community Multiscale Air Quality (CMAQ) modeling system was developed in order to support both air quality regulatory assessments by governmental agencies and scientific studies by research institutions [4]. CMAQ has been extensively applied over a wide range of meteorological conditions and geographical areas in order to address air quality issues related to CAPs such as ozone (O3) and fine particulate matter (PM2.5) during the past decades [5-15]. However, CMAQ only simulates CAPs, which hinders its application for HAPs. There is a growing awareness that CAPs and HAPs controls should be considered together because air quality issues in many areas of the US and abroad involve both types of pollutants [2]. The assessment of the model’s capability in representing HAPs together with CAPs is critical to the development of costeffective emission control strategies for both CAPs and HAPs. Accurate modeling of this complex MP system requires that a broad range of temporal and spatial scales of MP interactions be considered simultaneously. To address this issue and further advance the “one-atmosphere” modeling capability of CMAQ, an MP version of CMAQ (referred to as CMAQ-MP hereafter) has been developed by the US EPA to simulate O3, PM2.5, mercury (Hg), and other HAPs (or air toxics) in a single model framework.
Multiple full year simulations with CMAQ-MP hereafter have been conducted by the US EPA over domains that cover the entire US or a portion of continental US (CONUS) for 2002 at different horizontal grid resolutions [3]. In this work, a comprehensive model evaluation is performed by comparing simulated concentrations of O3, PM2.5 and its components, precursors of O3 and PM2.5, major air toxics, as well as Hg deposition with ground-based and satellite measurements. Likely reasons that influence prediction biases of major pollutants are identified. The seasonal photochemical characteristics are examined and the relative contributions of controlling processes to the formation and destruction of major CAPs and HAPs are quantified through process analysis (PA) tool imbedded in CMAQ to provide important information to the development of the effective emission control strategies. The objectives of this study are to examine the capability and performance of CMAQ-MP in reproducing temporal and spatial patterns of air pollutants, quantify the contributions of major atmospheric processes to these pollutants, guide further diagnostic evaluations for model improvement and further development, and build confidence in the utilization of CMAQMP to air quality regulatory and research communities. To our best knowledge, this is the first comprehensive performance evaluation and process analysis of CMAQMP that simulates both CAPs and HAPs. Previous modeling of HAPs focus on either one species (e.g., Hg [1618] or diesel PM [19]) using a version of CMAQ with Hg (i.e., CMAQ-Hg) based on the CB05CLHG gas-phase mechanism or a subset of HAPs species (e.g., some HAPs [20] or several trace metal HAPs [21]) using a version of CMAQ for HAPs modeling based on a different gasphase mechanism (i.e., SAPRC99TX3) from that used in CMAQ-HAPs (i.e., CB05CLTX) and that used in CMAQMP (CB05TXHG). CB05TXHG combines HAPs treatments in CB05CLTX with Hg treatments in CB05CLHG, providing a comprehensive treatment for all major HAPs.
2. Model Configurations, Observational Data, and Evaluation Protocols
2.1. Model System and Configurations
CMAQ-MP has been developed by the US EPA through modifying algorithms for gas-phase chemistry, aerosols, clouds, and emissions used in the previous Hg and HAPs versions of the CMAQ (i.e., CMAQ-Hg and CMAQHAPs [22,23]) and merging them into the default CMAQ v4.6. CMAQ-MP, which has almost the same air toxics treatments as in the newer version of CMAQ v4.7 and CMAQ v5.0 in this study, includes elemental Hg (Hg0), divalent gaseous Hg (Hg(II) or Hg2), particulate Hg (PHg), 31 additional gas-phase HAPs, 6 toxic metals, and diesel PM as well as CAPs in the base version of CMAQ (details about air toxic species can be found at http://www.cmaq-model.org/cmaqwiki/index.php?title=CMAQv4.7.1_Multipollutant_Model). The chemical reactions for chlorine, Hg, and HAPs were added with the Carbon Bond Mechanism 2005 (CB05 [24]) and implemented together into CMAQ. The gas-phase mechanism of CMAQ-MP consists of 219 reactions, which include 156 reactions from base CB05 mechanism, 21 reactions for chlorine chemistry, 38 reactions for gas-phase HAPs, and 4 reactions for Hg [23]. Those reactions for HAPs and Hg mainly involve the oxidations by radicals such as hydroxyl (OH) and nitrate (NO3) radicals. A modified version of aerosol module version 4 (AERO4) also contains the treatment of sea salt emissions. The vertical diffusion module associated with aerosol emissions is updated for CMAQ-MP aerosol simulations [13]. CMAQMP uses the dry deposition module adopted from CMAQHg. The aqueous-phase chemistry of Hg is largely based on CMAQ-Hg, which includes 7 aqueousphase kinetic and 6 equilibrium reactions. The aqueous-phase chemistry for other species such as SO2 is based on the Regional Acid Deposition Model (RADM).
In this study, CMAQ-MP is applied to three annual (2002) simulations conducted by the US EPA (US EPA, 2008) over a parent domain (CONUS) at a horizontal grid resolution of 36-km and two sub-domains (portions of the eastern US (EUS) and the western US (WUS)) at a finer grid resolution of 12-km, as shown in Figure 1. The vertical resolution for each domain includes 14 layers from the surface to approximately 100 hPa (at ~15 km) using a sigma-pressure coordinate system. The height of first model layer is ~38 m. The meteorological inputs for each domain are simulated separately by the US EPA using the 5th generation PSU/ NCAR mesoscale model (MM5) v3.6.3 for the 36-km CONUS domain and MM5 v3.7.2 for the 12-km EUS domain, and by the Western Regional Air Partnership (WRAP) using MM5 v3.6.2 for the 12-km WUS domain [25]. All the three MM5 simulations are conducted with the four dimensional data assimilation (FDDA) and use the Pleim-Xiu land surface model, Asymmetric Convective Model (ACM) planetary boundary layer (PBL) parameterization schemes, and the RRTM longwave and Dudhia shortwave radiation schemes. While the EPA simulations use the Reisner I scheme for microphysics and the KainFritsch II scheme for the subgrid or cumulus convection, the WRAP simulation uses the Reisner II scheme and the Betts-Miller scheme. The MM5 hourly meteorological outputs are converted to CMAQ compatible inputs with the Meteorology-Chemistry Interface Processor (MCIP) version 3.1. The emissions are generated with the Sparse Matrix Operator Kernel Emission system (SMOKE) version 2.3 based on the EPA’s 2002 National Emissions Inventory (NEI) v3.0 for all domains. The boundary conditions (BCONs) and initial conditions (ICONs) of the 36-km domain are provided by a global chemistry transport model, GEOS-Chem [3], for key CAPs and Hg species and those of the 12-km domains are taken from the 36-km simulation. For HAPs species, BCONs of the 36-km domain for formaldehyde (HCHO) and acetaldehyde (ALD2) are also from GEOS-Chem, but those for other species are static and based on scientific literatures and available field studies [1,26]. A ten-day spin-up period from 12/22 to 12/31 2001 is used to minimize the influence of the ICONs for each simulation.
2.2. Evaluation Protocols and Observational Data
Currently the model performance evaluation for most CAPs and related variables wet depositions has been guided by US EPA [27]. However, there are no recommended performance goals or objectives for evaluating HAPs. The recommended statistics for O3 or PM2.5 may not be appropriate for air toxics. Seigneur et al. [28] indicated that the model performance for HAPs may be relatively poor due to higher uncertainties in toxics emissions than in the emissions of CAPs. In this work, an
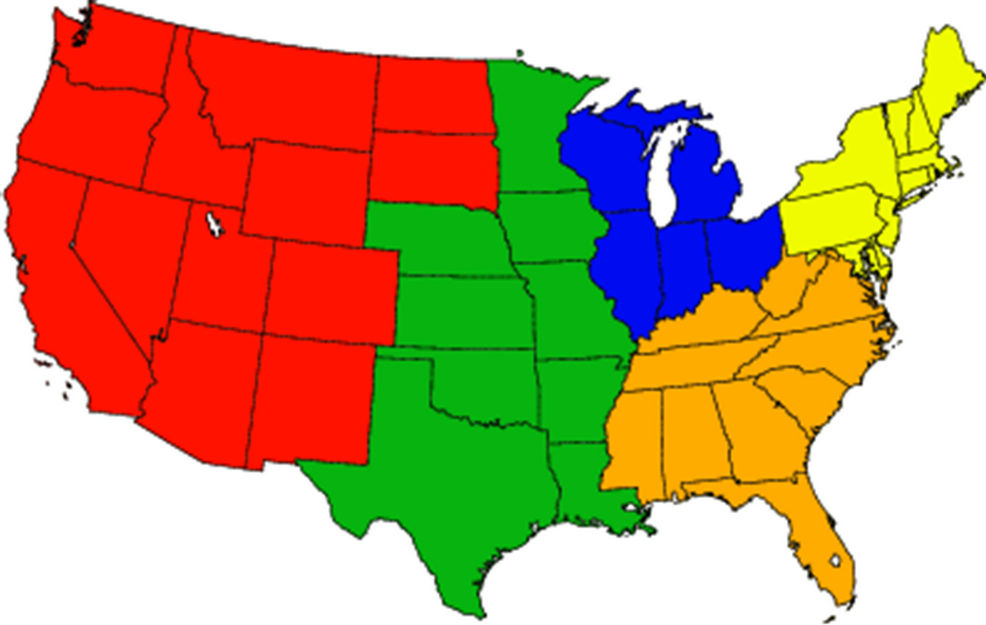
Figure 1. The CMAQ modeling domain. The black, red, and blue boxes denote domains over the 36-km continental US, the 12-km western US, and the 12-km eastern US, respectively (filled yellow, orange, blue, green, and red colors denote sub-regions northeast, southeast, Midwest, central and west for statistics).
operational model performance evaluation for O3, PM2.5 and its speciated components such as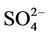 ,
, 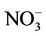 ,
, 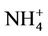 , EC, and OC, Hg wet deposition, and a selected set of HAPs is conducted using available routine surface monitoring data and satellite column data (Table 1). The surface data include those from the Clean Air Status and Trends Network (CASTNET), the Interagency Monitoring of Protected Visual Environments (IMPROVE), the Speciation Trends Network (STN), the Aerometric Information Retrieval System (AIRS)-Air Quality System (AQS), the Southeastern Aerosol Research and Characterization study (SEARCH), the National Acid Deposition Program (NADP), the Mercury Deposition Network (MDN), and the National Air Toxics Trends Stations (NATTS). Most of these networks are described in Eder and Yu [10] and Zhang et al. [6].
, EC, and OC, Hg wet deposition, and a selected set of HAPs is conducted using available routine surface monitoring data and satellite column data (Table 1). The surface data include those from the Clean Air Status and Trends Network (CASTNET), the Interagency Monitoring of Protected Visual Environments (IMPROVE), the Speciation Trends Network (STN), the Aerometric Information Retrieval System (AIRS)-Air Quality System (AQS), the Southeastern Aerosol Research and Characterization study (SEARCH), the National Acid Deposition Program (NADP), the Mercury Deposition Network (MDN), and the National Air Toxics Trends Stations (NATTS). Most of these networks are described in Eder and Yu [10] and Zhang et al. [6].
The satellite column data include the tropospheric CO columns from the Measurements of Pollution in the Troposphere (MOPITT) [29], the tropospheric NO2, HCHO columns, and their ratios (HCHO/NO2) from the Global Ozone Monitoring Experiment (GOME) [30], the tropospheric O3 residuals (TORs) from the Total Ozone Mapping Spectrometer/the Solar Backscattered Ultraviolet (TOMS/SBUV) [31], the AOD from the Moderate Resolution Imaging Spectroradiometer (MODIS) [32].
In addition to spatial plots, scatter plots, and time series plots, the model performance is examined using statistical metrics that follow Zhang et al. [6] including the mean bias (MB), correlation coefficient (R), the normalized mean bias (NMB), the normalized mean error (NME), and root mean square error (RMSE). The evaluation for surface predictions is conducted primarily using the EPA’s Atmospheric Model Evaluation Tool (AMET). AMET is a software package developed by EPA that can perform the operational evaluation of complex models. The column abundances of CO, NO2, HCHO, O3, and the ratios of column HCHO/NO2 are calculated using predicted concentrations from CMAQ and meteorologycal/ domain data (i.e., temperature, pressure, and layer thickness) from MM5 and converted into Dobson Unit (DU) for O3 and molecules·cm–2 for other species for comparison with satellite data. AODs are estimated based on CMAQ PM2.5 predictions using an empirical equation as described in Wang et al. [15] and Zhang et al. [8]. In addition, the column mass ratios of HCHO/NO2 simulated by CMAQ-MP are calculated and compared with observed ratios.
3. Evaluation of Model Performance
3.1. Meteorological Variables
Before initiating air quality simulations, it is important to identify the biases and errors associated with meteorological predictions. The MM5 model performance for 2002 MP modeling platform was evaluated separately from this study by Kemball-Cook et al. [25] and Dolwick
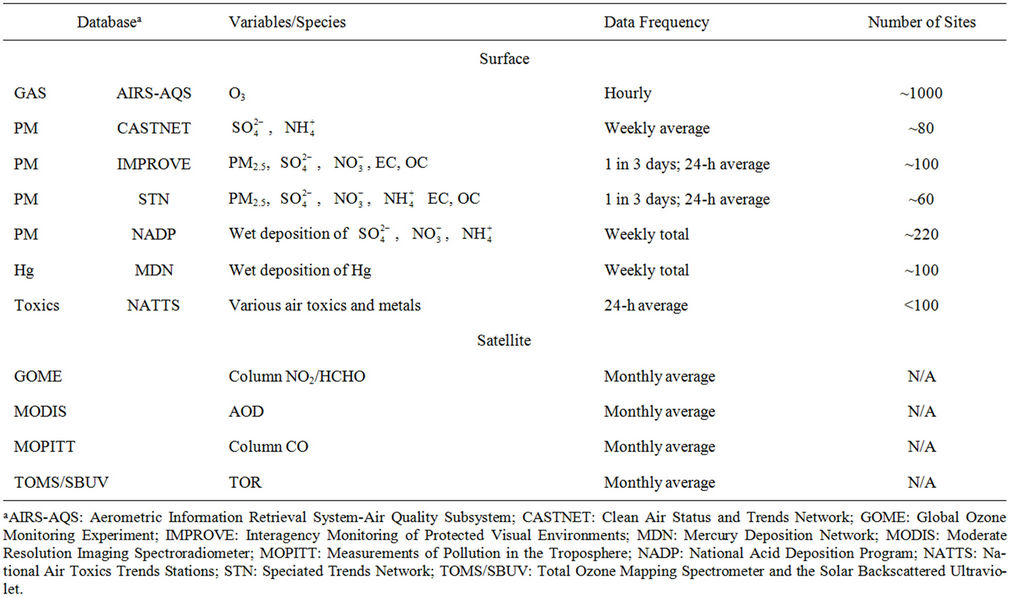
Table 1. Summary of observational databases used in the model evaluation.
et al. [33]. These evaluations show that the MM5 meteorological predictions over the three domains represent a good approximation of temperature and water vapor mixing ration with mean biases generally less than 1.5˚C and 0.1 g/kg. The model captures large-scale synoptic patterns such as high-pressure domes and upper-level troughs. However, cold bias of 2˚C - 3˚C on average exists in surface temperature predictions during winter, especially in January, from all three MM5 simulations, which may be due to the limitations of the PBL and land-surface schemes currently used in accurately simulating the air-land heat fluxes with the coarse grid resolution [25]. The effect of cold biases is the largest at night, which could overestimate the stability in the lowest layers and have a significant impact on chemical predictions [33]. MM5 is able to replicate the precipitation fairly accurately in spring, fall, and winter, but overestimates it in summer, likely due to the excessive convective cloud predicted by the model [25]. The model biases/errors for various variables over the Rocky Mountain and Great Lakes region are relatively larger than other regions due to complexity of terrains. Overall, the biases and errors associated with these meteorological simulations are generally within the range of past meteorological modeling results that have been used for air quality applications [3]. A rigorous performance testing demonstrates that the dynamic and thermodynamic fields generated by MM5 are quite sufficient for the 2002 MP modeling platform [33].
3.2. Criteria Air Pollutants at the Surface
Because of known differences between networks in terms of sampling protocols and measurement procedures, the evaluation for surface chemical predictions is conducted separately for individual network. For each network and pollutant, statistics are calculated for all sites in each domain and also with separate breakouts of five subregions (i.e., Midwest, northeast, southeast, central, and west of US) over the CONUS domain (as shown in Figure 1) or observed-predicted data pairs in monthly, seasonal, and annual averages. Since the CMAQ evaluation results for the 12-km and 36-km grids are fairly consistent especially for CAPs (see Table 2), our analyses focus primarily on CMAQ results at 36-km over CONUS in this section unless otherwise noted.
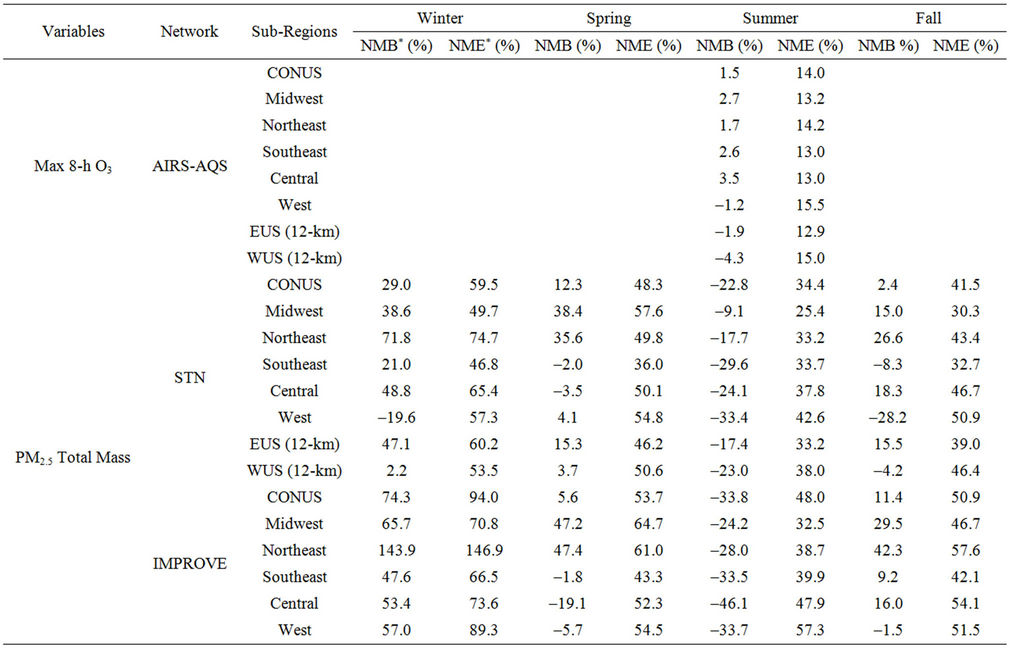
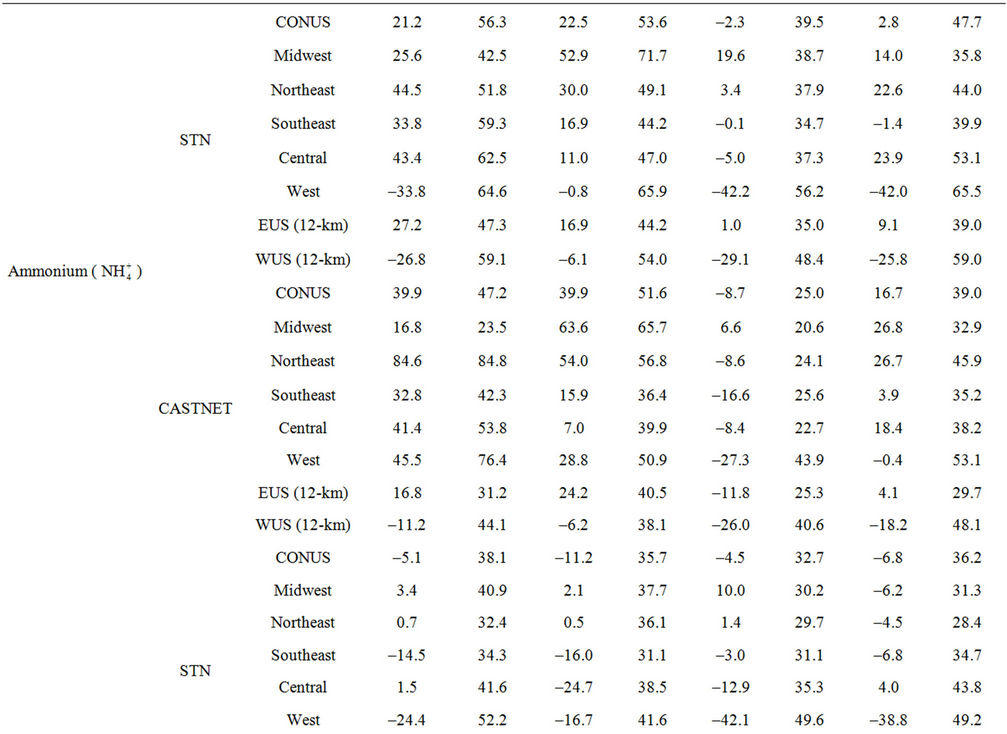
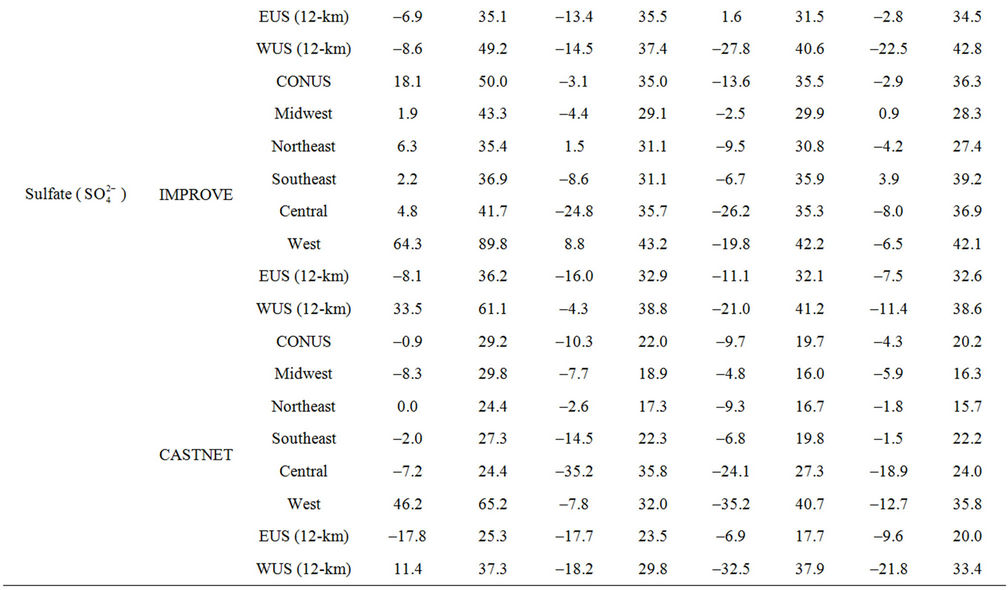
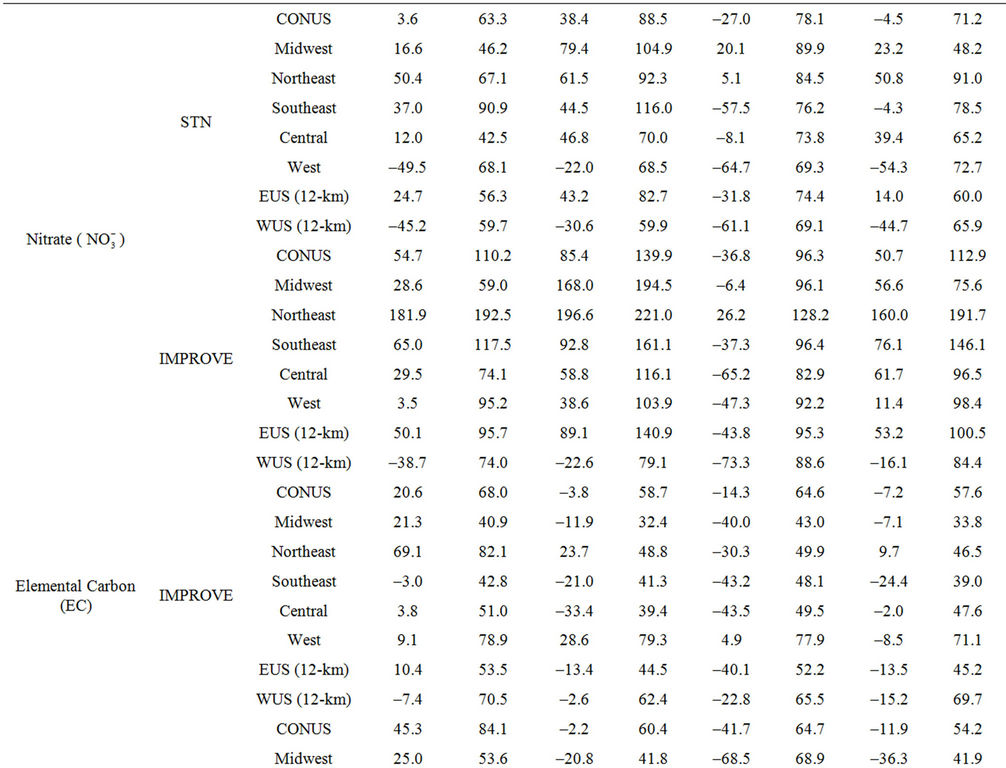
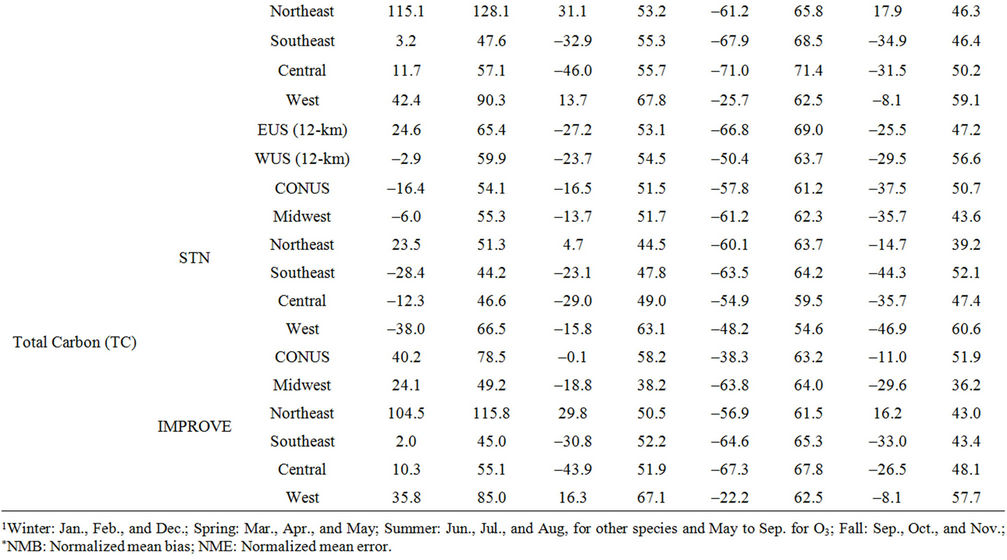
Table 2. Seasonal-mean model performance statistics for criteria air pollutants over CONUS and its 5 other sub-regions from the 36-km simulation and EUS and WUS from the 12-km simulation in 20021.
3.2.1. Ozone
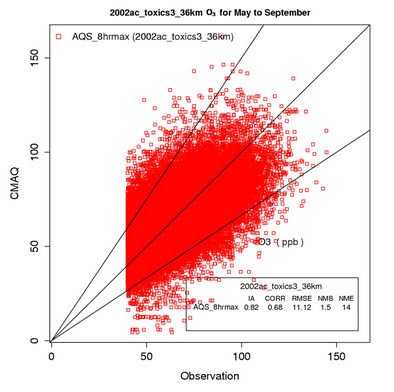
Figure 2(a) shows scatter plot of modeled and observed daily max 8-h O3 with a cut off value of 40 ppb (i.e., data pairs containing observed mixing ratios less or equal to 40 ppb are not used in the analysis) for the O3 season (i.e., May to September). As shown, CMAQ simulates max 8-h O3 mixing ratios quite well with R, NMB, and NME of 0.7%, 1.5%, and 14%, respectively and a vast majority of values within a factor of 1.5 of observations. Figure 2(b) shows the box plot of 25% and 75% quartiles (shading regions) along with the median values for diurnal O3 values during the entire O3 season at the AQS sites (i.e., urban and suburban areas) across the entire domain. As shown, the median simulated O3 values are fairly close to observations between 10:00 and 19:00, despite a systematic overprediction of nighttime and early morning O3. These findings are consistent with previous studies [10,12]. Although the capability of CMAQ in simulating nighttime O3 has been improved with an updated parameterization of the minimum KZ since CMAQ v4.5 (see release note at http://www.cmascenter.org/help/model_docs/cmaq/4.5/RELEASE_NOTES.txt), accurately simulating the evolution of nocturnal boundary layer remains difficult due to limitations of PBL and land-surface schemes in current models, and the use of a coarse horizontal resolution and vertical resolution in lower portion of PBL (e.g., ~38 m in depth for surface layer in this work).
Figures 2(c)-(e) show the spatial distribution of NMBs for max 8-h O3 with a cutoff value of 40 ppb over the CONUS at 36-km and WUS and EUS domains at 12-km for the O3 season in 2002. For CONUS at 36-km, CMAQ shows a good performance to capture the spatial variation of max 8-h O3 mixing ratios with NMBs of within ±10% and NMEs of less than 15% over majority (>80%) of AQS sites based on the suggested performance criteria by other studies [6,34]. CMAQ tends to moderately overpredict O3 mixing ratios along some coastal regions with NMBs of 20% - 30% (e.g., New England coast and Florida coast) and sometimes >30% (e.g., along Pacific coast in California). This can be attributed to a poor representation of coastal boundary layers [35,36]. There are also several small clusters of overpredictions (with NMBs ³20%) in the Midwest and southeastern US and a cluster of underpredictions (with NMBs of –25% to –15%) in some areas in southern California and Arizona. These large NMBs are likely due to the fact that the use of a coarse grid resolution of 36-km cannot accurately represent precursor emissions and elevated and/or complex terrains over those regions. As shown in Figures 2(d)-(e), the simulations at 12-km over WUS and EUS give lower NMBs over those regions (e.g., NMBs of –10% - 0% in most of the two domains).
Figure 2(f) shows the MBs for daily max 8-h O3 for the O3 season binned for the range of observed O3 values. CMAQ tends to reproduce O3 mixing ratios the best in the range of 40 - 60 ppb with MBs within 5 ppb but significantly underpredicts high mixing ratios (>80 ppb) and overpredicts low mixing ratios of O3 (<40 ppb). Those observed low O3 mixing ratios typically coincide with non-conducive meteorological conditions (e.g., high cloud cover and precipitation and cool temperature). The overestimation of low observed mixing ratios of O3 is due in part to the poor performance of CMAQ in simulating the nighttime O3 as discussed above. As shown in Tables 2 and 3, CMAQ shows overall excellent performance with very small domain-wide NMBs of 1.5%, 1.7%, 2.6%, 2.7%, 3.5%, and –1.2% for CONUS and its subregions including northeast, southeast, Midwest, central, and west US, respectively, during O3 season. The NMBs are –1.9% over EUS and –4.3% over WUS from the 12-km simulations. Although the discrepancies still exist for modeled and observed O3 mixing ratios, the results in this study demonstrate a moderate to significant improvement as compared with previous studies [8,10,12, 36] because of several factors. First, a relatively new version of CMAQ v4.6 with the newest CB05 chemical mechanism plus additional chloride reactions is used. Recent studies by Luecken et al. [37] and Yu et al. [38] showed that CB05 performs better in reproducing high O3 mixing ratios, especially in summer when compared with CB-IV and Statewide Air Pollution Research Center mechanism (SAPRC99) due to several updates in chemical species, reactions, and reaction rates. Second, a new option of PBL scheme, ACM2, is available in CMAQ v4.6 and used in this study. ACM2 includes both eddy diffusion and nonlocal schemes from the original ACM, which enables ACM2 to better represent the rise and fall of the convective boundary layer. Appel et al. [12] also compared O3 performance of CMAQ v4.5 with CMAQ v4.6 both with CB05 and found a better overall performance for max 8-h O3 by CMAQ v4.6, potentially due to the use of ACM2. Finally, the emission inventory used in this work is based on NEI 2002 v3, which represents the most comprehensive emission inventory upon its release and is more accurate than those used in previous studies.
3.2.2. PM2.5 and Its Compositions
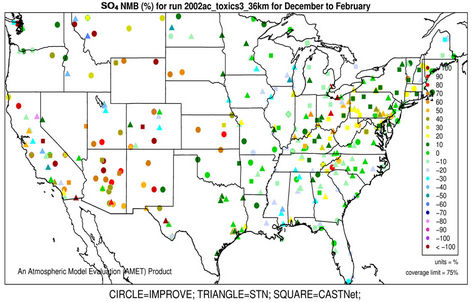
3.2.2.1. Sulfate Figures 3(a)-(b) show the spatial plots of NMBs for 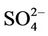 over the IMPROVE, STN, and CASTNET sites for winter (Jan., Feb., and Dec.) and summer (Jun., Jul., and Aug.) 2002 from the 36-km simulations over CONUS. In both winter and summer, CMAQ performs better over the eastern US than the western US with most of NMBs within ±20%. This is especially true in
over the IMPROVE, STN, and CASTNET sites for winter (Jan., Feb., and Dec.) and summer (Jun., Jul., and Aug.) 2002 from the 36-km simulations over CONUS. In both winter and summer, CMAQ performs better over the eastern US than the western US with most of NMBs within ±20%. This is especially true in
 (a)
(a)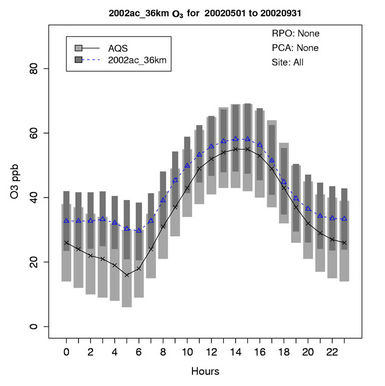 (b)
(b)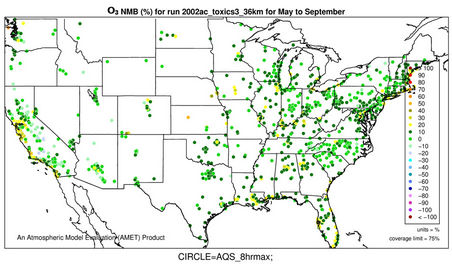 (c)
(c)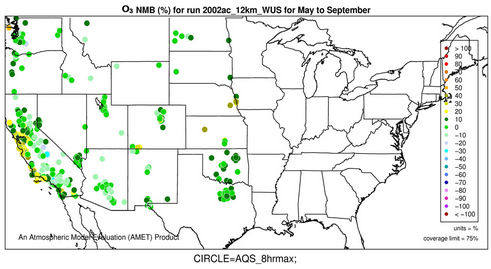 (d)
(d)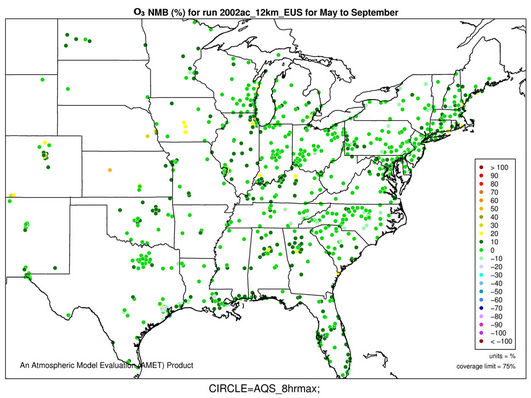 (e)
(e)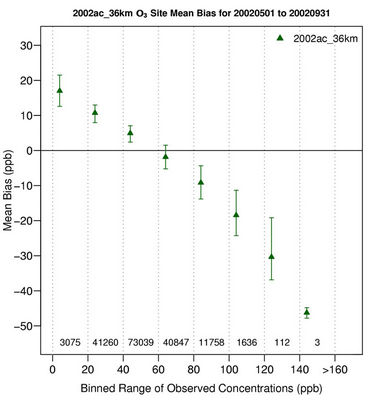 (f)
(f)
Figure 2. Comparison of the simulated and observed O3 concentrations at the AIRS-AQS monitoring sites during O3 season (i.e., May to September) in 2002. (a) Scatter plot of daily max 8-h O3 with a cut off value of 40 ppb (the 1:1, 1.5:1 and 1:1.5 lines are shown for reference); (b) Box plot of diurnal variation of median (the cross sign denotes AQS and the triangle sign denotes CMAQ) and inter-quartile ranges (light and dark shading denote AQS and CMAQ, respectively) for hourly average O3; Spatial distributions of NMBs for daily max 8-h O3 from (c) The 36-km simulation over CONUS, and the 12-km simulations over (d) WUS; and (e) EUS; (f) Median and inter-quartile range of MB binned by observed concentrations of daily max 8-h O3. The numbers above the X axis indicate the number of simulated/observed data pairs for each concentration bin.
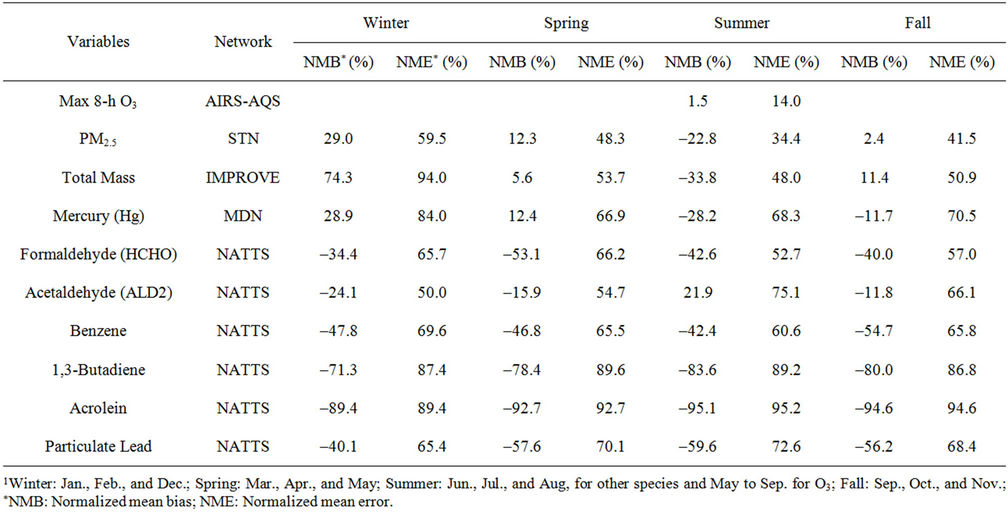
Table 3. Seasonal-mean model performance statistics for max 8-h O3, PM2.5, and selected hazardous air pollutants over CONUS in 20021.
summer when 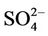 contributes the most to total PM2.5 mass concentrations in the eastern US, likely as the results of a better representation of emissions of SO2 and
contributes the most to total PM2.5 mass concentrations in the eastern US, likely as the results of a better representation of emissions of SO2 and 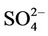 in the eastern US. Compared to the 2001 NEI that significantly underestimates SOX emissions in California (CA) [39] and possibly in other states in the western US during summer, the 2002 NEI showed much higher emissions in those regions in both summer and winter, indicating that the large negative NMBs (–60% to –20%) in
in the eastern US. Compared to the 2001 NEI that significantly underestimates SOX emissions in California (CA) [39] and possibly in other states in the western US during summer, the 2002 NEI showed much higher emissions in those regions in both summer and winter, indicating that the large negative NMBs (–60% to –20%) in 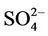 predictions in the western US are unlikely caused by underestimation in SOX emissions in summer but the large positive NMBs (30% - 100%) in this region may be caused by possible overestimation in SOX emissions in winter.
predictions in the western US are unlikely caused by underestimation in SOX emissions in summer but the large positive NMBs (30% - 100%) in this region may be caused by possible overestimation in SOX emissions in winter.
Table 2 summarizes the overall seasonal statistical performance of CMAQ for all PM2.5 species including 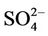 over different networks and sub-regions from three domains (i.e., CONUS, EUS, and WUS). The performance for
over different networks and sub-regions from three domains (i.e., CONUS, EUS, and WUS). The performance for  is the best among all PM2.5 species, with domain-wide NMBs typically within ±18% in different seasons in all sub-regions except for regions “Central” and “West” from the 36-km simulation and the region “WUS” from the 12-km simulation. The NMEs are moderate, ranging from 20% to 50% throughout the year. Several studies that used the 2001 NEI reported that CMAQ v4.4 underpredicted
is the best among all PM2.5 species, with domain-wide NMBs typically within ±18% in different seasons in all sub-regions except for regions “Central” and “West” from the 36-km simulation and the region “WUS” from the 12-km simulation. The NMEs are moderate, ranging from 20% to 50% throughout the year. Several studies that used the 2001 NEI reported that CMAQ v4.4 underpredicted 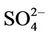 in winter and spring, overpredicted it in fall, either overpredicted or slightly underpredicted it in summer [8,11,13]. In contrast, our results show that CMAQ underestimates
in winter and spring, overpredicted it in fall, either overpredicted or slightly underpredicted it in summer [8,11,13]. In contrast, our results show that CMAQ underestimates 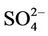 in almost all the seasons (particularly in summer) and overestimates it over the IMPROVE sites in winter, which are more consistent with the study of Luo et al. [40] that used CMAQ v4.7. This discrepancy is likely due to the updates in both convective cloud module and aerosol dry deposition module in CMAQ v4.6. Appel et al. [13] indicated that the use of ACM2-cloud scheme (in CMAQ v4.6 or later) over the RADM-cloud scheme (in CMAQ v4.4) may result in less aqueous production of
in almost all the seasons (particularly in summer) and overestimates it over the IMPROVE sites in winter, which are more consistent with the study of Luo et al. [40] that used CMAQ v4.7. This discrepancy is likely due to the updates in both convective cloud module and aerosol dry deposition module in CMAQ v4.6. Appel et al. [13] indicated that the use of ACM2-cloud scheme (in CMAQ v4.6 or later) over the RADM-cloud scheme (in CMAQ v4.4) may result in less aqueous production of 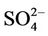 and the changes in aerosol dry deposition calculation in the new version of CMAQ may lead to higher dry deposition velocity and hence more
and the changes in aerosol dry deposition calculation in the new version of CMAQ may lead to higher dry deposition velocity and hence more 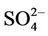 removal. Moreover, the CMAQ model bias in this study can be partially explained by the errors of MM5 in the predictions of precipitation and wet depositions. For example, MM5/ CMAQ tends to overestimate domain-wide precipitation and wet deposition of
removal. Moreover, the CMAQ model bias in this study can be partially explained by the errors of MM5 in the predictions of precipitation and wet depositions. For example, MM5/ CMAQ tends to overestimate domain-wide precipitation and wet deposition of 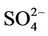 with NMBs of 42.5% and 13.1%, respectively, in summer and underestimate them with NMBs of –13.0% and –31.1%, respectively, in winter (figures not shown). Further, Luo et al. [40] reported that the convective precipitating cloud fraction and cloud water contents have been overestimated by CMAQ v4.6, which leads to an excessive scavenging of
with NMBs of 42.5% and 13.1%, respectively, in summer and underestimate them with NMBs of –13.0% and –31.1%, respectively, in winter (figures not shown). Further, Luo et al. [40] reported that the convective precipitating cloud fraction and cloud water contents have been overestimated by CMAQ v4.6, which leads to an excessive scavenging of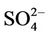 .
.
3.2.2.2. Nitrate Similar spatial plots of NMBs for 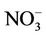 at the IMPROVE and STN sites are shown in Figures 3(c)-(d). In winter, CMAQ tends to overpredict
at the IMPROVE and STN sites are shown in Figures 3(c)-(d). In winter, CMAQ tends to overpredict 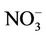 concentrations in the eastern US where NMBs often exceed 20% and it tends to underpredict in most of the western US. In summer, underpredictions of
concentrations in the eastern US where NMBs often exceed 20% and it tends to underpredict in most of the western US. In summer, underpredictions of 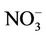 occur over almost all the CONUS domain. As shown in Table 2, CMAQ
occur over almost all the CONUS domain. As shown in Table 2, CMAQ
 (a)
(a)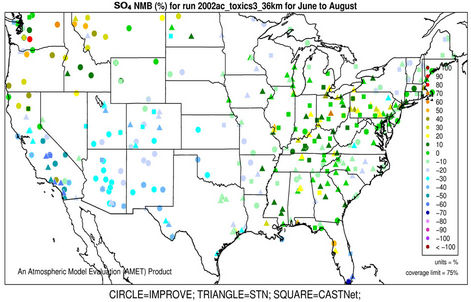 (b)
(b)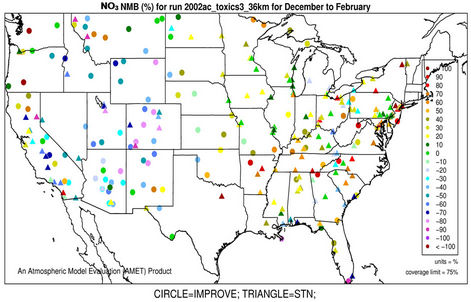 (c)
(c)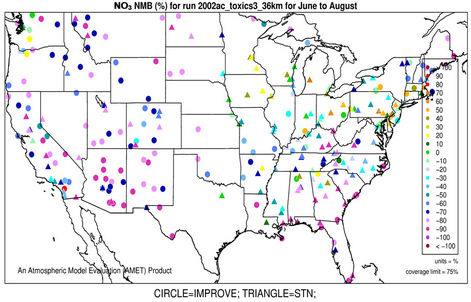 (d)
(d)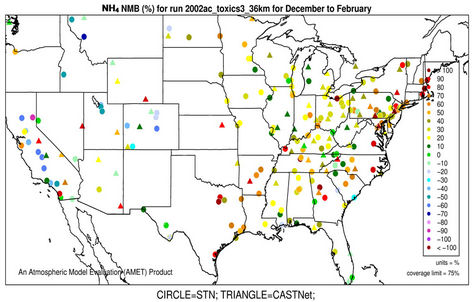 (e)
(e)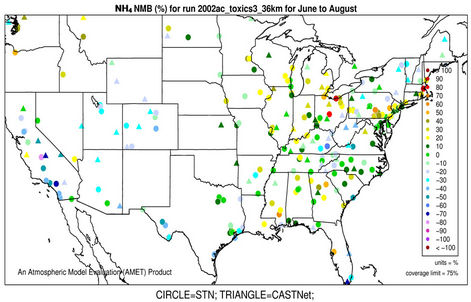 (f)
(f)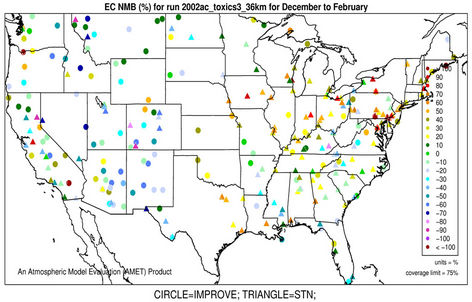 (g)
(g)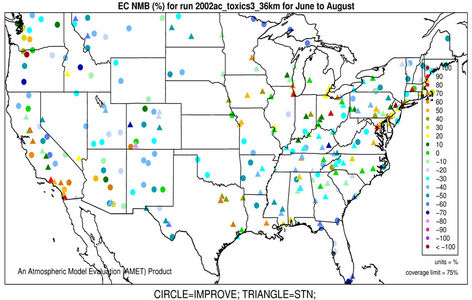 (h)
(h)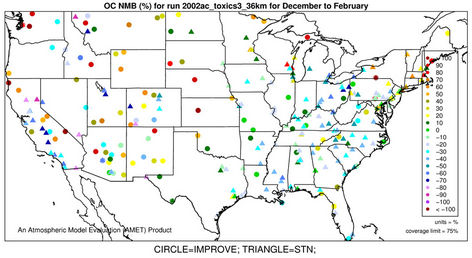 (i)
(i)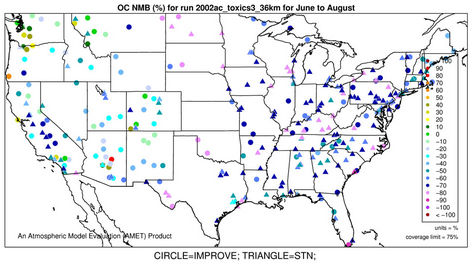 (j)
(j)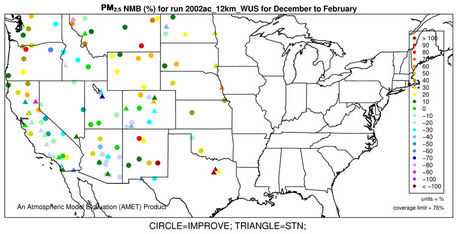 (k)
(k)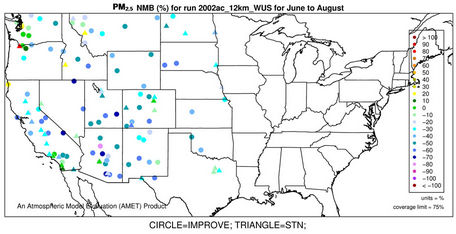 (l)
(l)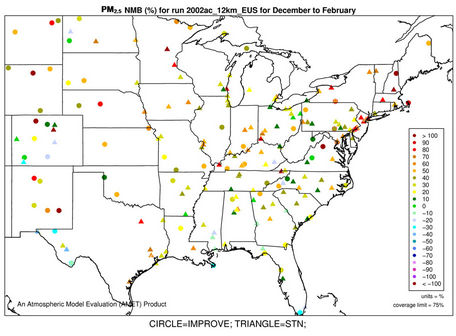 (m)
(m)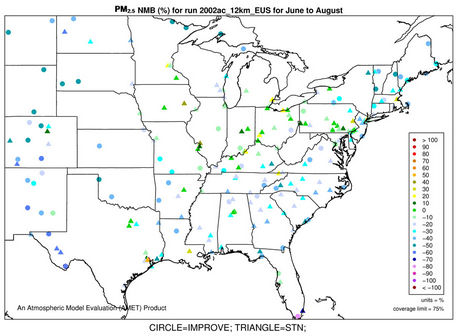 (n)
(n)
Figure 3. Spatial plots of NMBs for 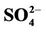 ((a) and (b)),
((a) and (b)),  ((c) and (d)),
((c) and (d)), 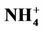 ((e) and (f)), EC ((g) and (h)), and OC ((i) and (j)) from the 36-km simulation over CONUS, and PM2.5 ((k)-(n)) from the 12-km simulation over WUS and EUS for winter (left panel) and summer (right panel) 2002.
((e) and (f)), EC ((g) and (h)), and OC ((i) and (j)) from the 36-km simulation over CONUS, and PM2.5 ((k)-(n)) from the 12-km simulation over WUS and EUS for winter (left panel) and summer (right panel) 2002.
performance for 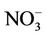 is much worse than that for
is much worse than that for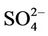 . NMBs and NMEs are much larger. Domain-wide NMBs and NMEs can be up to 85.4% and 139.9%, respectively. The model biases may be partially associated with the uncertainties in NH3 emissions, which are more rudimentary than those of other species such as NOX, particularly in its monthly variation that is poorly characterized [41]. Model performance with respect to
. NMBs and NMEs are much larger. Domain-wide NMBs and NMEs can be up to 85.4% and 139.9%, respectively. The model biases may be partially associated with the uncertainties in NH3 emissions, which are more rudimentary than those of other species such as NOX, particularly in its monthly variation that is poorly characterized [41]. Model performance with respect to 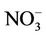 in this study suggests that NH3 emissions based on the 2002 NEI v3 are probably too low for summer and too high for other seasons, which was reported for previous versions of NEI in the literature [6] but remains in the 2002 NEI v3. The more accurate monthly-derived NH3 emissions by Carnegie Mellon University NH3 emission model are much higher in summer and lower in winter compared to the traditional NH3 emission inventories [14,42] and can be used to improve model performance in the future. Other important reasons include the high uncertainties in gas/particle partitioning simulated by ISORROPIA in CMAQ and the biases in the predictions of
in this study suggests that NH3 emissions based on the 2002 NEI v3 are probably too low for summer and too high for other seasons, which was reported for previous versions of NEI in the literature [6] but remains in the 2002 NEI v3. The more accurate monthly-derived NH3 emissions by Carnegie Mellon University NH3 emission model are much higher in summer and lower in winter compared to the traditional NH3 emission inventories [14,42] and can be used to improve model performance in the future. Other important reasons include the high uncertainties in gas/particle partitioning simulated by ISORROPIA in CMAQ and the biases in the predictions of 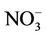 wet deposition fluxes. CMAQ tends to underestimate the
wet deposition fluxes. CMAQ tends to underestimate the  wet deposition amounts throughout the whole year with NMBs of –23.2%, –25.7%, –42.8%, and –19.2% for winter, spring, summer, and fall, respectively. Finally, the large model biases and errors in
wet deposition amounts throughout the whole year with NMBs of –23.2%, –25.7%, –42.8%, and –19.2% for winter, spring, summer, and fall, respectively. Finally, the large model biases and errors in 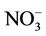 predictions could also be due in part to the uncertainties in the measurements, especially in summer. In fact, both modeled and observed
predictions could also be due in part to the uncertainties in the measurements, especially in summer. In fact, both modeled and observed 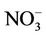 concentrations are very low in summer and the model biases are comparable to the uncertainty level (roughly ±0.5 μg∙m–3) of filter-based routine measurements [43].
concentrations are very low in summer and the model biases are comparable to the uncertainty level (roughly ±0.5 μg∙m–3) of filter-based routine measurements [43].
3.2.2.3. Ammonium As shown in Figures 3(e)-(f), since 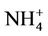 in the ambient atmosphere is generally present as (NH4)2SO4 (or NH4HSO4) and NH4NO3, the spatial pattern of
in the ambient atmosphere is generally present as (NH4)2SO4 (or NH4HSO4) and NH4NO3, the spatial pattern of 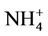 is more like the combined pattern of
is more like the combined pattern of 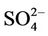 and
and 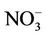 in winter when both (NH4)2SO4 and NH4NO3 concentrations are high and more similar to that of
in winter when both (NH4)2SO4 and NH4NO3 concentrations are high and more similar to that of 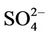 in summer when (NH4)2SO4 is dominant. In winter, CMAQ overpredicts
in summer when (NH4)2SO4 is dominant. In winter, CMAQ overpredicts 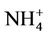 concentrations in the eastern US where NMBs often range from 20% to 60%. It underpredicts NH4+ concentrations in the most of the western US, where NMBs range from –60% to –20%. In summer, CMAQ shows a better performance over space, with slight overpredictions of
concentrations in the eastern US where NMBs often range from 20% to 60%. It underpredicts NH4+ concentrations in the most of the western US, where NMBs range from –60% to –20%. In summer, CMAQ shows a better performance over space, with slight overpredictions of 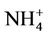 over the eastern US (with most NMBs of 0 to 40%) and slight underpredictions over the western US (with most NMBs of –40% to –20%), because
over the eastern US (with most NMBs of 0 to 40%) and slight underpredictions over the western US (with most NMBs of –40% to –20%), because 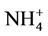 is dominant by (NH4)2SO4 and the performance of
is dominant by (NH4)2SO4 and the performance of 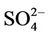 in summer is much better than that of
in summer is much better than that of 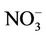 in winter. Table 2 shows a fairly good performance for
in winter. Table 2 shows a fairly good performance for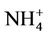 , which is slightly worse than
, which is slightly worse than 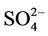 but better than
but better than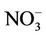 . The domain-wide NMBs and NMEs range from –2.3% to 39.9% and 25.0% to 56.3%, respectively, over different networks in different seasons. The statistics are consistent between STN and CAST-NET, with domain-wide positive NMBs in most seasons except for summer over both networks. As discussed above, the uncertainty associated with NH3 emissions is indicative of the main reason for the model bias of
. The domain-wide NMBs and NMEs range from –2.3% to 39.9% and 25.0% to 56.3%, respectively, over different networks in different seasons. The statistics are consistent between STN and CAST-NET, with domain-wide positive NMBs in most seasons except for summer over both networks. As discussed above, the uncertainty associated with NH3 emissions is indicative of the main reason for the model bias of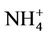 . Additionally, the underestimation of
. Additionally, the underestimation of 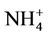 wet depositions throughout the whole year (NMBs are –47.5%, –26.4%, –8.9%, and –22.2% for winter, spring, summer, and fall, respectively) can explain in part the overestimation of NH4+ in most seasons.
wet depositions throughout the whole year (NMBs are –47.5%, –26.4%, –8.9%, and –22.2% for winter, spring, summer, and fall, respectively) can explain in part the overestimation of NH4+ in most seasons.
3.2.2.4. Elemental and Organic Carbon As shown in Figures 3(g)-(h), CMAQ moderately overpredicts EC at the IMPROVE and STN sites in the eastern US with NMBs generally between 20% and 50% and underpredicts it in the western US with NMBs between –50% and –20% in winter. As shown in Figures 3(i)-(j), CMAQ has the tendency to overpredict OC over the western US, Midwest, and New England areas especially for the IMPROVE sites. Underpredictions are also evident over most STN sites in the eastern US. While in summer, significant underpredictions are observed across the whole domain, particularly over the eastern US and the worst NMBs normally occurring over the STN sites.
The model seems to perform slightly better in winter (colder months) than summer (warmer months).
The seemingly worse performance at the STN sites for OC is due to the fact that the measurements are not blank corrected for carbon on the background filter (i.e., removing the adventitious carbon from the filter), which could add 20% - 40% to the observed OC concentrations [43]. In addition to that, STN BC and OC measurements use a different thermo-optical protocol compared with the IMPROVE network that may cause larger uncertainties in splitting BC/OC [39]. Therefore, only OC and EC concentrations from IMPROVE are used to evaluate the model performance in Table 2. As shown, CMAQ appears to moderately underpredict OC in summer and overpredict it in winter with domain-wide NMBs of –41.7% and 45.3%, respectively, and slightly underpredict in both spring and fall with NMBs of –2.2% and –11.9%, respectively. The errors associated with OC are relatively high with domain-wide NMEs ranging from 54.2% to 84.1%. On the other hand, the overall model performance for EC is much better than OC. The model performance for OC and EC shown here is somewhat similar to that of Tesche et al. [11] and Appel et al. [13], in which they also found that the largest underpredictions of OC and EC occur in the summer and fall. However, the overprediction of OC and EC in winter is more consistent with Karydis et al. [43]. Since the major component of organic aerosols in winter is POA. Both POA and EC are mainly affected by emissions, vertical mixing and deposition. The overprediction of OC and EC in winter is believed to be more related to the poor model representations of those processes. Some studies [10,44] indicated that the poor temporally-allocated wildfire emissions may contribute to the biases in OC and EC predictions. Several studies based on 1999 NEI v3 indicated underestimation in wildfire emissions [8,45]. The emissions of EC and POA from the 2002 NEI v3 are much lower than those from the 1999 v3, particularly in the western US, indicating a possible underestimation in wildfire emissions to a greater extent than the 1999 v3 in this region, due likely to the use of older fuel loading information (George Pouliot, US EPA, personal communication, 2011). During the summer months when SOA concentrations are more comparable with those of POA, the model underpredictions for OC could also be attributed to the underpredictions of photochemically-produced SOA aside from the uncertainties in the emissions of POA and SOA precursors [13]. This partly explains the worse model performance of OC, as compared with EC in summer. CMAQ v4.6 does not simulate SOA formation from the oxidation of several important precursors such as isoprene and sesquiterpenes, both of which may contribute substantially to the ambient OC concentrations [7,41]. In addition, the uncertainties associated with the measurement techniques of carbonaceous aerosols (e.g., OC and EC split) and the factor used to convert simulated organic matter (OM) to OC may also cause the discrepancies between simulations and observations [13].
3.2.2.5. PM2.5
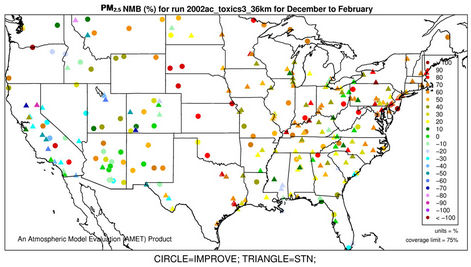
The accuracy of PM2.5 predictions in CMAQ is a composite of the accuracies of predictions of individual particulate species concentrations. Figures 4(a)-(b) and 3(k)- (n) show the spatial plots of NMBs for PM2.5 at the IMPROVE and STN sites from the 36-km simulation over CONUS and the 12-km simulations over WUS and EUS, respectively. CMAQ overpredicts PM2.5 in winter and underpredicts it in summer for all domains. In winter, the spatial variability of biases is more evident. The relatively high biases occur over the northeastern US, Great Lakes, and Midwest with NMBs generally >50% at 36-km and >30% at 12-km, indicating some improvements using a finer grid resolution of 12-km. The underprediction of PM2.5 in summer is more systematic with more than 95% of sites having negative biases. NMBs are typically larger (between –60% and –20%) in the western US than in the eastern US (–40% to –10%) at 36-km, with some improvement at the 12-km.
As shown in Figures 4(c)-(d), both CMAQ and observations show higher monthly PM2.5 concentrations at the STN sites than the IMPROVE sites throughout the year because most of the IMPROVE sites are located in remote and rural areas and the STN sites are located in more polluted urban areas. CMAQ underpredicts PM2.5 concentrations during the warmer months (i.e., April through September at the IMPROVE sites and May through August at the STN sites) but overpredicts during the colder months. Figures 4(e)-(f) show the stacked bar charts of modeled and observed average PM2.5 concentrations and the contributions of individual species concentrations (i.e.,  ,
, 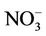 ,
, 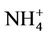 , total carbon (TC), and unspeciated PM2.5) to the total PM2.5 concentration at the STN sites in both winter and summer. In winter, TC is the most abundant (33.2%) PM2.5 component, followed by
, total carbon (TC), and unspeciated PM2.5) to the total PM2.5 concentration at the STN sites in both winter and summer. In winter, TC is the most abundant (33.2%) PM2.5 component, followed by 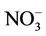 (21.2%),
(21.2%), 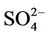 (19.8%), other unspeciated PM2.5 (14.3%) and
(19.8%), other unspeciated PM2.5 (14.3%) and 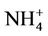 (11.6%) from the STN observations. However, CMAQ predicts the highest other unspeciated PM2.5 (36.7%), which contributes to the most to the PM2.5 overprediction in winter. The agreement between predicted and observed PM2.5 concentrations without accounting for the contribution of other unknown PM2.5 would be considerably better with a slight positive bias from CMAQ. In summer, both TC (34.2%) and
(11.6%) from the STN observations. However, CMAQ predicts the highest other unspeciated PM2.5 (36.7%), which contributes to the most to the PM2.5 overprediction in winter. The agreement between predicted and observed PM2.5 concentrations without accounting for the contribution of other unknown PM2.5 would be considerably better with a slight positive bias from CMAQ. In summer, both TC (34.2%) and 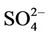 (29.9%) are the dominant PM2.5 component, following by other PM2.5 (21.2%),
(29.9%) are the dominant PM2.5 component, following by other PM2.5 (21.2%), 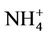 (9.9%), and
(9.9%), and 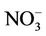 (4.8%) from the STN observations. CMAQ predicts concentrations of
(4.8%) from the STN observations. CMAQ predicts concentrations of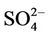 ,
, 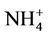 , and other PM2.5 quite well, but significantly underpredicts TC and
, and other PM2.5 quite well, but significantly underpredicts TC and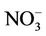 .
.
The reasons for this underprediction were discussed earlier in this section. Since the majority of the other unspeciated PM2.5 is primary aerosols, the model biases especially in winter are very likely due to errors in unspeciated primary emissions.
As shown in Tables 2 and 3, both NMBs and NMEs are relatively low over most sub-regions. Domain-wide NMBs range from –22.8% (summer) to 29% (winter) over the STN network and from –33.8% (summer) to 74.3% (winter) over the IMPORVE network over CONUS. NMEs are generally lower than 50% at both STN and IMPROVE sites throughout the year except for winter. There are currently no universally-accepted or EPA-recommended quantitative performance criteria for PM2.5. However, some specific model performance criteria have been recommended by other modeling studies [6,34]. Generally ±30% for model biases and 50% for model errors can be considered as satisfactory performance and the values below or beyond them should be considered as good and poor performance, respectively. The 2002 MP modeling platform demonstrates an overall good performance in predicting PM2.5 except for summer at the IMPROVE sites and winter at all sites. It also provides comparable or even better performance because of the state-of-science treatments in the model as well as more accurate model inputs.
3.3. Hazardous Air Pollutants at the Surface
3.3.1. Mercury
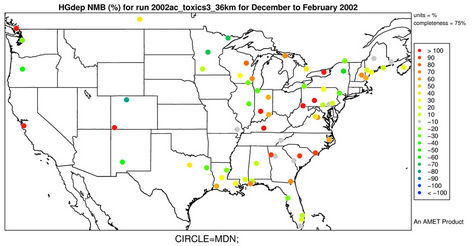
There were no routine networks existing with measurements of ambient Hg concentrations and dry depositions over the US back in 2002. MDN established by NADP was the only network that regularly monitored Hg wet deposition with most of its sites scattered throughout the remote areas in the US and Canada. The model evaluation will thus focus on the comparison of modeled Hg wet deposition against the MDN measurements, which is considered to be sufficient to provide a general concept of model performance for Hg [22,46]. Only sites where data are available more than half the weeks in a season are utilized for the seasonal performance evaluation in this study. Figures 5(a)-(b) display the spatial variation of NMBs for Hg wet deposition against data from the MDN network for winter and summer 2002. As shown, most MDN sites are clustered in the eastern and Midwest US. In winter, NMBs are much more scattered with NMBs from 10% to 50% occurring over the eastern US and some very high NMBs (>100%) occurring at several sites in both the western and eastern US. Some relatively small negative biases (NMBs of –20% to –10%) are observed in the eastern US and large negative biases (NMBs of about –60%) also occur in the Midwest. However, the overall trend for Hg wet deposition in CMAQ is
 (a)
(a)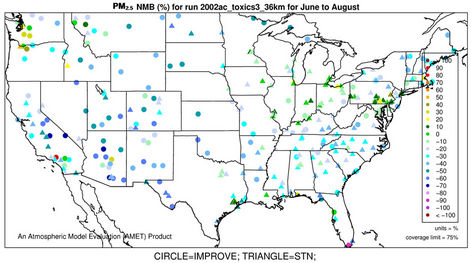 (b)
(b)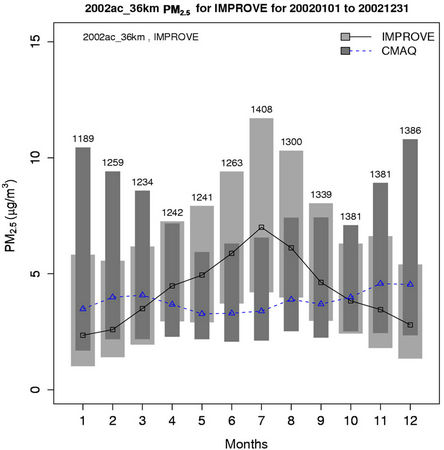 (c)
(c)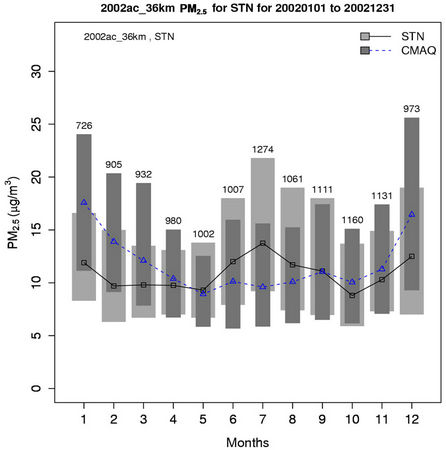 (d)
(d)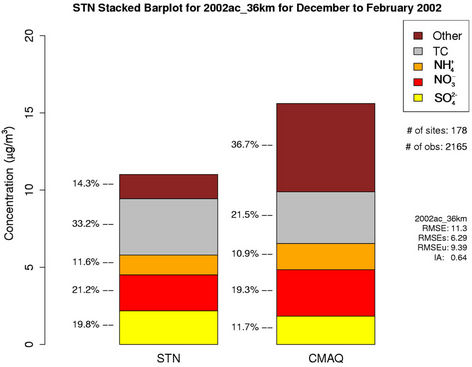 (e)
(e)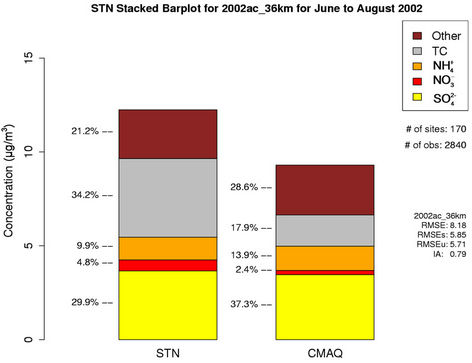 (f)
(f)
Figure 4. Comparison of the simulated and observed PM2.5 concentrations at the IMPROVE and STN sites in 2002. Spatial plots of NMBs from the 36-km simulation over CONUS for winter and summer in 2002 ((a) and (b)); Monthly box plot for total PM2.5 concentrations with 25% and 75% quartiles and median values over (c) the IMPORVE sites; and (d) the STN sites in 2002 (triangle and dark shading denote CMAQ, square and light shading denote observations, and the numbers above each bar indicate the number of simulated/observed data pairs for each month); Stacked bar charts of total mass concentrations of PM2.5 and its major components over the STN sits for (e) winter; and (f) summer in 2002. The percentages indicate the contribution of each species to the total PM2.5 mass.
 (a)
(a)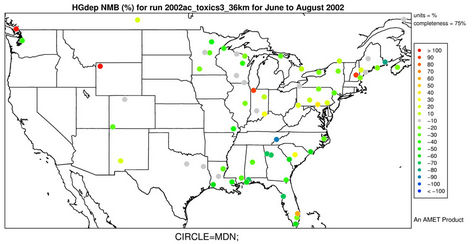 (b)
(b)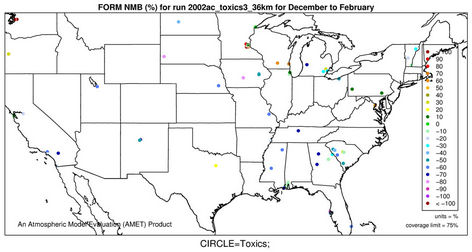 (c)
(c)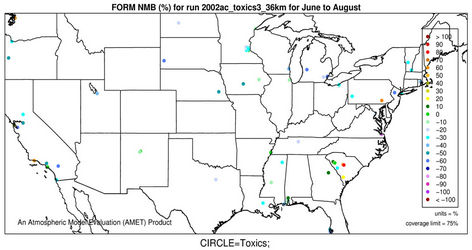 (d)
(d)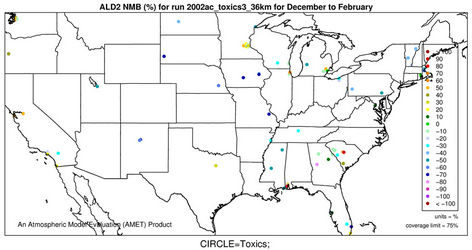 (e)
(e)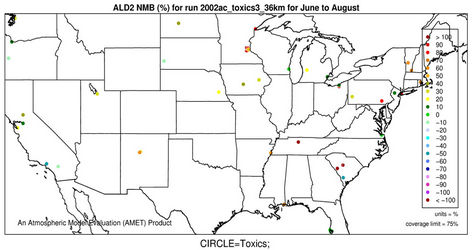 (f)
(f)
Figure 5. Spatial plots of NMBs for Hg wet deposition (top), formaldehyde mixing ratio (middle), and acetaldehyde mixing ratio (bottom) for winter (left panel) and summer (right panel) 2002.
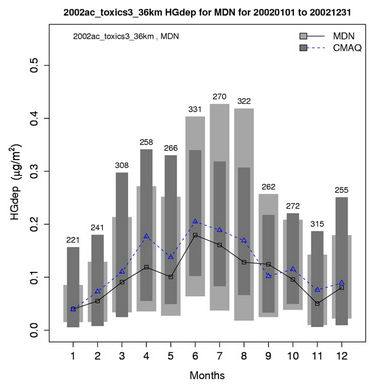
an overprediction in winter. In summer, the Hg wet deposition is generally underpredicted at more than 80% of the MDN sites, especially over the southern US with NMBs of –70% to –10%. For annual predictions of Hg wet deposition fluxes, more than half of data pairs are within the factor of 2 reference lines (figure not shown) with an R value of 0.45. As shown in Table 3 and Figure 6(a), CMAQ does reasonably well in simulating the monthly and seasonal Hg wet deposition over CONUS, with domain-wide seasonal NMBs of –28.2% to 28.9% and NMEs of 66.9% - 84.0%. The model performance is slightly better in spring and fall than in summer and winter.
The evaluation results of the present study are more in line with those from Gbor et al. [46] and Bullock et al. [18], and, show an improvement over those reported by Bullock and Brehme [22]. The Hg wet depositions in Bullock and Brehme [22] were significantly overpredicted for summer with an NMB of 60.2% and moderately overpredicted for spring with an NMB of 25.9%, compared to –28.2% and 12.4% in this study for summer and spring, respectively. The performance for precipitation is very similar between the two studies. The improvement of model performance is thus more likely related to the science updates in CMAQ-MP. These updates include: 1) The modification of the products and reaction rates for reactions of Hg0 with hydrogen peroxide (H2O2), O3, and hydroxyl radical (OH); 2) The explicit treatment of Hg0 between the air and various underlying surfaces (i.e., the dry deposition velocity is no longer zero as assumed in the previous Hg module); 3) The consideration of recycling or re-emitted Hg0 from the deposited Hg [47]. These updates are made to reflect the up-to-date science published in the peer-reviewed litera-
 (a)
(a)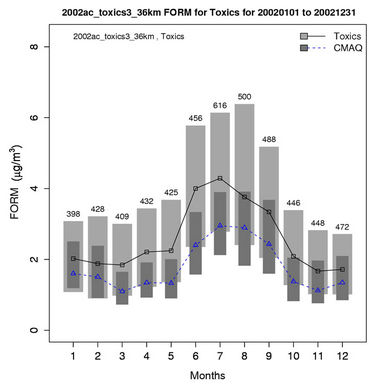 (b)
(b)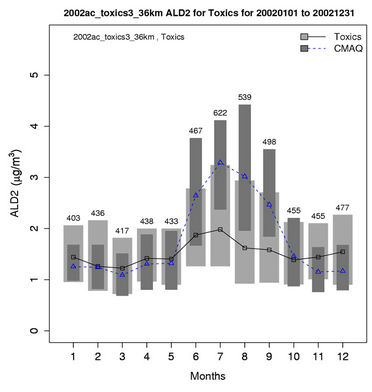 (c)
(c)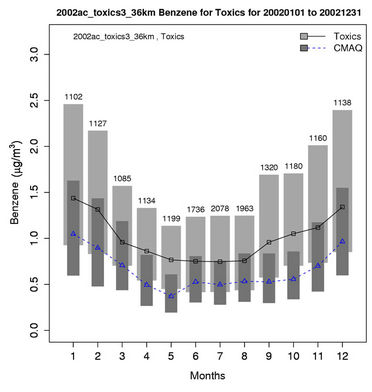 (d)
(d)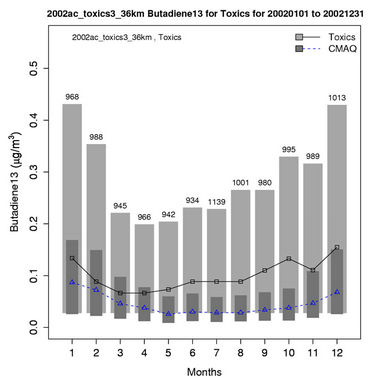 (e)
(e)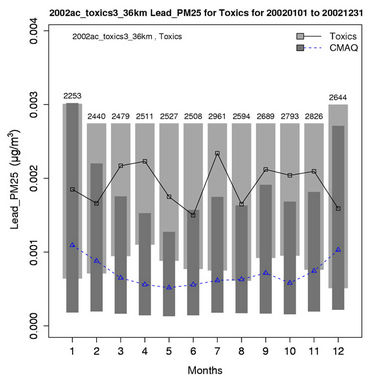 (f)
(f)
Figure 6. Monthly box plot for (a) Hg wet deposition, concentrations of (b) HCHO, (c) ALD2, (d) Benzene, (e) Butadiene13, and (f) Particulate lead with 25% and 75% quartiles and median values in 2002 (triangle and dark shading denote CMAQ, square and light shading denote observations, and numbers over each bar represent the numbers of observations).
ture [18]. Despite the model improvement, there still exist large discrepancies between CMAQ and MDN observations. The wet deposition of Hg is directly determined by the precipitation amount simulated by MM5 and the aqueous-phase concentrations of dissolved Hg(II) and absorbed PHg simulated by CMAQ. The model biases in Hg wet deposition predictions are thus determined by the errors in predicting those variables. However, as shown in the previous section, MM5 underpredicts precipitation in winter but overpredicts it in summer, which cannot help explain the overprediction of Hg wet deposition in winter and underprediction in summer. This means that the discrepancies between model and observations are more likely due to the predicted Hg(II) and PHg concentrations, which can be further attributed to the uncertainties in emission inputs, BCONs, and Hg chemistry treatments in the model. For example, as indicated by Gbor et al. [46], most modeling studies on Hg in the US have excluded a detailed treatment of Hg emissions from natural sources including vegetation, soil, and water. They estimated that the total natural mercury emission was 230 tons in 2002 based on their Hg natural emission model, while the anthropogenic emission was 126 tons based on the 1999 NEI. The total Hg emissions from the 2002 NEI are only 112 tons (the US EPA 2002 NEI booklet) which is predominated by anthropogenic emissions. Although the natural Hg emissions based on a modified Biogenic Emission Inventory System (BEIS) model [48] are also included in the 2002 MP modeling platform, the estimation may still be too low, especially, since Lin et al. [48] estimated that Hg emissions from vegetation ranged from 31 to 127 tons with the best estimation of 44 tons in 2001. This underestimation of natural Hg emissions is much more evident in the summer season during which meteorology, vegetation, and soil conditions favoring the generation of Hg emissions. A recent study by Pongprueksa et al. [17] showed that response of CMAQ to change of BCONs of Hg species, particularly Hg0, was strongly linear and they found an average of 1 ng·m–3 of Hg0 in BCONs could result in an increase of 0.81 ng·m–3 in the monthly average total Hg concentrations and 1270 ng·m–2 in the monthly average total deposition compared with clean condition of Hg0. This indicates that the uncertainties embedded in GEOSChem Hg simulation may contribute significantly to CMAQ predictions. Bullock et al. [18] also showed that CMAQ-Hg with BCONs from another CTM gave better performance than those from GEOS-Chem. It is known that the majority of Hg wet deposition are attributable to dissolved Hg(II), thus an accurate estimation of their concentrations is essential for accurate Hg wet deposition predictions. Gardfeldt and Jonsson [49] argued that Hg(II) reduction by HO2 in aqueous-phase chemistry, which is the most important chemical removal pathway for Hg(II)
in CMAQ, should not occur under ambient conditions. Lin et al. [16] and Pongprueksa et al. [17] tested this assumption by replacing the aqueous Hg(II)-HO2 reduction in CMAQ by two other different gas-phase reduction pathways (i.e., Hg(II) reduction by CO or photochemical-reduction of Hg(II)) separately. They found that those two new pathways generated more Hg wet deposition in summer and produced significantly better model agreement with the wet deposition measured by the MDN network. Finally, the missing reactions of Hg with other oxidants, such as bromines, in CMAQ may also contribute to the model uncertainties [50].
3.3.2. Other Air Toxics Compounds
1) There are two groups of gaseous HAPs species treated in CMAQ-MP. The first one, including HCHO, ALD2, 1,3-butadiene, and acrolein, can be generated or destroyed and then influence the concentrations of O3 and radicals via reactions. The second one, including the rest of species and serving as tracers, is only destroyed via chemical reactions with O3 and radicals but does not alter the concentrations of those oxidants. A modeling approach analogous to tracers in the gas-phase is used for the aerosol-phase HAPs such as diesel PM, lead, and chromium. The emissions of primary components of those species are tracked. Similar to EC, they are assumed to be chemically inert and only undergo microphysical and deposition processes, they therefore do not participate in cloud chemistry and have no effects on the rates of those processes (see CMAQ release note, http://www.cmascenter.org/help/model_docs/cmaq/4.6/HAZARDOUS_AIR_POLLUTANTS.txt). The approach taken above has its limitation. For example, Hutzell and Luecken [21] indicated that the hexavalent and trivalent states of chromium mass exchange might occur through chemistry within cloud droplets. However, the kinetics for that process is not well understood currently and will only be considered for future model development.
2) We therefore select 6 representative and also observationally available species including five gases and one aerosol species to assess the model performance of CMAQ-MP in predicting the HAPs. As shown in Figures 5(c)-(d), CMAQ-MP tends to underpredict HCHO at most NATTS sites in both winter and summer. Similar to the MDN sites, most NATTS sites are located in the eastern US and the model performance evaluation may not be representative for the western US. As shown in Figures 5(e)-(f), CMAQ-MP underpredicts ALD2 mixing ratios in winter while overpredicts them in summer at most sites. The NMBs of ALD2 in winter are similar to those of HCHO in spatial distributions but smaller in magnitude with a range of –70% to –30%. The NMBs in summer range from 20% to 60% with some extreme values occurring over Tennessee and South Carolina.
Figures 6(b)-(f) show the monthly concentrations between CMAQ and observations and Table 3 shows the seasonal statistics for HCHO, ALD2, benzene, 1,3-butadiene, acrolein, and particulate lead. The results show systematic underpredictions for most species except ALD2 throughout the year. No standard performance criteria are recommended by the US EPA and literature for HAPs modeling. Based on performance criteria used for O3 evaluation, the concentrations of HCHO and benzene are moderately-to-significantly underpredicted with NMBs of –53.1% (spring) to –34.4% (winter) for HCHO and –54.7% (fall) to –42.4% (summer) for benzene. That of ALD2 performs much better, with NMBs of –11.8% (fall) to 21.9% (summer). Based on performance criteria used for PM2.5 evaluation, the concentrations of particulate lead are also significantly underpredicted with NMBs of –40.1% (winter) to –59.6% (summer). Higher NMBs (generally –90% to –75%) and NMEs (>85%) occur for 1,3-butadiene and acrolein. The model performance for all species except for 1,3-butadiene in this study is consistent with or better than that reported by Luecken et al. [23]. For example, they reported NMBs of –52.0% and –39.0% for HCHO, –59.1% and –14.7% for ALD2, –39.1% and –69.8% for benzene, and –56.4% and –55.9% for 1,3-butadiene, for winter and summer, respectively. The larger underpredictions in the concentrations of 1,3-Butadiene are likely because that the CB05 mechanism used in this study generates more oxidants than SAPRC99 used by Luecken et al. [23] and includes additional chloride radicals. These additional oxidants and radicals will destroy more 1,3-Butadiene and result in smaller concentrations.
3) Overall, the model performance for HAPs is not as good as that for CAPs. Several factors may contribute to large model biases (mostly underpredictions) for HAPs. First, the grid resolution used in this study may be too coarse to resolve the sub-grid phenomena (such as urban canopies and sub-grid plumes) frequently associated with many HAP species as reported by other studies [51-53]. For example, Logue et al. [53] reported that most of air toxics compounds measured around Pittsburgh areas were characterized by short periods of elevated concentrations or plume events. Some local sources of emissions (e.g., HCHO) and the highly-reactive precursors (e.g., 1,3-butadiene with only a few hours of lifetime) may impact the monitors but not be captured in the grid average model predictions [23]. Ching et al. [51] also found that CMAQ predictions of air toxics are generally better (i.e., with higher values) at a horizontal grid resolution of 4-km than at 36-km. Second, errors in emission estimations of HAPs may contribute significantly to model biases, especially for those chemically nonreactive species (e.g., benzene and various metal particles). As indicated by Hutzell and Luecken [21], the uncertainties associated with HAPs emissions in the 2002 NEI are generally larger than those for CAPs. Note that most of HAPs emissions in the 2002 NEI are derived from Toxics Release Inventory (TRI). De Marchi and Hamilton [54] reported that the TRI underestimates lead emissions by as much as 50% and suggested that it may underestimate most other metal HAPs emissions since they normally share similar sources of emissions. Luecken et al. [23] also believed that the underestimation of precursor emissions (e.g., isoprene) may contribute to the negative biases for HCHO and ALD2 in CMAQ. Third, the assumption in chemical mechanism and aerosol module for HAPs in the current version of CMAQ-MP as described earlier in this section may play a role in the model underpredictions. In CB05, the rate of decay for most air toxic tracers is affected by OH and NO3 and it is difficult to determine how well CB05 reproduces their concentrations due to the lack of observations. In particular, CMAQ-MP performs poorly for those short-live and highly active HAPs (e.g., 1,3-butadiene and acrolein), further investigation of the reactions associated with those species is warranted. Finally, the errors from measurements such as sample handling, accuracy of analytical standards, and a lack of site density may also contribute to the model biases, but the impacts of these factors are believed to be smaller as compared with other reasons [55].
3.4. Column Variables
3.4.1. Column Mass of Gases
The model performance of the 2002 MP modeling platform above surface is further examined by evaluating seasonal CMAQ column predictions against available satellite measurements. The satellite measurements can provide substantial additional information with more complete spatial coverage and can also represent better the scale characteristics of model outputs that are averaged over a grid cell. The satellite dataset used in this study are all level-3 monthly-averaged data with various resolutions (i.e., 1 × 1.25 for TOMS/SBUV TOR, 1 × 1 for MOPITT CO column, 0.25 × 0.25 for GOME NO2 column, 0.5 × 0.5 for GOME HCHO column, and 1 × 1 for MODIS AOD). The satellite data with different resolutions are mapped to the Lambert conformal projection used in CMAQ using the bi-linear interpolation of the NCAR command language. The CMAQ model outputs are also processed and averaged at the same time of satellite overpasses in order to facilitate the comparison.
In terms of statistical performance (as shown in Table 4), CMAQ simulates TORs the best in fall and the worst in winter. Figure 7 shows the observed and simulated seasonal-mean TORs over the 36-km CONUS domain in 2002. The observed highest TORs occurred over the
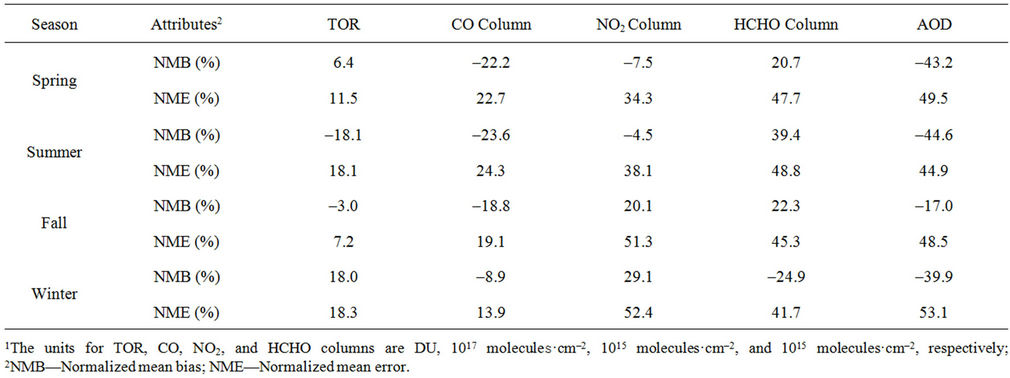
Table 4. Seasonal-mean performance statistics for column predictions over the 36-km CONUS domainin 20021.
North-eastern, Midwest, and Pacific coastal areas and the lowest TORs occurred over elevated terrains such as areas around Rocky Mountains in the US. CMAQ fails to capture the observed seasonal variations by TOMS/SBUV, i.e., simulated maximum and minimum TORs occur in spring and fall, respectively, but those observed ones occur in summer and winter, respectively. This discrepancy might be due to the BCONs for O3 used in CMAQ, especially in the upper layers that were provided by GEOS-Chem, which make the greatest contribution to TORs [8]. Other possible factors may include the uncertainties in both model treatments and the satellite retrieval algorithms. As pointed out by Tong and Mauzerall [56], the assumption of zero flux at the top layer of the model and the exclusion of the contribution of stratosphere-troposphere exchange (STE) of O3 limited the capability of CMAQ to reproduce O3 mixing ratios in the upper troposphere. Since TORs only represent about 10% of the total O3 columns in the atmosphere, they are very sensitive to errors in both retrievals of the total O3 column from TOMS and the stratospheric O3 column from SBUV (Fishman et al., 2003). One of the most important uncertainties in TOMS/SBUV data lies in the definition of tropopause. Stajner et al. [57] indicated that the differences of 1 - 2 km in tropopause altitudes can yield differences of 10% - 20% in tropospheric O3 columns (TOCs). They compared TOCs from four different definitions of tropopause. One of those tropopauses was determined from the lapse rate in the NECP/NCAR reanalysis, which is also used by TOMS/SBUV data. They found that the differences of TOCs from different tropopause definitions could be up to ~10 DU in summer and ~3 - 4 DU in winter over the US.
Figure 8 shows the observed and simulated seasonalmean tropospheric CO columns over the 36-km CONUS domain in 2002. Both MOPITT and CMAQ show high CO columns in winter and spring, especially over the CO source regions, notably the northeastern US, Great Lakes, and California. Both observed and simulated CO columns are also low over elevated altitude terrains (i.e., Rocky Mountains), which is similar to the TOR results. There are also observed elevated CO columns from MOPITT over the northeastern Pacific coastal region throughout the whole year and with maximum values in spring, which is attributed to the long-range transport of CO [15,58]. Nevertheless, CMAQ underpredicts CO columns throughout the whole year with NMBs ranging from –23.6% to –8.9% (see Table 4). CMAQ and MOPITT CO columns are better correlated in fall and summer with R values of 0.76 and 0.62, respectively, despite moderate underpredictions. Heald et al. [58] pointed out that the regional emissions, more specifically biomass burning emissions, could contribute significantly to elevated CO levels. The uncertainties in CO emissions used in this study could potentially be a major source of errors. The examination of seasonal CO emissions used in CMAQ shows that the CO emissions are the highest in winter, which accordingly contributes to the peak of simulated CO columns. On the other hand, the MOPITT CO observation (peaks in spring) shows that the CO emissions over CONUS, particularly in spring, might be too low. Other possible factors such as uncertainties in BCONs and MOPITT retrieval methods may also contribute to the discrepancies between model and satellite. For example, Heald et al. [58] indicated that the model bias in the vertical structure of CO (equivalent with BCONs or profile) could be an important source of model vs. MOPITT discrepancies. Emmons et al. [29] also showed positive biases (19%) of version 3 MOPITT retrievals over continents, as compared to oceans, and the bias may have been increasing over time.
Figure 9 shows the observed and simulated seasonalmean tropospheric NO2 columns over the 36-km CONUS domain in 2002. The spatial distribution and seasonal
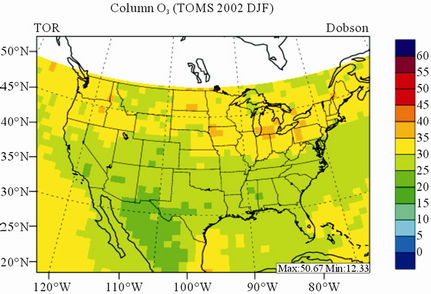
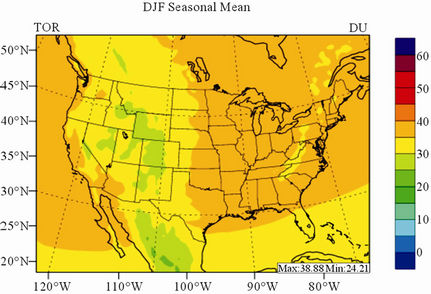
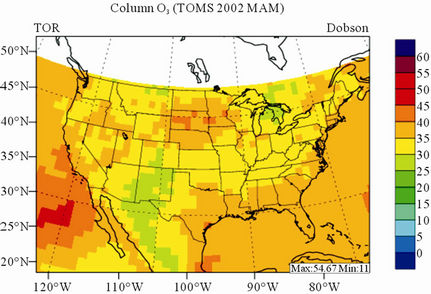
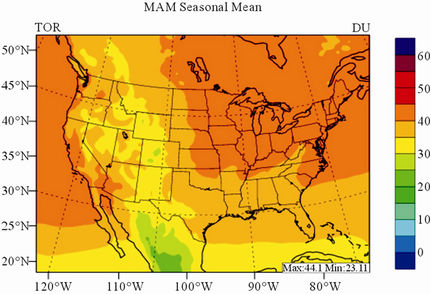
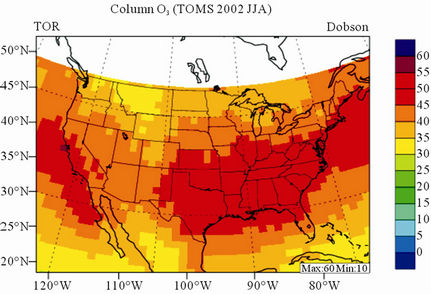
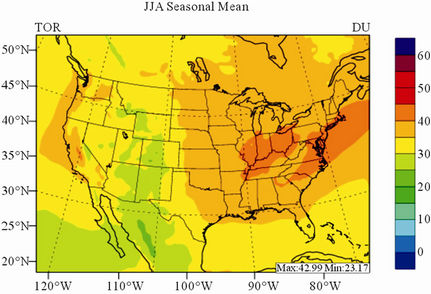
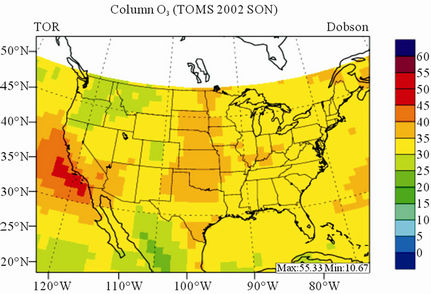
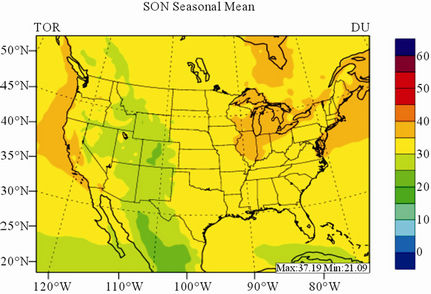
Figure 7. Spatial distributions of seasonal-mean TOR from TOMS/SBVU and CMAQ over CONUS in 2002. (a) TOMS/ SBUV 36-km; (b) CMAQ 36-km.
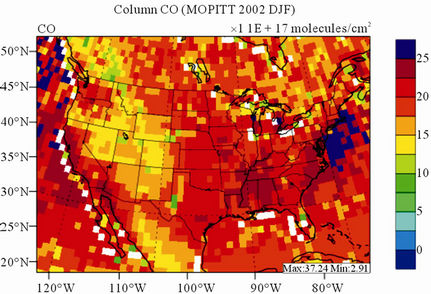
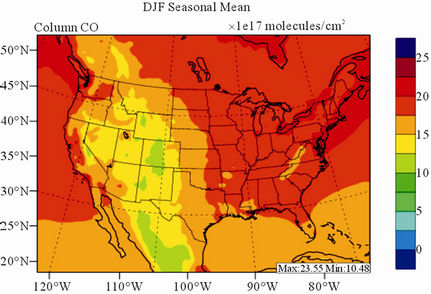
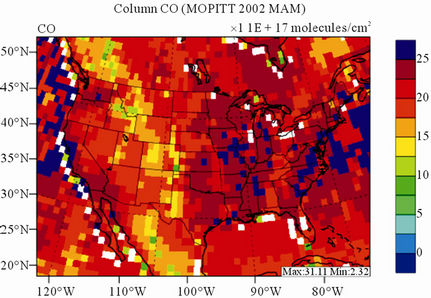
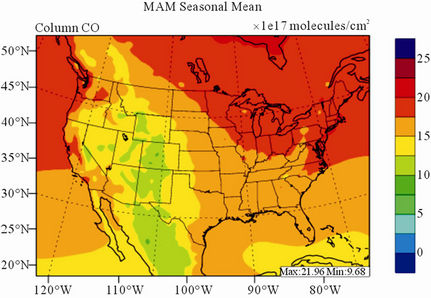
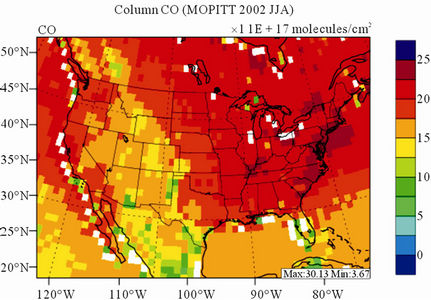
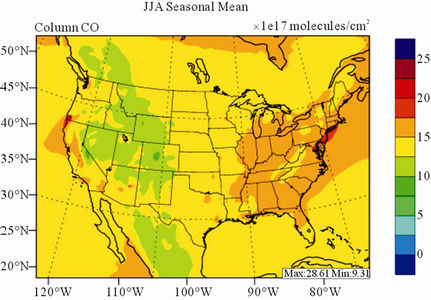
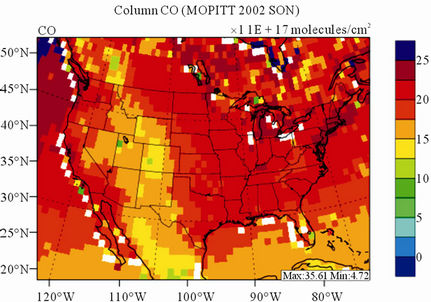
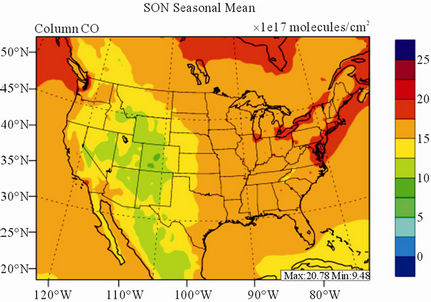
Figure 8. Spatial distributions of seasonal-mean tropospheric CO columns from MOPITT and CMAQ over CONUS in 2002. (a) MOPITT 36-km; (b) CMAQ 36-km.
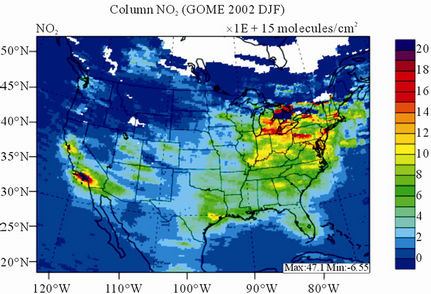
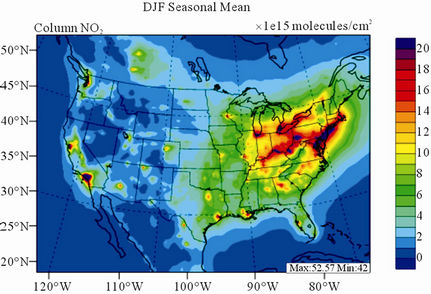
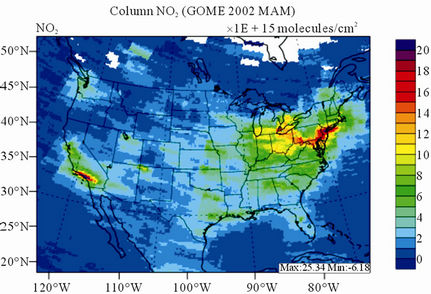
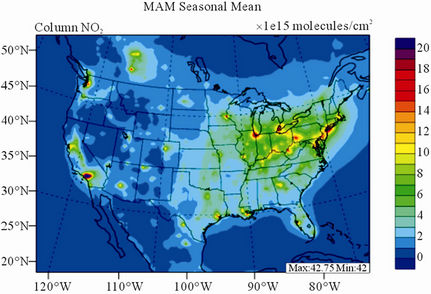

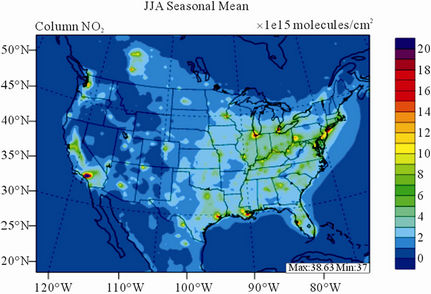
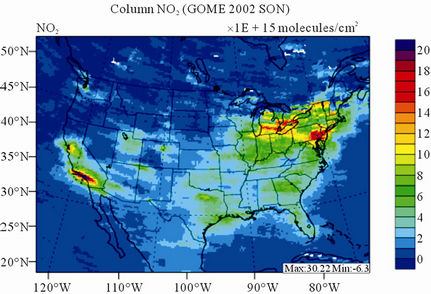
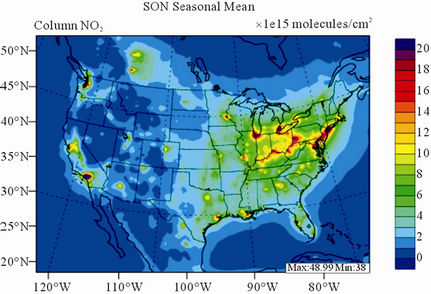
Figure 9. Spatial distributions of seasonal-mean tropospheric NO2 columns from GOME and CMAQ over CONUS in 2002. (a) MODIS 36-km; (b) CMAQ 36-km.
changes of GOME NO2 columns are generally well reproduced by CMAQ, with larger NO2 column amounts shown in winter than in other seasons and in the eastern US than in the western US. Both GOME and CMAQ show high NO2 columns over the industrialized and metropolitan areas throughout the whole year. Those areas are correlated very well with NOX emission source regions (figures not shown), which provides the rationale for many studies that used GOME NO2 columns as the constraints for emission inventories of NOX [59]. The NO2 columns over industrial source regions are the lowest in the summer due to a rapid loss by the reaction of NO2 with OH. The high winter NO2 columns are likely resulted from a combined effect of a decreased loss of NO2 via its reaction with OH and slightly increased emissions as compared to the summer [30]. CMAQ are well correlated with the GOME measurements throughout the whole year with R values of 0.74 to 0.85. The larger discrepancies (see Table 4) in fall and winter can be attributed to several factors including possible overestimation of NOX emissions in those seasons and uncertainties in model inputs, treatments, and satellite measurements and retrievals. Boersma et al. [60] and some other studies [59,61] showed that different NO2 column retrieval approaches may lead to ±5 × 1014 - 1 × 1015 molecules∙cm−2 for additive error and ±35% - 60% for relative error over polluted areas, particularly in winter. It is also worth noting that unlike TORs, the tropospheric NO2 columns are insensitive to the tropopause definition because the contributions to NO2 columns from the upper troposphere and lower stratosphere are negligibly small as compared to those from lower troposphere, especially over polluted regions [61]. This may partly explain the better performance of this study, since CMAQ typically gives more accurate predictions at lower altitudes [15]. Despite a small domainwide bias in spring and summer, the model performance in terms of both magnitude and spatial distribution can be potentially improved with more accurate emissions and model treatments. For example, there might also be missing sources of NOX emissions such as lightning emissions, which could be important in spring and summer. Estimations from other studies [62] show that the resultant NO2 columns produced by lightning can go up to (0.5 - 2.0) × 1015 molecules∙cm–2 over the southern US, the Gulf of Mexico, and western North Atlantic in May. As discussed in Zhang et al. [8], the plume-in-grid treatment in CMAQ for large US power plants can result in improved column NO2 performance in eastern US in summer.
Figure 10 shows the observed and simulated seasonalmean tropospheric HCHO columns over the 36-km CONUS domain in 2002. Both GOME and CMAQ show strong seasonal variations of HCHO columns with values of about a factor of two higher in summer than in winter.
GOME measurements show high HCHO columns over the southeastern US, particularly in summer, which is well captured by CMAQ despite some overpredictions. The spatial and temporal variability of HCHO columns over the southeastern US in the model correlates clearly with biogenic and biomass burning emissions (figures not shown here) and is believed to be largely driven by oxidation of biogenic VOCs (BVOCs) (e.g., isoprene and terpene) [63]. As shown in Table 4, CMAQ overpredicts HCHO columns in all seasons except for winter. This discrepancy could be in part due to the relatively high yield of HCHO from isoprene and terpene in the CB05 chemical mechanism, particularly in warm seasons and uncertainties in the emission inventory, particularly for biogenic emissions. More importantly, according to Stavrakou et al. [63], the GOME HCHO columns retrieved by Belgian Institute for Space Aeronomy (BIRA)/ Royal Netherlands Meteorological Institute (KNMI) used in this study are about 4 × 1015 molecules∙cm–2 (by 30%) lower in summer over the eastern US and about 2 × 1015 molecules∙cm–2 higher in winter over the US than another set of GOME columns retrieved by Harvard University [64], which used trace gas profiles from GEOSChem model and a different approach to calculate air mass factor. This indicates that the uncertainties in satellite retrievals may also be a contributor to the discrepancy between CMAQ and satellite HCHO columns.
3.4.2. AOD
Figure 11 shows observed and simulated seasonal-mean AODs over the 36-km CONUS domain in 2002. Both MODIS and CMAQ AODs show consistent seasonal variations with the highest values in summer and the lowest in winter. They, however, display quite different spatial distributions over the CONUS domain with the most noticeable differences in the western US. There is a persistently high level of AODs that is up to 0.6 in summer and spring over the northwestern US, western US, and northern Mexico observed by MODIS in 2002. In contrast, CMAQ AODs are much lower (by a factor of 3 - 4) over those regions with only up to about 0.15 in summer. In addition, CMAQ did not reproduce elevated AODs (with values of up to 0.3) over Pacific and off the Pacific coast observed by MODIS in spring as the results of trans-Pacific transport of Asian air pollutants and dust storms, potentially due to the errors in lateral BCONs. CMAQ does predict the enhanced AODs in summer over the eastern US observed by MODIS although they are lower by a factor of two than MODIS. Statistically, CMAQ underpredicts AODs for all seasons with NMBs of –44.6% to –17.0%. These findings are consistent with those of Zhang et al. [8]. Several possible reasons may explain the discrepancies between MODIS and CMAQ AODs. First, the lack of model treatment of mineral dust
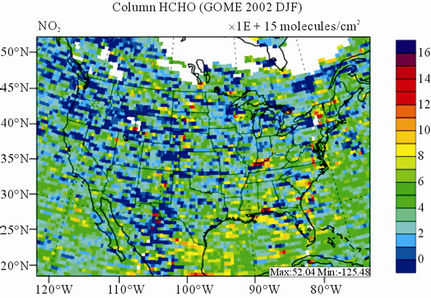
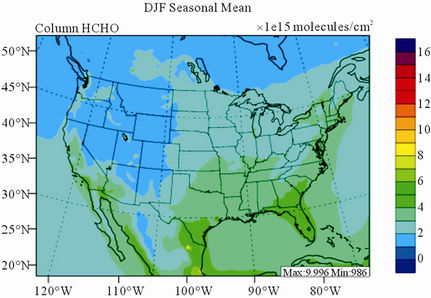

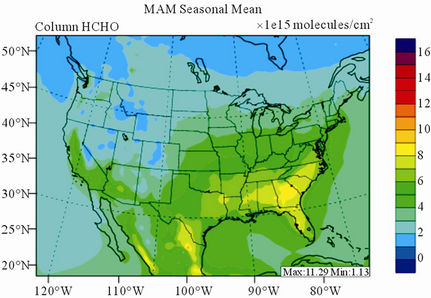
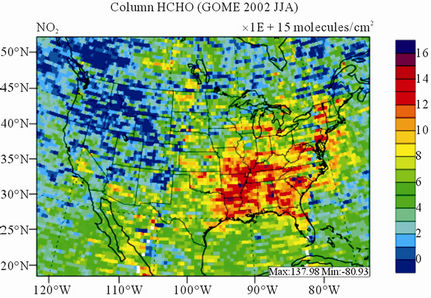
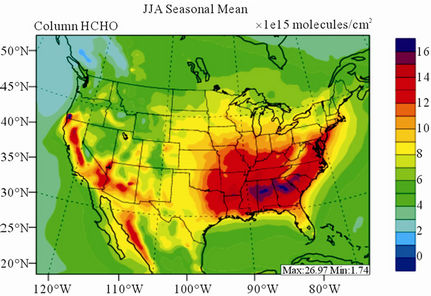
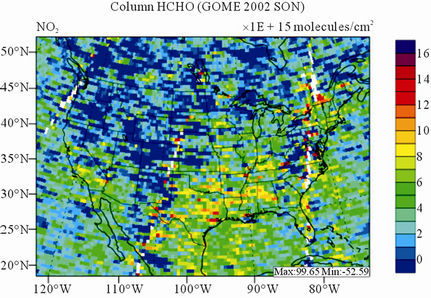
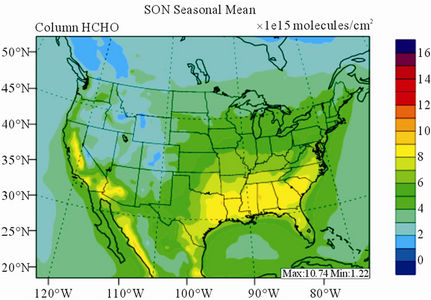
Figure 10. Spatial distributions of seasonal-mean tropospheric HCHO columns from GOME and CMAQ over CONUS in 2002. (a) GOME 36-km; (b) CMAQ 36-km.
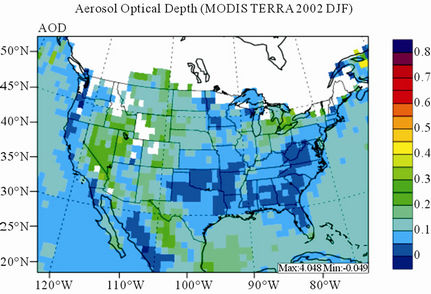
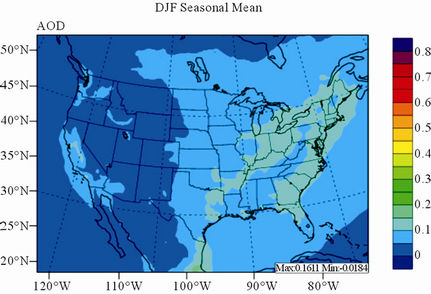
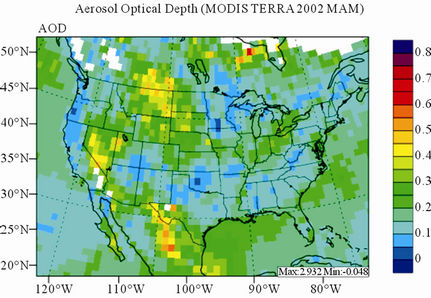
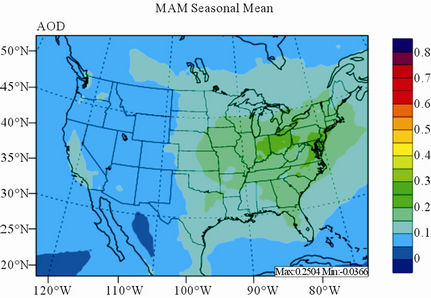
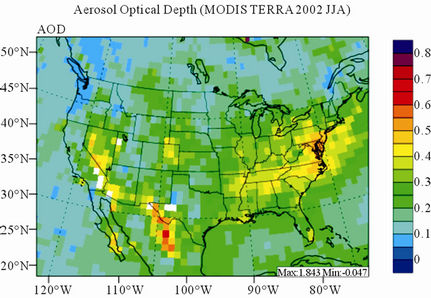
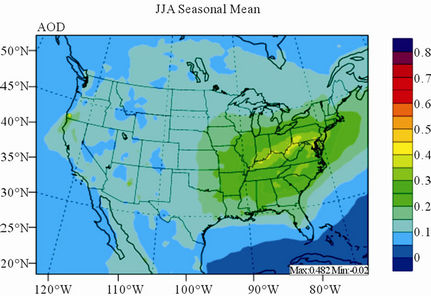
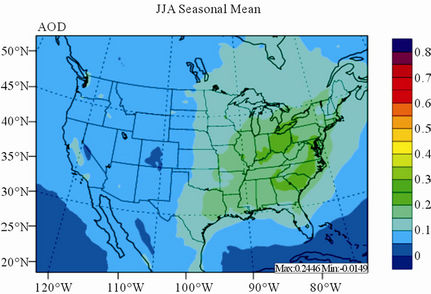
Figure 11. Spatial distributions of seasonal-mean AODs from MODIS and CMAQ over CONUS in 2002. (a) MODIS 36-km; (b) CMAQ 36-km.
in CMAQ may lead to the underprediction of AODs over the arid areas of the western US. Second, the inaccurate predictions of PM2.5 concentrations, particularly the underprediction of 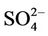 and OC (as shown in Section 3.2) over the southeastern US can contribute significantly to the underestimate of AOD in the eastern US. Third, there are uncertainties in BCONs of PM2.5 and its components. Kaufman et al. [65] derived the background AODs to be 0.052 at 500 nm over the Pacific Ocean by using Aerosol Robotic network (AERONET) data. However, the averaged CMAQ AODs over the Pacific Ocean in this work are only from 0.015 to 0.039 in different seasons. This reflects that the BCONs for PM2.5 species might be too low from GEOS-Chem. Fourth, uncertainties exist in the empirical equations and the associated parameters for the AOD calculation. For example, the equations used in this study do not explicitly consider the contribution of
and OC (as shown in Section 3.2) over the southeastern US can contribute significantly to the underestimate of AOD in the eastern US. Third, there are uncertainties in BCONs of PM2.5 and its components. Kaufman et al. [65] derived the background AODs to be 0.052 at 500 nm over the Pacific Ocean by using Aerosol Robotic network (AERONET) data. However, the averaged CMAQ AODs over the Pacific Ocean in this work are only from 0.015 to 0.039 in different seasons. This reflects that the BCONs for PM2.5 species might be too low from GEOS-Chem. Fourth, uncertainties exist in the empirical equations and the associated parameters for the AOD calculation. For example, the equations used in this study do not explicitly consider the contribution of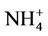 . They also completely exclude the other fine-mode inorganic aerosols and coarse-mode aerosols (e.g., soils and sea salts). A set of modified empirical equations are being developed and will be applied in the future work to improve the model-derived AODs (Wang and Zhang, Implementation of dust emission and heterogeneous chemistry into the Community Multiscale Air Quality Model and an initial application to April 2001 Asian dust storm episode, manuscript in review). Finally, similar to other satellite data, there are limitations and uncertainties in the MODIS data used in this work. For example, according to Remer et al. [32], the uncertainty of MODIS monthly AODs (denoted as τ) can be up to ±0.05 ± 0.15τ over land because of clouds and surface reflectance. More recently, Drury et al. [66] found that there are some errors in the surface reflectance estimates in MODIS operational AOD products used in this study, which can lead to high biases of AODs especially over the western and central US. Their results by using improved AOD retrieval algorithm showed more consistent pattern as our CMAQ AODs in summer.
. They also completely exclude the other fine-mode inorganic aerosols and coarse-mode aerosols (e.g., soils and sea salts). A set of modified empirical equations are being developed and will be applied in the future work to improve the model-derived AODs (Wang and Zhang, Implementation of dust emission and heterogeneous chemistry into the Community Multiscale Air Quality Model and an initial application to April 2001 Asian dust storm episode, manuscript in review). Finally, similar to other satellite data, there are limitations and uncertainties in the MODIS data used in this work. For example, according to Remer et al. [32], the uncertainty of MODIS monthly AODs (denoted as τ) can be up to ±0.05 ± 0.15τ over land because of clouds and surface reflectance. More recently, Drury et al. [66] found that there are some errors in the surface reflectance estimates in MODIS operational AOD products used in this study, which can lead to high biases of AODs especially over the western and central US. Their results by using improved AOD retrieval algorithm showed more consistent pattern as our CMAQ AODs in summer.
4. Process Analysis
Two process analysis approaches are embedded in CMAQ and they are integrated process rate (IPR) analysis and integrated reaction rate (IRR) analysis. IPRs assess the net effects of each atmospheric process simulated in CMAQ while IRRs calculate the rates of change of species concentration due to individual gas-phase reactions and track the chemical transformation pathways. Both IPRs and IRRs have been used to study various issues such as O3 chemistry and transport [9,67,68], regional and long range transport of air pollutants [9,15], and controlling processes/process budgets of different air pollutants [69,70]. In this section, the relative contributions of controlling processes to the formation and destruction of the selected CAPs and HAPs species are quantified through IPR analysis and the seasonal photochemical characteristics are examined through IRR analysis for January (representing winter) and July (representing summer) 2002.
4.1. IPR Analysis
The original outputs of IPRs are combined to represent several major processes including horizontal transport (sum of horizontal advection and diffusion), vertical transport (sum of vertical advection and diffusion), gasphase chemistry, aerosol processes (the net effect of gas-to-particle mass transfer and coagulation), emissions, dry deposition, and cloud processes (the net effect of cloud attenuation of photolytic rates, convective and nonconvective mixing and scavenging by clouds, aqueousphase chemistry, and wet deposition). The process contribution can be either positive or negative, indicating build-up or removal, respectively, of a species concentration due to a specific process.
Figure 12 depicts the process budgets for selected CAPs species including NOX, O3, 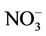 , and PM2.5 in PBL over different sub-regions. The process budgets for NOX in both months show very similar variation with major contribution coming from emissions and major removal by chemistry. The contribution from transport seems to be higher in winter, indicating the higher wind speed in cold season. The emission rates for NOX are the highest over Midwest and the lowest over the western US in both months. The removal rate of NOX due to gas-phase chemistry is comparable between winter and summer, due to different reasons. In winter, the removal of NOX is mainly caused by the strong titration of O3, but in summer, NOX is mainly removed by radicals. In contrast, the processes contributing to O3 show a strong seasonality, with much higher formation of O3 from gasphase chemistry over all sub-regions in summer than in winter. In summer, the highest chemistry production over Midwest is consistent with the highest precursor emissions (e.g., NOX). The vertical transport and dry deposition are two major removal processes for O3 over all sub-regions. As expected, the contribution from chemistry is much weaker in winter. The horizontal/vertical transport instead plays more important role in the O3 budgets. The high values of O3 build-up from vertical transport and removal from horizontal transport over the western US indicate the persistent period of high pressure system locating over the western US in January 2002 that transports more O3 from the free troposphere to the PBL and horizontally out of western US. The opposite vertical transport for O3 over the western US in summer indicates the low pressure system and downward turbulent transport. For
, and PM2.5 in PBL over different sub-regions. The process budgets for NOX in both months show very similar variation with major contribution coming from emissions and major removal by chemistry. The contribution from transport seems to be higher in winter, indicating the higher wind speed in cold season. The emission rates for NOX are the highest over Midwest and the lowest over the western US in both months. The removal rate of NOX due to gas-phase chemistry is comparable between winter and summer, due to different reasons. In winter, the removal of NOX is mainly caused by the strong titration of O3, but in summer, NOX is mainly removed by radicals. In contrast, the processes contributing to O3 show a strong seasonality, with much higher formation of O3 from gasphase chemistry over all sub-regions in summer than in winter. In summer, the highest chemistry production over Midwest is consistent with the highest precursor emissions (e.g., NOX). The vertical transport and dry deposition are two major removal processes for O3 over all sub-regions. As expected, the contribution from chemistry is much weaker in winter. The horizontal/vertical transport instead plays more important role in the O3 budgets. The high values of O3 build-up from vertical transport and removal from horizontal transport over the western US indicate the persistent period of high pressure system locating over the western US in January 2002 that transports more O3 from the free troposphere to the PBL and horizontally out of western US. The opposite vertical transport for O3 over the western US in summer indicates the low pressure system and downward turbulent transport. For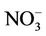 , the aerosol process is the
, the aerosol process is the
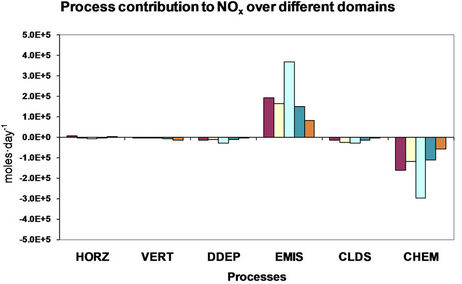 (a)
(a)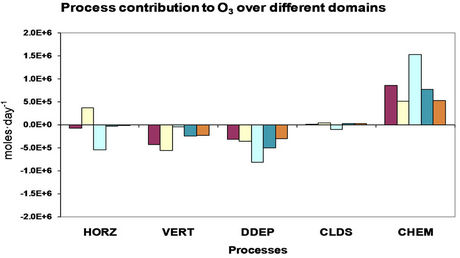 (b)
(b)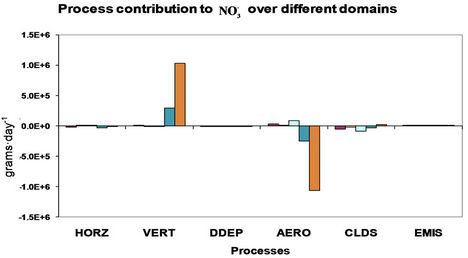 (c)
(c)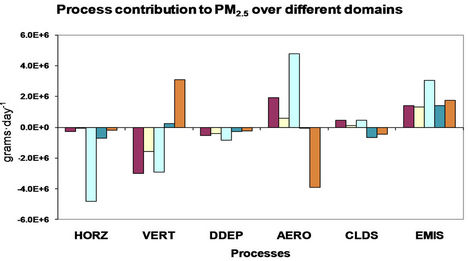 (d)
(d)
Figure 12. The monthly-mean contributions of individual processes to the concentrations of selected criteria air pollutants: (a) NOX, (b) O3, (c) , and (d) PM2.5 over different sub-regions of CONUS domain in January (left panel) and July (right panel) 2002. HORZ, VERT, DDEP, CLDS, CHEM, AERO, EMIS denote the processes of horizontal transport, vertical transport, dry deposition, cloud process, chemistry, aerosol chemistry, and emissions, respectively.
, and (d) PM2.5 over different sub-regions of CONUS domain in January (left panel) and July (right panel) 2002. HORZ, VERT, DDEP, CLDS, CHEM, AERO, EMIS denote the processes of horizontal transport, vertical transport, dry deposition, cloud process, chemistry, aerosol chemistry, and emissions, respectively.
dominant contributor over most sub-regions in winter. While in summer the higher temperature prevents HNO3 from condensing onto the existing particle surface to form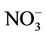 , although the HNO3 concentrations are higher. In particular, over the central and western US, aerosol process removes significant amount of
, although the HNO3 concentrations are higher. In particular, over the central and western US, aerosol process removes significant amount of 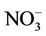 via evaporation and desorption. Besides aerosol process, horizontal/vertical transport and cloud processes over most sub-regions in winter and vertical transport over the central/western US in summer also play important roles in the
via evaporation and desorption. Besides aerosol process, horizontal/vertical transport and cloud processes over most sub-regions in winter and vertical transport over the central/western US in summer also play important roles in the 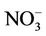 budget. The processes contributing to PM2.5 also show a strong seasonality. The overall emissions are comparable between two months with higher emission contributions over northeastern, southeastern, and Midwest US in winter and higher emission contributions over central and western US in summer. The removal of PM2.5 due to dry deposition is higher in summer than winter due to the general higher dry deposition velocity of aerosols over more vegetated areas. The changes of PM2.5 due to other processes are complicated over different subregions in both months. For example, the aerosol process tends to remove PM2.5 over the northeastern US and southeastern US, where ocean grid cells are included in the IPR calculation in winter because of a negative contribution to aerosol process of particulate-phase chloride (figure not shown) due to the fact that the reaction NaCl(s) + HNO3(g) ® NaNO3(s) + HCl(g) is favorable in winter. The negative budget of PM2.5 due to aerosol processes over the western US in summer is mainly due to the loss of
budget. The processes contributing to PM2.5 also show a strong seasonality. The overall emissions are comparable between two months with higher emission contributions over northeastern, southeastern, and Midwest US in winter and higher emission contributions over central and western US in summer. The removal of PM2.5 due to dry deposition is higher in summer than winter due to the general higher dry deposition velocity of aerosols over more vegetated areas. The changes of PM2.5 due to other processes are complicated over different subregions in both months. For example, the aerosol process tends to remove PM2.5 over the northeastern US and southeastern US, where ocean grid cells are included in the IPR calculation in winter because of a negative contribution to aerosol process of particulate-phase chloride (figure not shown) due to the fact that the reaction NaCl(s) + HNO3(g) ® NaNO3(s) + HCl(g) is favorable in winter. The negative budget of PM2.5 due to aerosol processes over the western US in summer is mainly due to the loss of 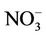 and SOA (figures not shown), both of which have relatively low precursor emissions and high removal rates due to gas-particle equilibrium favoring their volatility to the gas phase over that region. Similar to most of other species, horizontal/vertical transport are also important for PM2.5.
and SOA (figures not shown), both of which have relatively low precursor emissions and high removal rates due to gas-particle equilibrium favoring their volatility to the gas phase over that region. Similar to most of other species, horizontal/vertical transport are also important for PM2.5.
Figure 13 depicts the process budgets for selected HAPs species Hg(II), PHg, HCHO, and particulate lead in PBL over different sub-regions. The gas-phase chemistry, emission, and horizontal/vertical transport (except horizontal transport in the Midwest and western US and vertical transport in the central US) dominate the production of Hg(II) and dry deposition and cloud processes dominate the removal of Hg(II) over most sub-regions in both months, but the magnitude of IPR for each process has a strong seasonality. For example, the IPRs of chemistry are much higher in summer because of higher oxidant levels. The IPR of dry deposition is comparable to that of cloud processes in both months, indicating that the wet deposition may also contribute significantly to the removal of Hg(II). The signs of IPRs for horizontal/vertical transports are more diverse in winter than summer, indicating a much different wind field pattern in some regions in winter. The IPRs of emissions for PHg also indicate that the major sources of Hg are located in the northeastern and Midwest US. The IPRs for PHg also show a strong seasonality, especially for aerosol and cloud processes and to a lesser extent for horizontal and vertical transport. The aerosol process contributes to the formation of PHg and the cloud process contributes to the removal. The contributions from both processes are much higher in summer due to higher concentrations of oxidants, which lead to higher aqueousand particulate-phase oxidation of Hg. To a lesser extent, the remaining processes also play some roles in the PHg budgets. The IPRs for HCHO show a strong seasonality with much higher contributions in summer than winter. Both emission and chemistry contribute to the formation of HCHO. The IPRs for chemistry, however, are about 5 to 10 times higher over different sub-regions in summer than winter, resulting from much higher direct and precursor emissions and rates of formation from precursors due to a stronger oxidation capability. The vertical transport, dry deposition, and cloud processes are the major processes to remove HCHO from the atmosphere. Unlike other HAPs, the seasonality for particulate lead is not evident. Emission is the major or only source for the build-up of lead over almost all sub-regions, indicating that the uncertainties in emission inventory may contribute significantly to model biases as discussed in the previous section (see Table 3). Cloud processes act as a major removal process for particulate lead followed by horizontal transport, vertical transport, and dry deposition. The contribution from aerosol processes is zero due to the assumption of chemical inertia of lead in CMAQ. The vertical transport for particulate lead and PHg plays a different role, indicating that the long-range transport of PHg is more important than particulate lead.
4.2. IRR Analysis
CB05 used in this study include 219 reactions. The IRRs of those reactions are grouped into 43 products according to the reactions for radical initiation, propagation, production, and termination (see Table 1 from Zhang et al. [9] for most products). Figure 14 shows the monthlymean spatial distributions of photochemical indicators of surface layer 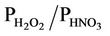 and column HCHO/NO2 predicted by CMAQ and column HCHO/NO2 observed by GOME satellite. As reported by Zhang et al. (2009b), several photochemical indicators have been proposed in the past in order to determine the NOX- or VOC-limited O3 chemistry in the regional modeling studies [71,72]. The ratio between the production rates of H2O2 and HNO3 (
and column HCHO/NO2 predicted by CMAQ and column HCHO/NO2 observed by GOME satellite. As reported by Zhang et al. (2009b), several photochemical indicators have been proposed in the past in order to determine the NOX- or VOC-limited O3 chemistry in the regional modeling studies [71,72]. The ratio between the production rates of H2O2 and HNO3 (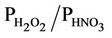 ) has been widely used in chemical indicator analysis due to its robust theoretical background [71].
) has been widely used in chemical indicator analysis due to its robust theoretical background [71].  less than 0.2 typically indicates a VOC-limited O3 chemistry and a larger value indicates a NOX-limited O3 chemistry [9]. As shown in Figure 14(a), during winter, most regions over US except for some areas over the western US have VOC-
less than 0.2 typically indicates a VOC-limited O3 chemistry and a larger value indicates a NOX-limited O3 chemistry [9]. As shown in Figure 14(a), during winter, most regions over US except for some areas over the western US have VOC-
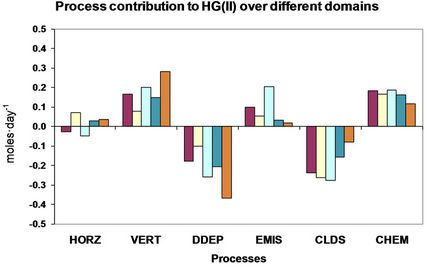 (a)
(a)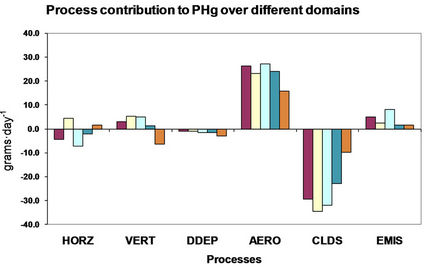 (b)
(b)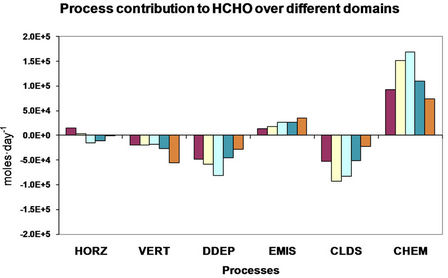 (c)
(c)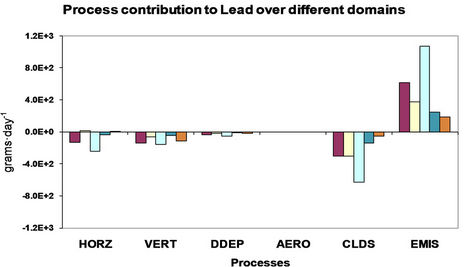 (d)
(d)
Figure 13. The monthly-mean contributions of individual processes to the concentrations of selected hazardous air pollutants: (a) Hg(II); (b) PHg; (c) HCHO; and (d) Particulate lead over different sub-regions of CONUS domain in January (left panel) and July (right panel) 2002. HORZ, VERT, DDEP, CLDS, CHEM, AERO, EMIS denote the processes of horizontal transport, vertical transport, dry deposition, cloud process, chemistry, aerosol chemistry, and emissions, respectively.
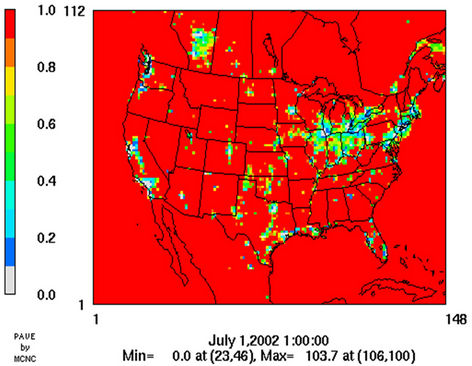 (a)
(a)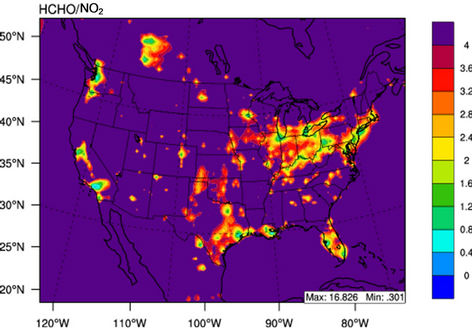 (b)
(b)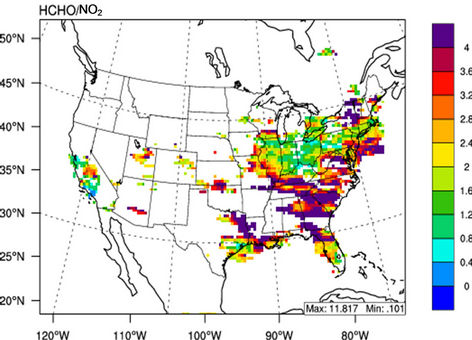 (c)
(c)
Figure 14. The monthly-mean spatial distributions of photochemical indicators of (a) surface layer 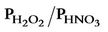 and (b) column HCHO/NO2 predicted by CMAQ and (c) column HCHO/NO2 observed by GOME satellite in January (left panel) and July (right panel) 2002. Blank in GOME observations represents no data available.
and (b) column HCHO/NO2 predicted by CMAQ and (c) column HCHO/NO2 observed by GOME satellite in January (left panel) and July (right panel) 2002. Blank in GOME observations represents no data available.
limited O3 chemistry due to high NOX and low BVOC emissions. By contrast, while the major cities and industry areas remain VOC-limited chemistry, all other areas (mostly rural and remote areas) change to NOX-limited O3 chemistry in summer. The results shown here are overall consistent with those reported by Zhang et al. [9] and Liu et al. [70]. In order to verify the robustness of 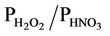 as a photochemical indicator, we also calculate the column ratio of HCHO/NO2, another indicator recommended by Martin et al. [72]. The rationale to use two column species to indicate the surface photochemistry is due to that the bulk of their columns are within the lower mixed layer over polluted regions and the columns are closely related to VOC and NOX emissions [72,73]. Another reason is that there are space-based observations of both tropospheric HCHO and NO2 column mass and the modeled ratio of HCHO/NO2 can be further examined by largescale and long term satellite observations. The transition value for column HCHO/NO2 originally used by Martin et al. [72] was 1, but Duncan et al. [73] suggested values of 1.2 - 2.2, above which O3 chemistry is VOC-limited. As shown in Figures 14(a)-(b), the spatial pattern of VOCvs. NOX-limited areas indicated by column HCHO/NO2 predicted by CMAQ is very similar to that of
as a photochemical indicator, we also calculate the column ratio of HCHO/NO2, another indicator recommended by Martin et al. [72]. The rationale to use two column species to indicate the surface photochemistry is due to that the bulk of their columns are within the lower mixed layer over polluted regions and the columns are closely related to VOC and NOX emissions [72,73]. Another reason is that there are space-based observations of both tropospheric HCHO and NO2 column mass and the modeled ratio of HCHO/NO2 can be further examined by largescale and long term satellite observations. The transition value for column HCHO/NO2 originally used by Martin et al. [72] was 1, but Duncan et al. [73] suggested values of 1.2 - 2.2, above which O3 chemistry is VOC-limited. As shown in Figures 14(a)-(b), the spatial pattern of VOCvs. NOX-limited areas indicated by column HCHO/NO2 predicted by CMAQ is very similar to that of 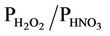 in both months, if a transition value of 1.6 is used for column HCHO/NO2. Comparing with satellite observations (Figure 14(c)), CMAQ demonstrates a promising accuracy in reproducing the spatial variation of column HCHO/NO2 in most areas, despite some discrepancies in some areas (e.g., in Texas and northern Mexico in January and in the Ohio valley in July), which can be attributed to the uncertainties in both model predicttions and satellite measurements. The above findings indicate that both
in both months, if a transition value of 1.6 is used for column HCHO/NO2. Comparing with satellite observations (Figure 14(c)), CMAQ demonstrates a promising accuracy in reproducing the spatial variation of column HCHO/NO2 in most areas, despite some discrepancies in some areas (e.g., in Texas and northern Mexico in January and in the Ohio valley in July), which can be attributed to the uncertainties in both model predicttions and satellite measurements. The above findings indicate that both 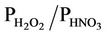 and column HCHO/NO2 are robust indicators for development and assessment of various precursor emission reduction strategies for O3 control.
and column HCHO/NO2 are robust indicators for development and assessment of various precursor emission reduction strategies for O3 control.
5. Summary and Conclusions
This study presents a comprehensive evaluation and analysis of several full year simulations over contiguous US domains using the US EPA’s the multiple-pollut-ant version of CMAQ v4.6 (i.e., the 2002 MP modeling platform). Model evaluation is performed by comparing simulated concentrations of O3, PM2.5, and its components, precursors O3 and PM2.5, and major air toxics as well as the Hg deposition with the measurements collected from ground-based monitoring networks and satellites. Our results show that CMAQ simulates well the spatial and seasonal variation of O3, especially during the O3 season and gives the best agreement with observed O3 mixing ratio range of 40 - 60 ppb. These results demonstrate a moderate to great improvement in O3 predictions compared to the previous studies for several reasons including the newest CB05 gas-phase chemistry mechanism with chloride related reactions, a new PBL scheme ACM2, and new emission inventories. Model performance for PM2.5 and its components is satisfactory or marginally-satisfactory. CMAQ predicts 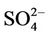 the best among all PM2.5 components, with a slight improvement compared with previous study which is likely attributed to updates in both convective cloud module and aerosol dry deposition module in CMAQ. The uncertainty associated with NH3 emissions is found to be indicative of the main reason for the model bias of
the best among all PM2.5 components, with a slight improvement compared with previous study which is likely attributed to updates in both convective cloud module and aerosol dry deposition module in CMAQ. The uncertainty associated with NH3 emissions is found to be indicative of the main reason for the model bias of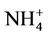 . The performance for
. The performance for 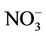 remains poor, despite some improvements in terms of statistics as compared to earlier studies. OC underpredictions are much worse than those of EC, particularly in summer, because of underpredictions of photochemically-produced SOA, in addition to uncertainties in the emissions of POA and SOA precursors in summer. CMAQ shows a satisfactory performance in predicting PM2.5 that is comparable to or even better than previous studies due to several model updates, although it overpredicts PM2.5 in winter mainly due to overpredictions in concentrations of other unknown PM2.5, and underpredicts it in summer mainly due to underpredictions in OC concentrations.
remains poor, despite some improvements in terms of statistics as compared to earlier studies. OC underpredictions are much worse than those of EC, particularly in summer, because of underpredictions of photochemically-produced SOA, in addition to uncertainties in the emissions of POA and SOA precursors in summer. CMAQ shows a satisfactory performance in predicting PM2.5 that is comparable to or even better than previous studies due to several model updates, although it overpredicts PM2.5 in winter mainly due to overpredictions in concentrations of other unknown PM2.5, and underpredicts it in summer mainly due to underpredictions in OC concentrations.
The overall model performance for HAPs is worse than CAPs due to several reasons. For example, the emission inventory for HAPs is not as accurate as that of CAPs, the model treatments for HAPs species are not as mature as those for CAPS, and there is a lack of routine measurements of HAPs. However, CMAQ does reasonably well in simulating seasonal Hg wet deposition, with consistent or even better performance as compared with previous studies because of several model updates. The model performance is slightly better in spring and fall than in summer and winter. The evaluation results for selected air toxics show a systematic underprediction for most species except for ALD2 throughout the year due to several reasons, including the incapability of the coarse grid resolution in resolving the high-level plume event, the underestimation of emissions for most of HAPs, and the simplified assumption of HAPs chemistry in current CMAQ-MP. The overall model performance in the 2002 MP modeling platform is fairly good for HCHO and ALD2, moderately good for benzene and particulate lead, and very poor for 1,3-butadiene and acrolein.
The spatial distribution and seasonal variations of GOME NO2 columns are generally well reproduced by CMAQ, with a good correlation throughout the year. Despite moderate underpredictions, CMAQ reasonably captures high MOPITT CO columns over source regions. Although relatively small NMBs for simulated TORs, CMAQ fails to capture the observed seasonal variations, likely due to uncertainties in the upper BCONs for O3 used in CMAQ. Moderate-to-significant overpredictions of HCHO columns from CMAQ occur in all seasons except for winter. CMAQ underpredicts MODIS AODs and fails to capture spatial distributions for all seasons. Several possible reasons for model biases in column predictions are identified. These include inaccurate seasonal allocation, underestimation of emissions, inaccurate BCONs in higher altitudes, lack of model treatments such as mineral dust or plume-in-grid process, as well as limitations and errors in satellite data retrievals.
The IPRs of the process analysis show that emissions are important sources for NOX, PM2.5, and many of HAPs such as Hg(II), PHg, and particulate lead over almost all the sub-regions in both seasons. Gas-phase chemistry is the dominant contributor to both HCHO and O3 especially in summer, however, it removes NOX significantly in both seasons. Aerosol processes contribute significantly to PHg formation and also play important but complex roles in the formation/removals of 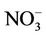 and PM2.5. Cloud processes remove most of HAPs significantly over all the sub-regions. The role of dry deposition is relatively more important for O3, HCHO, and Hg(II) especially in summer. Horizontal and vertical transport play important role for most of species, indicating the importance of accurate prediction of wind fields on air pollutants. The IPR results suggest that improving model treatments of those dominant processes may help to improve the model performance. The IRRs show a dominant NOX-limited chemistry in most areas but VOC-limited chemistry over urban and industry areas in summer and VOC-limited chemistry in winter over most of US, consistent with previous modeling studies and GOME satellite observations. The results indicate that integrated NOX/VOC emission controls should be considered over different regions in different seasons.
and PM2.5. Cloud processes remove most of HAPs significantly over all the sub-regions. The role of dry deposition is relatively more important for O3, HCHO, and Hg(II) especially in summer. Horizontal and vertical transport play important role for most of species, indicating the importance of accurate prediction of wind fields on air pollutants. The IPR results suggest that improving model treatments of those dominant processes may help to improve the model performance. The IRRs show a dominant NOX-limited chemistry in most areas but VOC-limited chemistry over urban and industry areas in summer and VOC-limited chemistry in winter over most of US, consistent with previous modeling studies and GOME satellite observations. The results indicate that integrated NOX/VOC emission controls should be considered over different regions in different seasons.
As illustrated in this study, the predictions of CAPs and HAPs from the 2002 MP 36-km and 12-km simulations are within the range or better than those reported in several recent EPA applications. This attests its scientific capability in assessing O3 and PM2.5 as well as air toxics for the purposes of the NAAQS Final Rule. The model evaluation also identifies several key areas for potential model improvements, thus providing guidance for sensitivity studies and further model development and improvement efforts and directions in the future.
6. Acknowledgements
This work was supported by the US EPA’s ICAP project, the National Research Initiative Competitive Grant No. 2008-35112-18758 from the USDA Cooperative State Research, Education, and Extension Service Air Quality Program, and the US EPA’s Science to Achieve Results (STAR) grant #R833863. Thanks are due to Sharon Phillips and Carey Jang, the US EPA, for some technical guidance and helpful discussions; Wyat Appel, the US EPA, for providing help on AMET analysis; Jack Fishman and his colleagues, NASA Langley Research Center, US, for providing the TOMS/SBUV and MOPITT CO satellite data; NASA MODIS Adaptive Processing System, for providing MODIS AOD data; TEMIS of the European Space Agency for providing GOME NO2 and HCHO data. Thanks are also due to George Pouliot of the US EPA for helpful discussions on uncertainties in emissions.
REFERENCES
- M. C. McCarthy, H. R. Hafner and S. A. Montzka, “Background Concentrations of 18 Air Toxics for North America,” Journal of the Air & Waste Management Association, Vol. 56, No. 1, 2006, pp. 3-11. doi:10.1080/10473289.2006.10464436
- R. Scheffe, B. Hubbell, T. Fox, V. Rao and W. Pennell, “The Rationale for a Multipollutant, Multimedia Air Quality Management Framework,” Environmental Managers, 2007, pp. 14-20.
- US EPA, “Air Quality Modeling Platform for the Ozone National Ambient Air Quality Standard Final Rule Regulatory Impact Analysis, Office of Air Quality Planning and Standards,” EPA, Research Triangle Park, EPA-454/ R-08-003, 2008.
- D. W. Byun and K. L. Schere, “Review of the Governing Equations, Computational Algorithms, and Other Components of the Models-3 Community Multi-Scale Air Quality (CMAQ) Modeling System,” Applied Mechanics Reviews, Vol. 59, No. 2, 2006, pp. 51-77. doi:10.1115/1.2128636
- Y. Zhang, B. Pun, S.-Y. Wu, K. Vijayaraghavan and C. Seigneur, “Application and Evaluation of Two Air Quality Models for Particulate Matter for a Southeastern US Episode,” Journal of the Air & Waste Management Association, Vol. 54, No. 12, 2004, pp. 1478-1493. doi:10.1080/10473289.2004.10471012
- Y. Zhang, P. Liu, B. Pun and C. Seigneur, “A Comprehensive Performance Evaluation of MM5-CMAQ for the Summer 1999 Southern Oxidants Study Episode—Part I: Evaluation Protocols, Databases, and Meteorological Predictions,” Atmospheric Environment, Vol. 40, No. 26, 2006, pp. 4825-4838. doi:10.1016/j.atmosenv.2005.12.043
- Y. Zhang, J.-P. Huang, D. K. Henze and J. H. Seinfeld, “Role of Isoprene in Secondary Organic Aerosol Formation on a Regional Scale,” Journal of Geophysical Research, Vol. 112, 2007, Article ID: D20207. doi:10.1029/2007JD008675
- Y. Zhang, K. Vijayaraghavan, X.-Y. Wen, H. E. Snell and M. Z. Jacobson, “Probing into Regional O3 and PM Pollution in the US: Part I: A 1-Year CMAQ Simulation and Evaluation Using Surface and Satellite Data,” Journal of Geophysical Research, Vol. 114, 2009, Article ID: D22 304. doi:10.1029/2009JD011898
- Y. Zhang, X.-Y. Wen, K. Wang, K. Vijayaraghavan and M. Z. Jacobson, “Probing into Regional O3 and PM Pollution in the US: Part II: An Examination of Formation Mechanisms through a Process Analysis Technique and Sensitivity Study,” Journal of Geophysical Research, Vol. 114, 2009, Article ID: D22305. doi:10.1029/2009JD011900
- B. Eder and S. Yu, “A Performance Evaluation of the 2004 Release of Models-3 CMAQ,” Atmospheric Environment, Vol. 40, No. 26, 2006, pp. 4811-4824. doi:10.1016/j.atmosenv.2005.08.045
- T. W. Tesche, R. Morris, G. Tonnesen, D. McNally, J. Boylan and P. Brewer, “CMAQ/CAMx Annual 2002 Performance Valuation over the Eastern US,” Atmospheric Environment, Vol. 40, No. 26, 2006, pp. 4906-4919. doi:10.1016/j.atmosenv.2005.08.046
- K. W. Appel, A. B. Gilliland, G. Sarwar and R. C. Gilliam, “Evaluation of the Community Multiscale Air Quality (CMAQ) Model Version 4.5: Sensitivities Impacting Model Performance: Part I—Ozone,” Atmospheric Environment, Vol. 41, No. 40, 2007, pp. 9603-9615. doi:10.1016/j.atmosenv.2007.08.044
- K. W. Appel, P. V. Bhave, A. B. Gilliland, G. Sarwar and S. J. Roselle, “Evaluation of Community Multiscale Air Quality (CMAQ) Model Version 4.5: Sensitivities Impacting Model Performance: Part II—Particulate Matter,” Atmospheric Environment, Vol. 42, No. 24, 2008, pp. 6057- 6066.
- S.-Y. Wu, S. Krishnan, Y. Zhang and V. Aneja, “Modeling Atmospheric Transport and Fate of Ammonia in North Carolina—Part I: Evaluation of Meteorological and Chemical Predictions,” Atmospheric Environment, Vol. 42, No. 14, 2008, pp. 3419-3436. doi:10.1016/j.atmosenv.2007.04.031
- K. Wang, Y. Zhang, C. Jang, S. Phillips and B. Wang, “Modeling Intercontinental Air Pollution Transport over the Trans-Pacific Region in 2001 Using Community Multiscale Air Quality Modeling System,” Journal of Geophysical Research, Vol. 114, 2009, Article ID: D04307. doi:10.1029/2008JD010807
- C. J. Lin, P. Pongprueksa, S. E. Lindberg, S. O. Pehkonen, D. Byun and C. Jang, “Scientific Uncertainties in Atmospheric Mercury Models I: Model Science Evaluation,” Atmospheric Environment, Vol. 40, No. 16, 2006, pp. 2911- 2928.
- P. Pongprueksa, C. J. Lin, S. E. Lindberg, C. Jang, T. Braverman, O. R. Bullock, T. C. Ho and H. Chu, “Scientific Uncertainties in Atmospheric Mercury Models III: Boundary and Initial Conditions, Model Grid Resolution, and Hg(II) Reduction Mechanism,” Atmospheric Environment, Vol. 42, No. 8, 2008, pp. 1828-1845. doi:10.1016/j.atmosenv.2007.11.020
- O. R. Bullock, et al., “An Analysis of Simulated Wet Deposition of Mercury from the North American Mercury Model Intercomparison Study,” Journal of Geophysical Research, Vol. 114, 2009, Article ID: D08301. doi:10.1029/2008JD011224
- L. A. Díaz-Robles, J. S. Fu, G. D. Reed and A. J. DeLucia, “Seasonal Distribution and Modeling of Diesel Particulate Matter in the Southeast US,” Environment International, Vol. 35, No. 6, 2009, pp. 956-964. doi:10.1016/j.envint.2009.04.005
- ENVIRON, “Development, Application, and Evaluation of an Advanced Photochemical Air Toxics Modeling System,” ENVIRON, CRC Project A-42-2, Novato, 2002.
- W. T. Hutzell and D. J. Luecken, “Fate and Transport of Emissions for Several Trace Metals over the United States,” Science of the Total Environment, Vol. 396, No. 2-3, 2008, pp. 164-179. doi:10.1016/j.scitotenv.2008.02.020
- O. R. Bullock and K. A. Brehme, “Atmospheric Mercury Simulation with CMAQ Model: Formulation Description and Analysis of Wet Deposition Results,” Atmospheric Environment, Vol. 36, No. 13, 2002, pp. 2135-2146. doi:10.1016/S1352-2310(02)00220-0
- D. J. Luecken, W. T. Hutzell and G. L. Gipson, “Development and Analysis of Air Quality Modeling Simulations for Hazardous Air Pollutants,” Atmospheric Environment, Vol. 40, No. 26, 2006, pp. 5087-5096. doi:10.1016/j.atmosenv.2005.12.044
- G. Yarwood, S. Rao, M. Yocke and G. Z. Whitten, “Updates to the Carbon Bond Mechanism: CB05,” Report to the US Environmental Protection Agency, RT-04-00675, 2005.
- S. Kemball-Cook, Y. Jia, C. Emery, R. Morris, Z. Wang and G. Tonnesen, “Annual 2002 MM5 Meteorological Modeling to Support Regional Haze Modeling of the Western United States,” Report for the Western Regional Air Partnership (WRAP), Denver, 2005.
- M. A. Witt, R. Baker and T. D. Jickells, “Atmospheric Trace Metals over the Atlantic and South Indian Oceans: Investigation of Metal Concentrations and Lead Isotope Ratios in Coastal and Remote Marine Aerosols,” Atmospheric Environment, Vol. 40, No. 28, 2006, pp. 5435-5451. doi:10.1016/j.atmosenv.2006.04.041
- US EPA, “Guidance on the Use of Models and other Analyses for Demonstrating Attainment of Air Quality Goals for Ozone, PM2.5, and Regional Haze,” The US Environmental Protection Agency, Research Triangle Park, EPA-454/B-07-002, 2007.
- C. Seigneur, K. Lohman and B. Pun, “Critical Review of Air Toxics Modeling Current Status and Key Issues,” CRC A-42-Phase I, Coordinating Research Council, Inc., Alpharetta, 2002.
- L. K. Emmons, D. P. Edwards, M. N. Deeter, J. C. Gille, T. Campos, P. Nedelec, P. Novelli and G. Sachse, “Measurements of Pollution in The Troposphere (MOPITT) Validation through 2006,” Atmospheric Chemistry and Physics, Vol. 9, No. 5, 2009, pp. 1795-1803. doi:10.5194/acp-9-1795-2009
- G. J. Velders, M. C. Granier, R. W. Portmann, K. Pfeilsticker, M. Wenig, T. Wagner, U. Platt, A. Richter and J. P. Burrows, “Global Tropospheric NO2 Column Distributions: Comparing the Three-Dimensional Model Calculations with GOME Measurements,” Journal of Geophysical Research, Vol. 106, No. D12, 2001, pp. 12643-12666. doi:10.1029/2000JD900762
- J. Fishman, A. E. Wozniak and J. K. Creilson, “Global Distribution of Tropospheric Ozone from Satellite Measurements Using the Empirically Corrected Tropospheric Ozone Residual Technique: Identification of the Regional Aspects of Air Pollution,” Atmospheric Chemistry and Physics, Vol. 3, No. 4, 2003, pp. 893-907. doi:10.5194/acp-3-893-2003
- L. A. Remer, et al., “The MODIS Aerosol Algorithm, Products, and Validation,” Journal of Atmospheric Sciences, Vol. 62, No. 4, 2005, pp. 947-973. doi:10.1175/JAS3385.1
- P. Dolwick, R. Gilliam, L. Reynolds and A. Huffman, “Regional and Local-Scale Evaluation of 2002 MM5 Meteorological Fields for Various Air Quality Modeling Applications,” The 6th Annual CMAS Conference, Chapel Hill, 1-3 October 2007.
- R. E. Morris, B. Koo, D. McNally, T. W. Tesche and G. Tonnesen, “Application of Multiple Models to Simulation Fine Particulate in the Southeastern US,” National RPO Modeling Meeting, Denver, 25-26 May 2004.
- R. C. Gilliam, C. Hogrefe and S. T. Rao, “New Methods for Evaluating Meteorological Models Used in Air Quality Applications,” Atmospheric Environment, Vol. 40, No. 26, 2006, pp. 5073-5086. doi:10.1016/j.atmosenv.2006.01.023
- S. C. Yu, R. Mathur, K. Schere, D. Kang, J. Pleim and T. L. Otte, “A Detailed Evaluation of the Eta-CMAQ Forecast Model Performance for O3, Its Related Precursors, and Meteorological Parameters during the 2004 ICARTT Study,” Journal of Geophysical Research, Vol. 112, 2007, Article ID: D12S14. doi:10.1029/2006JD007715
- D. J. Luecken, S. Phillips, G. Sarwar and C. Jang, “Effects of Using the CB05 vs. SAPRC99 vs. CB4 Chemical Mechanism on Model Predictions: Ozone and Gas-Phase Photochemical Precursor Concentrations,” Atmospheric Environment, Vol. 42, No. 23, 2008, pp. 5805-5820. doi:10.1016/j.atmosenv.2007.08.056
- S. Yu, R. Mathur, G. Sarwar, D. Kwang, D. Tong, G. Pouliot and J. Pleim, “Eta-CMAQ Air Quality Forecasts for O3 and Related Species Using Three Different Photochemical Mechanisms (CB4, CB05, SAPRC-99): Comparisons with Measurements during the 2004 ICARTT Study,” Atmospheric Chemistry and Physics, Vol. 10, No. 6, 2010, pp. 3001-3025.
- Y. Zhang, Y.-S. Chen, G. Sarwar and K. Schere, “Impact of Gas-Phase Mechanisms on WRF/Chem Predictions: Mechanism Implementation and Comparative Evaluation,” Journal of Geophysical Research, Vol. 117, 2011, Article ID: D01301. doi:10.1029/2011JD015775
- C. Luo, Y. Wang, S. Mueller and E. Knipping, “Evaluation of Sulfate Simulations Using CMAQ Version 4.6,” The 8th Annual CMAS Conference, Chapel Hill, 19-21 October 2009.
- R. E. Morris, D. E. McNally, T. W. Tesche, G. Tonnesen, J. W. Boylan and P. Brewer, “Preliminary Evaluation of the Community Multiscale Air Quality Model for 2002 over the Southeastern United States,” Journal of the Air & Waste Management Association, Vol. 55, No. 11, 2005, pp. 1694-1708. doi:10.1080/10473289.2005.10464765
- R. W. Pinder, P. J. Adams, S. N. Pandis and A. B. Gilliland, “Temporally Resolved Ammonia Emission Inventories: Current Estimates, Evaluation Tools, and Measurement Needs,” Journal of Geophysical Research, Vol. 111, 2006, Article ID: D16310. doi:10.1029/2005JD006603
- V. A. Karydis, A. P. Tsimpidi and S. N. Pandis, “Evaluation of a Three-Dimensional Chemical Transport Model (PMCAMx) in the Eastern United States for All Four Seasons,” Journal of Geophysical Research, Vol. 112, 2007, Article ID: D14211. doi:10.1029/2006JD007890
- C. Wiedinmyer, B. Quayle, C. Geron, A. Belote, D. McKenzie, X. Zhang, S. O’Neill and K. Klos Wynne, “Estimating Emissions from Fires in North America for Air Quality Modeling,” Atmospheric Environment, Vol. 40, No. 19, 2006, pp. 3419-3432. doi:10.1016/j.atmosenv.2006.02.010
- B. Roy, G. A. Pouliot, A. Gilliland, T. Pierce, S. Howard, P. V. Bhave and W. Benjey, “Refining Fire Emissions for Air Quality Modeling with Remotely Sensed Fire Counts: A Wildfire Case Study,” Atmospheric Environment, Vol. 41, No. 3, 2007, pp. 655-665. doi:10.1016/j.atmosenv.2006.08.037
- P. K. Gbor, D. Wen, F. Meng, F. Yang and J. J. Sloan, “Modeling of Mercury Emission, Transport and Deposition in North America,” Atmospheric Environment, Vol. 41, No. 6, 2007, pp. 1135-1149. doi:10.1016/j.atmosenv.2006.10.005
- J. O. Bash, “Description and Initial Simulation of a Dynamic Bidirectional Air Surface Exchange Model for Mercury in Community Multiscale Air Quality (CMAQ) Model,” Journal of Geophysical Research, Vol. 115, 2010, Article ID: D06305. doi:10.1029/2009JD012834
- C. J. Lin, S. E. Lindberg, T. C. Ho and C. Jang, “Development of a Processor in BEIS3 for Estimating Vegetative Mercury Emission in the Continental United States,” Atmospheric Environment, Vol. 39, No. 39, 2005, pp. 7529-7540.
- K. Gardfeldt and M. Jonsson, “Is Bimolecular Reduction of Hg(II) Complexes Possible in Aqueous Systems of Environmental Importance,” Journal of Physical Chemistry A, Vol. 107, No. 22, 2003, pp. 4478-4482. doi:10.1021/jp0275342
- D. Obrist, E. Tas, M. Peleg, V. Matveev, X. Fain, D. Asaf and M. Luria, “Bromine-Induced Oxidation of Mercury in the Mid-Latitude Atmosphere,” Nature Geoscience, Vol. 4, 2010, pp. 22-26. doi:10.1038/ngeo1018
- J. Ching, T. Pierce, T. Palma and W. Hutzell, “Application of Fine Scale Air Toxics Modeling with CMAQ and HAPEM5,” The 3rd Annual CMAS Conference, Chapel Hill, 18-20 October 2004.
- V. Isakov, J. S. Irwin and J. Ching, “Using CMAQ for Exposure Modeling and Characterizing the Subgrid Variability for Exposure Estimates,” Journal of Applied Meteorology, Vol. 46, No. 9, 2007, pp. 1354-1371. doi:10.1175/JAM2538.1
- J. M. Logue, K. E. Huff-Hartz, A. T. Lamb, N. M. Donahue and A. L. Robinson, “High Time-Resolved Measurements of Organic Air Toxics in Different Source Regimes,” Atmospheric Environment, Vol. 43, No. 39, 2009, pp. 6205-6217. doi:10.1016/j.atmosenv.2009.08.041
- S. De Marchi and J. T. Hamilton, “Assessing the Accuracy of Self Reported Data: An Evaluation of the Toxics Release Inventory,” Journal of Risk and Uncertainty, Vol. 32, No. 1, 2006, pp. 57-76. doi:10.1007/s10797-006-6666-3
- G. C. Pratt, Y. Chun, D. Bock, J. L. Adgate, G. Ramachandran, T. H. Stock, M. Morandi and K. Sexton, “Comparing Air Dispersion Model Predictions with Measured Concentrations of VOCs in Urban Communities,” Environmental Science and Technology, Vol. 38, No. 7, 2004, pp. 1949-1959. doi:10.1021/es030638l
- D. Q. Tong and D. L. Mauzerall, “Spatial Variability of Summertime Tropospheric Ozone over the Continental United States: Implications of an Evaluation of the CMAQ Model,” Atmospheric Environment, Vol. 40, No. 17, 2006, pp. 3041-3056. doi:10.1016/j.atmosenv.2005.11.058
- I. Stajner, et al., “Assimilated Ozone from EOS-Aura: Evaluation of the Tropopause Region and Tropospheric Columns,” Journal of Geophysical Research, Vol. 113, 2008, Article ID: D16S32. doi:10.1029/2007JD008863
- C. L. Heald, et al., “Asian Outflow and Trans-Pacific Transport of Carbon Monoxide and Ozone Pollution: An Integrated Satellite, Aircraft, and Model Perspective,” Journal of Geophysical Research, Vol. 108, 2003, pp. 4804-4816. doi:10.1029/2003JD003507
- R. V. Martin, D. J. Jacob, K. Chance, T. P. Kurosu, P. I. Palmer and M. J. Evans, “Global Inventory of Nitrogen Oxide Emissions Constrained by Space-Based Observations of NO2 Columns,” Journal of Geophysical Research, Vol. 108, No. D17, 2003, pp. 4537-4548. doi:10.1029/2003JD003453
- [61] K. F. Boersma, H. J. Eskes and E. J. Brinksma, “Error Analysis for Tropospheric NO2 Retrieval from Space,” Journal of Geophysical Research, Vol. 109, 2004, Article ID: D04311. doi:10.1029/2003JD003962
- [62] T. P. C. van Noije, et al., “Multi-Model Ensemble Simulations of Tropospheric NO2 Compared with GOME Retrievals for the Year 2000,” Atmospheric Chemistry and Physics, Vol. 6, No. 10, 2006, pp. 2943-2979. doi:10.5194/acp-6-2943-2006
- [63] Y. Choi, Y. Wang, T. Zeng, D. Cunnold, E.-S. Yang, R. Martin, K. Chance, V. Thouret and E. Edgerton, “Springtime Transitions of NO2, CO, and O3 over North America: Model Evaluation and Analysis,” Journal of Geophysical Research, Vol. 113, 2008, Article ID: D20311. doi:10.1029/2007JD009632
- [64] T. Stavrakou, J.-F. Müller, I. De Smedt, M. Van Roozendael, G. R. van der Werf, L. Giglio and A. Guenther, “Evaluating the Performance of Pyrogenic and Biogenic Emission Inventories against One Decade of Space-Based Formaldehyde Columns,” Journal of Physical Chemistry, Vol. 9, No. 3, 2009, pp. 1037-1060. doi:10.5194/acp-9-1037-2009
- [65] K. V. Chance, P. I. Palmer, R. J. D. Spurr, R. V. Martin, T. P. Kurosu and D. J. Jacob, “Satellite Observations of Formaldehyde over North America from GOME,” Geophysical Research Letters, Vol. 27, No. 21, 2000, pp. 3461- 3464. doi:10.1029/2000GL011857
- [66] Y. J. Kaufman, A. Smirnov, B. N. Holben and O. Dubovik, “Baseline Maritime Aerosol: Methodology to Derive the Optical Thickness and Scattering Properties,” Geophysical Research Letters, Vol. 28, No. 17, 2001, pp. 3251-3254. doi:10.1029/2001GL013312
- [67] E. Drury, D. J. Jacob, J. Wang, R. J. D. Spurr and K. Chance, “Improved Algorithm for MODIS Satellite Retrievals of Aerosol Optical Depths over Western North America,” Journal of Geophysical Research, Vol. 113, 2008, Article ID: D16204. doi:10.1029/2007JD009573
- [68] Y. Zhang, K. Vijayaraghavan and C. Seigneur, “Evaluation of Three Probing Techniques in a Three-Dimensional Air Quality Model,” Journal of Geophysical Research, Vol. 110, 2005, Article ID D02305. doi:10.1029/2004JD005248
- [69] X.-H. Liu, Y. Zhang, J. Xing, Q. Zhang, K. Wang, D. G. Streets, C. J. Jang, W.-X. Wang and J.-M. Hao, “Understanding of Regional Air Pollution over China Using CMAQ: Part II. Process Analysis and Ozone Sensitivity to Precursor Emissions,” Atmospheric Environment, Vol. 44, 2010, pp. 3719-3727. doi:10.1016/j.atmosenv.2010.03.036
- [70] S. Yu, R. Mathur, K. Schere, D. Kang and D. Tong, “A Study of the Ozone Formation by Ensemble Back Trajectory-Process Analysis Using the Eta-CMAQ Forecast Model over the Northeastern US during the 2004 ICARTT Period,” Atmospheric Environment, Vol. 43, No. 2, 2009, pp. 355-363. doi:10.1016/j.atmosenv.2008.09.079
- [71] P. Liu, Y. Zhang, S. Yu and K. Schere, “Use of a Process Analysis Tool for Diagnostic Study on Fine Particulate Matter Predictions in the US—Part II: Analyses and Sensitivity Simulations,” Atmospheric Pollution Research, Vol. 2, 2011, pp. 61-71. doi:10.5094/APR.2011.008
- [72] G. S. Tonnesen and R. L. Dennis, “Analysis of Radical Propagation Efficiency to Assess Ozone Sensitivity to Hydrocarbons and NOX 1. Local Indicators of Instantaneous Odd Oxygen Production Sensitivity,” Journal of Geophysical Research, Vol. 105, No. D7, 2000, pp. 9213- 9225. doi:10.1029/1999JD900371
- [73] R. V. Martin, A. M. Fiore and A. Van Donkelaar, “SpaceBased Diagnosis of Surface Ozone Sensitivity to Anthropogenic Emissions,” Geophysical Research Letters, Vol. 31, 2004, Article ID: L06120. doi:10.1029/2004GL019416
- [74] B. Duncan, Y. Yoshida, C. Retscher, K. Pickering and E. Celarier, “The Sensitivity of US Surface Ozone Formation to NOX and VOCs as Viewed from Space,” The 8th Annual CMAS Conference, Chapel Hill, 19-21 October 2009.
NOTES
*Corresponding author.